
Abstract
Regulated cell death is one major factor to ensure homoeostasis in multicellular organisms. For decades, apoptosis was considered as the sole form of regulated cell death, whereas necrosis was believed to be accidental and unregulated. Due to this view, research on necrosis was somewhat neglected, especially in the field of anti-cancer treatment. However, new interest in necrosis has been sparked by the recent discovery of different forms of necrosis that show indeed regulated pathways. More and more studies now address the molecular pathways of regulated necrosis and its connections within the cellular signaling networks. Necroptosis, a subform of regulated necrosis, has so far hardly been focused on with regard to a future treatment of cancer patients and may emerge as a novel and effective approach to eliminate tumor cells. However, and similar to apoptosis, tumor cells can develop resistances against necroptosis to ensure their own survival. In this context, new molecules that enhance necroptosis are currently being identified to overcome such resistances. This review discusses cancer and necroptosis as friends or foes, i.e. the options to exploit necroptosis in anti-cancer therapies (“foes”), but also potential limitations that may block or actually cause necroptosis to act in a protumoral manner (“friends”). The balance between these two possible roles will determine whether necroptosis can indeed be used as a promising tool for early diagnosis of tumors, prevention of metastasis and anti-cancer treatment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The precise balance between proliferation and cell death is critical for the survival of any multicellular organism [1]. This homoeostasis is affected by nearly every extrinsic and intrinsic stimulus and has to be maintained by multiple molecular mechanisms. Regulated cell death (RCD) represents one of these major mechanisms and is a critical element in the prevention of many human diseases, especially tumor formation. The first description of RCD dates back to 1964, when Richard Lockshin observed that intersegmental muscle cell death was triggered by hormones [2]. Later, this form of RCD was termed apoptosis and it became clear that apoptosis is a caspase-dependent type of RCD [3]. In contrast to apoptosis, which now represents the most well-studied type of RCD, necrosis was believed to be an accidental, non-regulated type of cell death that occurs upon cellular insults by physical stresses like unphysiologic temperature, pH and osmotic shocks [4]. This conception has however changed over the last years with the discovery that types of necrosis exist that are tightly regulated [5–7]. These types of RCD are summarized under the term “regulated necrosis” (RN) and are morphologically characterized by cellular swelling, cytoplasmic granulation and finally, cell membrane leakage with release of cellular components into the surrounding [8]. In the last years, RN has increasingly moved into the center of cell death research, and been in the focus of many studies. According to the various molecular pathways by which the different forms of RN can be executed, various forms of RN have been defined, e.g., necroptosis, parthanatos, oxytosis, ferroptosis, ETosis, NETosis, pyronecrosis and pyroptosis [9]. This review focuses on necroptosis, a form of RN that can be triggered, e.g., by the family of death receptors such as CD95 and the receptors for tumor necrosis factor (TNF; TNF-R1), TNF-related apoptosis inducing ligand (TRAIL, TRAIL-R1/2), TNF-related weak inducer of apoptosis (TWEAK; Fn14), as well as on the relevance of necroptosis for anti-cancer therapy.
Death receptor-induced necroptosis
Binding of a ligand to its respective death receptor can result in pro- as well as anti-apoptotic signaling (e.g., activation of NF-κB). Pro-apoptotic signaling is characterized by the formation of a death-inducing signaling complex (DISC), the activation of caspase-8 and the downstream executioner caspases-3 and -7. In the above scenarios, active caspase-8 suppresses necroptosis by cleavage of the kinases RIPK1 and RIPK3. In contrast, when the activity of caspase-8 is compromised (e.g., by caspase inhibitors like zVAD-fmk), necroptosis is initiated by formation of the “necrosome”, a complex that contains the proteins FADD, procaspase-8, RIPK1 and RIPK3 (Fig. 1). It has been shown that RIPK1 and RIPK3 are indispensable for necrosome formation. Subsequently, the mixed lineage kinase domain like protein (MLKL) is phosphorylated at threonine 357 and serine 358 by RIPK3, which is critical for further downstream events to execute necroptosis [10–12]. A more detailed view on the molecular pathways of both apoptosis and necroptosis can be found in other reviews in this issue and elsewhere [12].

Molecular pathway of necroptosis triggered by the family of death receptors. Upon caspase-8 inhibition (e.g. by caspase inhibitors such as zVAD-fmk), necroptosis is initiated by formation of the necrosome and subsequent phosphorylation of mixed lineage kinase domain like protein (MLKL). Necroptosis can be inhibited by pharmacological substances like necrostatin 1s (NEC1s) and necrosulfonamide (NSA)
Cancer and necroptosis: foes? Relevance of necroptosis for anti-cancer therapy
In contrast to apoptosis, necroptosis is thought to represent a proinflammatory form of RCD [13]. Therefore, necroptosis-induced inflammatory responses might be beneficial for anti-cancer therapy, e.g., it has recently been demonstrated that polyinosinic-polycytidylic acid (polyI:C)-induced necroptosis can back up immune effector-mediated tumor elimination in vivo [14]. Likewise, it has been shown that necroptotic cancer cells release interleukin-1α, which activates dendritic cells to produce IL-12, a cytokine critical for anti-tumor responses [15]. Importantly, both interleukin-1α release as well as activation of dendritic cells were strictly dependent on RIPK3 expression in the tumor cells. With regard to inflammation, a key feature of necroptotic cells is the release of damage-associated molecular patterns (DAMPs) which can activate both innate and adaptive immunity (reviewed in [16–18]). Especially those DAMPs might have a beneficial effect on anti-cancer therapy once immunogenic necroptosis is induced [19]. Notably, Yatim and co-workers have recently reported that an efficient induction of antitumoral immunity requires robust cross-priming of CD8-positive effector T cells by dendritic cells, and that this cross-priming critically depends on the release of inflammatory DAMPs by necroptotic cells as well as on a simultaneous inflammatory activation of NF-κB signaling in the dying cells [20].
Recently, a multitude of various compounds has been described to induce necroptosis especially in order to eliminate tumor cells [21]. Several drugs that are used for induction of apoptosis in cancer cells also have the potential to induce necroptosis, e.g., staurosporine [22] or cisplatin [23, 24]. Since caspase-8 is a key factor for necroptotic signaling by controlling RIPK1-RIPK3 cleavage [25, 26], these normally pro-apoptotic drugs could be employed under caspase-compromised conditions by chemical application of synthetic broad-range caspase inhibitors (i.e., zVAD-fmk or caspase-inhibiting drugs being already in clinical trials, i.e., IDN-6556 (Emricasan, Conatus Pharmaceuticals), IDN-5370 (CGP 82630, Idun Pharmaceuticals), IDN-7866 (CGP 82630, IDUN/Novartis) [27]). Another strategy to induce caspase-independent cell death may be the use of those caspase inhibitors in combination with antagonists of inhibitor of apoptosis (IAP) proteins [i.e., Smac mimetics that are already in clinical use such as AT-406 (Ascenta Therapeutics) or TL32711 (Tetralogic Pharmaceuticals)] [28]. It was shown that the IAP antagonist/Smac mimetic LWB242 (Novartis Pharma) was able to sensitize colon cancer cells to irinotecan-induced cell death through activation of necroptotic pathways [29]. Further, Smac mimetics were used in combination with TNF to trigger apoptosis in HT-29 cells, which could be switched to RIPK3-dependent necroptosis upon caspase inhibition [30]. Under those conditions, cancer cell-based drug resistance to particular anti-apoptotic drugs might be bypassed by activating alternative necroptotic pathways as a combinatory therapy with Smac mimetics. A similar synergistic effect to overcome apoptosis resistance of acute myeloid leukemia cells by induction of necroptosis was shown for Smac mimetics in combination with the demethylating agents 5-azacytidine or 5-aza-2′-deoxycytidine [31]. Moreover, drug-resistant cancer cells can be re-sensitized to necroptotic cell death by various inhibitors alone or in combination with chemotherapeutic drugs. For example, inhibition of glycogen synthase kinase-3 alpha (GSK3A) or beta (GSK3B) and administration of DNA-damaging agents, i.e., 5-fluorouracil led in consequence to RIPK1-dependent necroptosis in p53-null colon cancer cells [32]. Independently, it was reported that in apoptosis-resistant leukemic cells bearing the BCR-ABL fusion protein, administration of the tyrosine kinase inhibitor imatinib (Glivec, Gleevec; Novartis) elicited caspase-independent necroptosis that was executed by the serine protease HtrA2/Omi [33]. Likewise, in apoptosis-resistant esophageal cancer cells, the chemotherapeutic drug cisplatin that is known to induce apoptosis is also able to trigger RIPK3-dependent necroptosis [34]. Moreover, it is suggested that RIPK3 might be used as a potential marker for predicting the sensitivity in apoptosis-resistant and advanced esophageal cancer towards cisplatin-induced necroptosis [24].
Interestingly, naturally occurring substances that are able to induce apoptosis such as shikonin, a naphthoquinone derivative used in Chinese traditional medicine and isolated from purple gromwell (Lithospermum purpurocaerula), and its analog alkannin reportedly induce necroptosis in specific cancer cells, i.e., glioma cells [35] or multiple myeloma [36]. Importantly, this type of necroptotic cell death seems to be cancer cell specific, as these compounds are inhibiting tumor-specific pyruvate kinase-M2 (PKM2) [37]. Further, natural compounds such as neoalbaconol (NA), extracted from ningyotake mushroom (Albatrellus confluens) inhibit cIAP1/2 in order to induce RIPK1–RIPK3-dependent necroptosis through generation of TNF and reactive oxygen species (ROS) [38, 39]. NA targets the 3-phosphoinositide-dependent protein kinase 1 pathway and thereby activates necroptosis besides autophagy and apoptosis through independent pathways. Notably, drugs used for treatment unrelated to induction of cell death possess anti-tumoral activity through activation of necroptotic pathways. For example, 5′-benzylglycinyl-amiloride (UCD38B), a derivate of amyloride, a potassium-sparing diuretic normally used for treatment of hypertension and congestive heart failure was shown to induce caspase-independent necroptosis in malignant glioma cells [40]. Similarly, a combination of metformin (Glucophage; Merck), an antidiabetic drug with multiple molecular mechanisms of action [i.e., on the mitochondrial respiratory chain (complex I), on AMP-activated protein kinase (AMPK), on cyclic adenosine monophosphate (cAMP), on protein kinase A (PKA) and on mitochondrial glycerophosphate dehydrogenase (GPD)] and simvastatin (Zocor; MSD), a low density lipoprotein (LDL)-lowering drug, induced RIPK1-RIPK3-dependent necroptosis in prostate cancer cells [41]. Likewise, the combination of the purine analog azathioprine and buthionine sulfoximine, an inductor of oxidative stress, triggered caspase-independent necrosis associated with glutathione (GSH) depletion and JNK activation in a broad range of hepatoma and colon carcinoma cell lines [42]. Also, the inhibition of proliferative factors such as NF-κB itself by treatment with BAY 11-7082 or by the presence of an undegradable form of IκBα was found to enhance necroptosis in glioblastoma during 5-aminolevulinic acid-based photodynamic therapy [43]. Recently, several additional compounds were reported to exert their antitumor activity via induction of caspase-independent forms of cell death, e.g., Polyphenon E®, a standardized green tea extract in prostate cancer [44], selenium compounds (selenite, selenodiglutathione and seleno-DL-cystine) in ovarian carcinoma [45], compound C (also called dorsomorphin) in glioma cells [46], BI2536 [an inhibitor of mitotic polo-like kinase 1 (Plk1)] in prostate cancer [47], and bacitracin-mediated inhibition of the protein disulfide isomerases PDIA4 and PDIA6 in lung carcinoma [48]. However, with regard to the question whether these latter compounds indeed elicit their anti-tumoral activities via necroptosis, further studies are required, since some data also point to a possible involvement of autophagy and/or apoptosis.
Another emerging strategy to induce necroptosis in cancer cells may involve the manipulation of intracellular sphingolipid levels. For example, the sphingolipid ceramide has been reported as a key downstream mediator of necroptosis [49, 50]. Demonstrating the potential of ceramide for the necroptotic elimination of tumor cells, inhibition of the enzyme acid sphingomyelinase (A-SMase), a ceramide-producing enzyme, provided significant protection from death receptor-induced necroptosis in multiple different tumor cell lines [24]. As another example, the sphingosine-1-phosphate analogue FTY720 (fingolimod, Gilenya; Novartis) was identified to induce RIPK1-dependent necroptosis in lung cancers [51]. Similarly, interaction with lipid formation and cholesterol homeostasis by administration of 24(S)-hydroxycholesterol (24S-OHC) can induce necroptosis under conditions of caspase inhibition in neuroblastoma cells [52].
The cytokine TRAIL is known to selectively induce apoptosis in cancer cells in vitro and in vivo while showing no toxicity in normal cells. Unfortunately, despite initial encouraging results of apoptosis-based treatment strategies in clinical phase I and phase II studies, neither TRAIL nor TRAIL receptor agonists elicited significant antitumoral effects in subsequent phase IIb studies [53]. Nevertheless, TRAIL may represent a promising new option for the necroptotic elimination of tumor cells. In a recent study, it was shown that a broad range of cancer cells can be eliminated through TRAIL-induced necroptosis [24]. Moreover, it was shown that homoharringtonine, an already FDA-approved anti-leukemia drug sensitizes tumor cells for TRAIL-induced necroptosis when caspases are inhibited [54], similarly as TRAIL receptor ligation can trigger necroptosis after exposing cancer cells to an acidic extracellular milieu [55].
Notably, necroptosis can also be triggered by inhibition of caspase-8 via several viral anti-apoptotic proteins, i.e., M45 from murine cytomegalovirus (CMV) and competitors of RIP homotypic interaction motif (RHIM)-containing proteins from herpes simplex virus (HSV) [56], CrmA from cowpox virus, p35 and p49 from baculoviruses [27] or via viral and bacterial RNA and DNA [16]. This has been previously employed in a strategy to eliminate cervical cancer cells by application of polyinosinic:polycytidylic acid (PolyIC), a synthetic analog of viral dsRNA, that triggered RIPK3-dependent necroptosis cervical cancer cells [15].
Although proteins involved in necroptosis are not reported to contribute to apoptosis-based chemotherapy [29], proteins that are involved in apoptosis may play significant role in the regulation of necroptosis. For example, MEF cells deficient for the pro-apoptotic proteins BAX and BAK or cells overexpressing Bcl-XL were protected from necroptosis [57]. Conversely, cells deficient for transforming growth factor β (TGF-β)-activated kinase 1 (TAK1) are dying through RIPK1-RIPK3-dependent necroptosis when treated with TNF [58]. Therefore, regulation of TAK1 activity might be important for the development of necroptosis-based cancer therapies. Further in death receptor-mediated necroptosis, the regulatory subunit of IκB kinase, NF-κB essential modifier (NEMO) appears to function downstream of RIPK1-RIPK3 [57]. Therefore, NEMO inhibits necroptosis through its binding to ubiquitinated RIPK1 and in consequence by restraining RIPK1 from engaging the necroptotic death pathway [59].
Of note, it has been reported that necroptosis can also be induced by nanoparticles, i.e. Nec-1 treatment significantly reduced gold nanoparticle-induced cell death in human A549 lung adenocarcinoma epithelial cells with low antioxidant capacity, while treatment with zVAD-fmk did not abolish cell death [60]. But also other metal-based nanoparticles such as ZnO and Ag have been reported to trigger necroptosis in cancer cells which mostly resulted in elevated levels of ROS (reviewed in [61]). However, additional studies are required to strengthen the concept of nanoparticle-induced necroptosis, e.g. employing more specific necroptosis inhibitors like Nec-1s and Western blots to validate the presence of phosphorylated MLKL in the dying cells.
Cancer and metastasis
Progression of cancers to the metastatic state is the most destructive step and responsible for almost all deaths of cancer patients [62]. The metastatic spreading of the tumor often leads to a massive reduction of the available therapeutic options. In this multistage process, malignant cells spread from the initial tumor into the body and affect distant organs [63]. Although there is a wide heterogeneity, the process of metastasis can be roughly divided into five basic steps: local invasion → intravasation → dissemination → extravasation → colonization [64]. In the final step, the metastatic cell proliferates to form new micro- and/or macrometastases with an estimated efficiency of about 0.01 % of circulating metastatic cells [65]. This relative low efficiency is connected to various factors such as loss of cell–cell-contacts, exposition to ROS, the recognition and destruction of metastatic cells by the immune system and the lack of metabolites (e.g. growth factors) at the new site of colonization. All these events can finally result in the destruction of the metastatic tumor cell by regulated cell death, including apoptosis and necroptosis. The functional roles of the major known apoptotic participants in this context have been extensively studied and are reviewed elsewhere [66]. Notably, it has been reported that pro-apoptotic cytokines such as CD95 ligand, TNF and TRAIL can enhance the migration and invasion of tumor cells via anti-apoptotic signaling cascades, thereby limiting the efficacy of apoptosis-based tumor therapies [53, 67–71]. So far, similar “metastasis-stimulating” effects have not been reported for necroptosis.
Necroptosis and metastasis
The role of necroptosis in metastasis is poorly investigated. To date, only few reports link this type of RCD with metastasis formation. It has been shown that the molecule shikonin massively reduced metastasis of osteosarcoma by induction of RIPK1- and RIPK3-dependent necroptosis [72]. The same effect of shikonin was also reported in C6 and U87 glioma cells, where oxidative stress additionally participated in the induction of RIPK1-dependent necroptosis [35]. As previously reported, ROS have been implicated as important mediators of necroptosis [73], although their role may not be universal [50]. Especially RIPK3 has been reported to regulate ROS under necroptotic conditions [74, 75] and thereby participates in anti-metastatic reactions by mediating oxidative stress that is critical for the killing of ECM-detached (metastatic) cells. This suggests that, similar to apoptosis, necroptosis may need to be actively inhibited by cancer cells to ensure successful tumor progression and metastasis.
Although most metastatic cells from the primary tumor are destined to die, a fraction of surviving cancer cells is still able to macrometastasize. This process may be supported by the autophagic machinery that can improves the fitness of these metastatic cells under stressful conditions [66]. Especially induction of apoptosis may promote a myriad of signal transduction elements, which are in cross-talk to antagonize or assist autophagy, e.g., the processing activity of caspases can elicit autophagy through cleavage of Atg4D [76]. Therefore, the sole induction of apoptosis may be insufficient for the successful elimination of metastatic tumor cells when taking into account the sophisticated interactions of the various forms of RCD and possible anti-metastatic functions of autophagy [66]. On the other hand, in apoptosis-compromised cells, autophagy was described to enhance other types of RCD such as caspase-1-dependent pyroptosis or necroptosis [76]. Accordingly, the induction of autophagy by GX15-070 (obatoclax; Cephalon), a drug that is already in clinical phase II studies in patients with myelofibrosis [77], was described as a promising strategy to trigger RIPK1-RIPK3-dependent necroptosis [78], although this compound can simultaneously elicit apoptosis and autophagy [79]. Further, employing obatoclax to induce autophagy-dependent necroptosis was successful in the elimination of apoptosis-resistant acute lymphoblastic leukemia (ALL). It was observed that obatoclax and rapamycin effectively restored the response to the chemotherapeutic glucocorticoid drug dexamethasone in glucocorticoid-resistant ALL by triggering non-apoptotic cell death [80]. In some cell lines, such as lung and bladder cancer cells, the novel chalcone derivative chalcone-24 (Chal-24) induced autophagy-dependent inhibition of IAPs and in consequence formation of the ripoptosome and induction of RIPK1-RIPK3-dependent necroptosis [81]. It was shown that the main cytotoxicity induced by Chal-24 was elicited with no detectable activation of caspase-8 or caspase-3, and without cleavage of PARP1. In line, the use of caspase inhibitor zVAD-fmk to suppress apoptosis showed only a minimal protection on Chal-24-induced cell death. As a control, inhibition of autophagy effectively suppressed the association of FADD and caspase-8 with RIP1 and thereby formation of the ripoptosome [81].
Despite the above advances, the most effective strategy to combat cancer metastasis currently available is still the early diagnosis of cancer, before metastatic spread has occurred [64]. In this regard, several reports indicate that components of the necroptotic signaling pathway may be useful as potential markers to predict the further course of the disease. For example, the expression of MLKL can obviously serve as a prognostic biomarker in patients with early-stage resected pancreatic adenocarcinoma. In a recent study, low expression of MLKL was associated with decreased overall survival after gemcitabine-based chemotherapy [82]. In an independent investigation, low expression of MLKL was similarly associated with low disease-free survival and overall survival in patients with in ovarian carcinomas [83]. Additionally, it has been shown that a monoclonal antibody that specifically recognizes phosphorylated MLKL can be used to detect necroptotic cells in human liver biopsy samples from patients suffering from drug-induced liver injury [84]. Therefore, in the future, it may be worthwhile to look for pMLKL as a potential indicator of treatment efficacy also in biopsies from tumor patients undergoing necroptosis-based treatment regimes. Although activating mutations for MLKL have been reported, e.g., in adenocarcinomas, the gene for human MLKL is rarely mutated. The highest observed rates were 2.14 % in skin samples, 1.51 % in stomach samples, and 1.44 % in samples from the large intestine, with an overall presence of mutations in 101 out of 29,480 samples, corresponding to an average rate of 0.34 %, according to the Catalogue of Somatic Mutations in Cancer (COSMIC) [85]. This is much less than what is observed in genes like TP53 (28,794 out of 111,137 samples, corresponding to 25.9 %), making MLKL a good target for highly specific antibodies. Besides MLKL, RIPK3 was recently described as another potential biomarker for necroptosis-sensitive tumor cell lines [24]. Taken together, therapeutic induction of necroptosis will most likely not be sufficient to prevent cancer metastasis by itself, but may nevertheless represent an important component of future combination therapies [24]. Additionally, components of the necroptosis machinery may serve as markers to better predict the future course of cancer in patients.
Cancer and necroptosis: friends? Limitations of necroptosis in cancer therapy
Induction of regulated/programmed cell death is often considered to be beneficial, e.g., during the development of an organism. Unfortunately, RCD may also be “beneficial” for the growth and metastasis of tumors. In a recent study [86], the authors point to the exciting concept that inside the central core of a tumor, so far unknown mechanisms of RN cause the death of the tumor cells. By this mechanism, the tumor could induce neovascularization and thereby promote its own growth. Another important aspect to consider is the potential of necroptosis to elicit an inflammatory response [87]. Swelling of the necroptotic cell due to leakage of the cell membrane and the final disruption of the cell releases intracellular components such as pro-inflammatory molecules into the surrounding and thereby promotes inflammatory responses [9]. These pro-inflammatory effects of necroptosis might trigger the damage of healthy tissue, causing severe side effects which would prohibit a corresponding therapeutic approach. In addition, the damage of healthy tissue may also promote cancer cell spreading. Along these lines, mice with FADD-deficient keratinocytes developed skin inflammation caused by keratinocyte-specific RIPK3-dependent necroptosis [88]. In another mouse model with genetic deficiency of FADD, the animals displayed elevated levels of necrotic intestinal epithelial cells and also developed spontaneous colitis and ileitis [89]. In the same study, RIPK3 deficiency prevented epithelial cell death and inflammation. In line with those murine studies, first lines of evidence suggest that necroptosis is also involved in inflammatory intestinal diseases in humans, e.g., in Crohn’s disease [90, 91].
Moreover, although clinical data are still lacking, it is very probable that induction of necroptosis will not always be sufficient to elicit an efficient anti-cancer response. As observed for apoptosis-based treatment strategies, cancer cells may acquire mutations that inactivate or alter the expression of components of the necroptotic signaling machinery. Also, the sensitivity of cancer cells to necroptosis may depend on external conditions such as the availability of oxygen or nutrients. For example, under hypoxic conditions and in the absence of glucose, RIPK1-RIPK3-dependent necroptosis was increased in human colorectal carcinoma cells, but was suppressed when glucose was present, most likely because of pyruvate scavenging of mitochondrial free radicals [92]. Likewise, mutations or loss of RIPK1 and/or RIPK3 might foil the necroptotic control of cancer, e.g., certain genetic variations of RIPK3 (single nucleotide polymorphisms) were found to correlate with a higher risk of non-Hodgkin lymphoma [93]. Similarly, RIPK3 was downregulated in leukemia cells from patients with acute myeloid leukemia (AML), and re-expression of RIPK3 restored the sensitivity of these cells to necroptosis [94]. Multiple analyses suggest that RIPK3 deficiency is positively selected during tumor growth/development as RIPK3 expression is often silenced in cancer cells, e.g., breast cancer cells due to genomic methylation [95]. In this regard, treatment of cancer patients with hypomethylating agents may restore RIPK3 expression [95], and thereby promote sensitivity to RIPK3-dependent necroptosis. Further, molecules that regulate the necrosome signaling complex may also influence the efficacy of necroptosis in cancer cells. The protein phosphatase 1B (Ppm1b) was identified as a RIPK3 phosphatase that can restrict necroptosis through dephosphorylation of RIPK3 [96]. Importantly, metastatic prostate cancer cells express lower levels of Ppm1b [97], implicating that they exhibit an increased sensitivity for induction of necroptosis. Likewise, a cytosolic complex of heat shock protein 90 (Hsp90) and co-chaperone CDC37 was identified to be required for RIPK3 activation [98]. Many malignant cells depend on overexpression of the CDC37 oncoprotein that mediates carcinogenesis by stabilizing the compromised structures of mutant and/or overexpressed oncogenic kinases [99]. Therefore, naturally (or therapy-induced) CDC37-overexpressing tumor cells may also be more susceptible for elimination by necroptosis. Downregulation of Makorin Ring Finger Protein 1 (MKRN1), an E3 ligase that regulates the ubiquitin-dependent degradation of FADD, enhanced formation of the necrosome when caspases were simultaneously inhibited. In consequence, severe defects in tumor growth were observed in a breast cancer xenograft model upon treatment with TRAIL, suggesting that depletion of MKRN1 increases the sensitivity of cancer cells to TRAIL-mediated necroptotic and apoptotic cell death [100]. In addition to the above modulators of necroptosis, the deubiquitinase CYLD, an enzyme that directly regulates the ubiquitination status of RIPK1 might enhance necroptosis [101] and facilitate necroptosis-based anti-cancer therapies. In line, inactivating CYLD mutations have been found in epidermal cancer cells that promote skin tumor progression [102]. Conversely, Toll-like receptor-mediated downregulation of CYLD protected from necroptosis in macrophages [103]. All these examples clearly illustrate the need to develop more pathway-specific drugs in order to enhance the execution of necroptosis during anti-cancer therapy [29].
Conclusions and perspectives
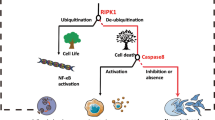
Many types of cancer like glioblastoma, melanoma or pancreatic ductal adenocarcinoma often show resistance towards apoptosis (reviewed in [104]), demonstrating an unmet urgent clinical need for alternative treatment strategies. In this review, we focused on the potential advantages and pitfalls of necroptosis as an alternative anti-cancer strategy (Fig. 2). Although the study of necroptosis in cancer therapy is clearly still in its infancy, interest in this field has been sparked and we expect significant progress and breakthroughs in the near future, hopefully directly for the benefit of patients suffering from cancer.

Schematic overview on the relationship of cancer and necroptosis as friend or foe. Events listed under “friend?” are beneficial for the survival of the whole tumor (e.g., induction of neovascularization) or for the survival of the individual cancer cell itself (e.g., downregulation/lack of key components of necroptosis). The induction of autophagy can further improve the invasion efficacy of metastatic cells by strengthening their fitness against necroptosis. Compounds that induce necroptosis in cancer cells or at least sensitize them for necroptosis are listed under “foe?”. In addition to those chemicals and naturally occurring substances, viral anti-apoptotic proteins (e.g., M45) and nanoparticles (e.g., ZnO and Ag) have also been shown to induce necroptosis in cancer cells. Since the sphingolipid ceramide has been reported as a key downstream mediator of necroptosis, the manipulation of intracellular sphingolipid levels may also represent a feasible target to induce necroptosis in cancer cells. The promotion of inflammatory responses by necroptosis may either be beneficial for the tumor (e.g., by promoting cancer cell spreading), or may contribute to a more efficient clearance of tumor cells by the immune system via the release of cytokines and DAMPs
References
Lockshin RA, Zakeri Z (2007) Cell death in health and disease. J Cell Mol Med 11(6):1214–1224. doi:10.1111/j.1582-4934.2007.00150.x
Lockshin RA, Williams CM (1964) Programmed cell death. 2. Endocrine potentiation of the breakdown of the intersegmental muscles of silkmoths. J Insect Physiol 10(4):643–649. doi:10.1016/0022-1910(64)90034-4
Ellis HM, Horvitz HR (1986) Genetic-control of programmed cell-death in the nematode C. elegans. Cell 44(6):817–829. doi:10.1016/0092-8674(86)90004-8
Golstein P, Kroemer G (2007) Cell death by necrosis: towards a molecular definition. Trends Biochem Sci 32(1):37–43. doi:10.1016/j.tibs.2006.11.001
Laster SM, Wood JG, Gooding LR (1988) Tumor necrosis factor can induce both apoptic and necrotic forms of cell-lysis. J Immunol 141(8):2629–2634
Vercammen D, Vandenabeele P, Beyaert R, Declercq W, Fiers W (1997) Tumour necrosis factor-induced necrosis versus anti-Fas-induced apoptosis in L929 cells. Cytokine 9(11):801–808. doi:10.1006/cyto.1997.0252
Kawahara A, Ohsawa Y, Matsumura H, Uchiyama Y, Nagata S (1998) Caspase-independent cell killing by Fas-associated protein with death domain. J Cell Biol 143(5):1353–1360. doi:10.1083/jcb.143.5.1353
Ziegler U, Groscurth P (2004) Morphological features of cell death. News Physiol Sci 19:124–128. doi:10.1152/nips.01519.2004
Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P (2014) Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Bio 15(2):134–146. doi:10.1038/nrm3737
Christofferson DE, Yuan JY (2010) Necroptosis as an alternative form of programmed cell death. Curr Opin Cell Biol 22(2):263–268. doi:10.1016/j.ceb.2009.12.003
Sun LM, Wang HY, Wang ZG, He SD, Chen S, Liao DH, Wang L, Yan JC, Liu WL, Lei XG, Wang XD (2012) Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148(1–2):213–227. doi:10.1016/j.cell.2011.11.031
Fuchs Y, Steller H (2015) Live to die another way: modes of programmed cell death and the signals emanating from dying cells. Nat Rev Mol Cell Bio 16(6):329–344. doi:10.1038/nrm3999
Pasparakis M, Vandenabeele P (2015) Necroptosis and its role in inflammation. Nature 517(7534):311–320. doi:10.1038/nature14191
Takemura R, Takaki H, Okada S, Shime H, Akazawa T, Oshiumi H, Matsumoto M, Teshima T, Seya T (2015) PolyI:C-induced, TLR3/RIP3-dependent necroptosis backs up immune effector-mediated tumor elimination in vivo. Cancer Immunol Res 3(8):902–914. doi:10.1158/2326-6066.CIR-14-0219
Schmidt SV, Seibert S, Walch-Ruckheim B, Vicinus B, Kamionka EM, Pahne-Zeppenfeld J, Solomayer EF, Kim YJ, Bohle RM, Smola S (2015) RIPK3 expression in cervical cancer cells is required for PolyIC-induced necroptosis, IL-1 alpha release, and efficient paracrine dendritic cell activation. Oncotarget 6(11):8635–8647
Kaczmarek A, Vandenabeele P, Krysko DV (2013) Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity 38(2):209–223. doi:10.1016/j.immuni.2013.02.003
Linkermann A, Green DR (2014) Necroptosis. New Engl J Med 370(5):455–465. doi:10.1056/Nejmra1310050
Inoue H, Tani K (2014) Multimodal immunogenic cancer cell death as a consequence of anticancer cytotoxic treatments. Cell Death Differ 21(1):39–49. doi:10.1038/cdd.2013.84
Guo ZS, Liu Z, Bartlett DL (2014) Oncolytic immunotherapy: dying the right way is a key to eliciting potent antitumor immunity. Front Oncol 4:74. doi:10.3389/fonc.2014.00074
Yatim N, Jusforgues-Saklani H, Orozco S, Schulz O, Barreira da Silva R, Reis e Sousa C, Green DR, Oberst A, Albert ML (2015) RIPK1 and NF-kappaB signaling in dying cells determines cross-priming of CD8(+) T cells. Science 350(6258):328–334. doi:10.1126/science.aad0395
Fulda S (2014) Therapeutic exploitation of necroptosis for cancer therapy. Semin Cell Dev Biol 35:51–56. doi:10.1016/j.semcdb.2014.07.002
Dunai ZA, Imre G, Barna G, Korcsmaros T, Petak I, Bauer PI, Mihalik R (2012) Staurosporine induces necroptotic cell death under caspase-compromised conditions in U937 cells. PLoS One 7(7):e41945. doi:10.1371/journal.pone.0041945
McCabe KE, Bacos K, Lu D, Delaney JR, Axelrod J, Potter MD, Vamos M, Wong V, Cosford ND, Xiang R, Stupack DG (2014) Triggering necroptosis in cisplatin and IAP antagonist-resistant ovarian carcinoma. Cell Death Dis 5:e1496. doi:10.1038/cddis.2014.448
Voigt S, Philipp S, Davarnia P, Winoto-Morbach S, Röder C, Arenz C, Trauzold A, Kabelitz D, Schütze S, Kalthoff H, Adam D (2014) TRAIL-induced programmed necrosis as a novel approach to eliminate tumor cells. BMC Cancer 14:74. doi:10.1186/1471-2407-14-74
Vercammen D, Beyaert R, Denecker G, Goossens V, Van Loo G, Declercq W, Grooten J, Fiers W, Vandenabeele P (1998) Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J Exp Med 187(9):1477–1485. doi:10.1084/jem.187.9.1477
Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, Bodmer JL, Schneider P, Seed B, Tschopp J (2000) Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol 1(6):489–495. doi:10.1038/82732
Callus BA, Vaux DL (2007) Caspase inhibitors: viral, cellular and chemical. Cell Death Differ 14(1):73–78. doi:10.1038/sj.cdd.4402034
Fulda S, Vucic D (2012) Targeting IAP proteins for therapeutic intervention in cancer. Nature Rev Drug Discov 11(2):109–124. doi:10.1038/nrd3627
Moriwaki K, Bertin J, Gough PJ, Orlowski GM, Chan FK (2015) Differential roles of RIPK1 and RIPK3 in TNF-induced necroptosis and chemotherapeutic agent-induced cell death. Cell Death Dis 6:e1636. doi:10.1038/cddis.2015.16
He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X (2009) Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 137(6):1100–1111. doi:10.1016/j.cell.2009.05.021
Steinhart L, Belz K, Fulda S (2013) Smac mimetic and demethylating agents synergistically trigger cell death in acute myeloid leukemia cells and overcome apoptosis resistance by inducing necroptosis. Cell Death Dis 4:e802. doi:10.1038/cddis.2013.320
Grassilli E, Ianzano L, Bonomo S, Missaglia C, Cerrito MG, Giovannoni R, Masiero L, Lavitrano M (2014) GSK3A is redundant with GSK3B in modulating drug resistance and chemotherapy-induced necroptosis. PLoS One 9(7):e100947. doi:10.1371/journal.pone.0100947
Okada M, Adachi S, Imai T, Watanabe K, Toyokuni SY, Ueno M, Zervos AS, Kroemer G, Nakahata T (2004) A novel mechanism for imatinib mesylate-induced cell death of BCR-ABL-positive human leukemic cells: caspase-independent, necrosis-like programmed cell death mediated by serine protease activity. Blood 103(6):2299–2307. doi:10.1182/blood-2003-05-1605
Xu Y, Lin Z, Zhao N, Zhou L, Liu F, Cichacz Z, Zhang L, Zhan Q, Zhao X (2014) Receptor interactive protein kinase 3 promotes cisplatin-triggered necrosis in apoptosis-resistant esophageal squamous cell carcinoma cells. PLoS One 9(6):e100127. doi:10.1371/journal.pone.0100127
Huang CJ, Luo YA, Zhao JW, Yang FW, Zhao HW, Fan WH, Ge PF (2013) Shikonin kills glioma cells through necroptosis mediated by RIP-1. PLoS One 8(6):e66326. doi:10.1371/journal.pone.0066326
Wada N, Kawano Y, Fujiwara S, Kikukawa Y, Okuno Y, Tasaki M, Ueda M, Ando Y, Yoshinaga K, Ri M, Iida S, Nakashima T, Shiotsu Y, Mitsuya H, Hata H (2015) Shikonin, dually functions as a proteasome inhibitor and a necroptosis inducer in multiple myeloma cells. Int J Oncol 46(3):963–972. doi:10.3892/ijo.2014.2804
Chen J, Xie J, Jiang Z, Wang B, Wang Y, Hu X (2011) Shikonin and its analogs inhibit cancer cell glycolysis by targeting tumor pyruvate kinase-M2. Oncogene 30(42):4297–4306. doi:10.1038/onc.2011.137
Yu X, Deng Q, Li W, Xiao L, Luo X, Liu X, Yang L, Peng S, Ding Z, Feng T, Zhou J, Fan J, Bode AM, Dong Z, Liu J, Cao Y (2015) Neoalbaconol induces cell death through necroptosis by regulating RIPK-dependent autocrine TNFalpha and ROS production. Oncotarget 6(4):1995–2008
Deng Q, Yu X, Xiao L, Hu Z, Luo X, Tao Y, Yang L, Liu X, Chen H, Ding Z, Feng T, Tang Y, Weng X, Gao J, Yi W, Bode AM, Dong Z, Liu J, Cao Y (2013) Neoalbaconol induces energy depletion and multiple cell death in cancer cells by targeting PDK1-PI3-K/Akt signaling pathway. Cell Death Dis 4:e804. doi:10.1038/cddis.2013.324
Pasupuleti N, Leon L, Carraway KL 3rd, Gorin F (2013) 5-Benzylglycinyl-amiloride kills proliferating and nonproliferating malignant glioma cells through caspase-independent necroptosis mediated by apoptosis-inducing factor. J Pharmacol Exp Ther 344(3):600–615. doi:10.1124/jpet.112.200519
Babcook MA, Sramkoski RM, Fujioka H, Daneshgari F, Almasan A, Shukla S, Nanavaty RR, Gupta S (2014) Combination simvastatin and metformin induces G1-phase cell cycle arrest and Ripk1- and Ripk3-dependent necrosis in C4-2B osseous metastatic castration-resistant prostate cancer cells. Cell Death Dis 5:e1536. doi:10.1038/cddis.2014.500
Hernandez-Breijo B, Monserrat J, Ramirez-Rubio S, Cuevas EP, Vara D, Diaz-Laviada I, Fernandez-Moreno MD, Roman ID, Gisbert JP, Guijarro LG (2011) Preclinical evaluation of azathioprine plus buthionine sulfoximine in the treatment of human hepatocarcinoma and colon carcinoma. World J Gastroenterol 17(34):3899–3911. doi:10.3748/wjg.v17.i34.3899
Coupienne I, Bontems S, Dewaele M, Rubio N, Habraken Y, Fulda S, Agostinis P, Piette J (2011) NF-kappaB inhibition improves the sensitivity of human glioblastoma cells to 5-aminolevulinic acid-based photodynamic therapy. Biochem Pharmacol 81(5):606–616. doi:10.1016/j.bcp.2010.12.015
Rizzi F, Naponelli V, Silva A, Modernelli A, Ramazzina I, Bonacini M, Tardito S, Gatti R, Uggeri J, Bettuzzi S (2014) Polyphenon E(R), a standardized green tea extract, induces endoplasmic reticulum stress, leading to death of immortalized PNT1a cells by anoikis and tumorigenic PC3 by necroptosis. Carcinogenesis 35(4):828–839. doi:10.1093/carcin/bgt481
Wallenberg M, Misra S, Wasik AM, Marzano C, Björnstedt M, Gandin V, Fernandes AP (2014) Selenium induces a multi-targeted cell death process in addition to ROS formation. J Cell Mol Med 18(4):671–684. doi:10.1111/jcmm.12214
Liu X, Chhipa RR, Nakano I, Dasgupta B (2014) The AMPK inhibitor compound C is a potent AMPK-independent antiglioma agent. Mol Cancer Ther 13(3):596–605. doi:10.1158/1535-7163.MCT-13-0579
Deeraksa A, Pan J, Sha Y, Liu XD, Eissa NT, Lin SH, Yu-Lee LY (2013) Plk1 is upregulated in androgen-insensitive prostate cancer cells and its inhibition leads to necroptosis. Oncogene 32(24):2973–2983. doi:10.1038/onc.2012.309
Tufo G, Jones AW, Wang Z, Hamelin J, Tajeddine N, Esposti DD, Martel C, Boursier C, Gallerne C, Migdal C, Lemaire C, Szabadkai G, Lemoine A, Kroemer G, Brenner C (2014) The protein disulfide isomerases PDIA4 and PDIA6 mediate resistance to cisplatin-induced cell death in lung adenocarcinoma. Cell Death Differ 21(5):685–695. doi:10.1038/cdd.2013.193
Thon L, Mathieu S, Kabelitz D, Adam D (2006) The murine TRAIL receptor signals caspase-independent cell death through ceramide. Exp Cell Res 312(19):3808–3821. doi:10.1016/j.yexcr.2006.08.017
Thon L, Möhlig H, Mathieu S, Lange A, Bulanova E, Winoto-Morbach S, Schütze S, Bulfone-Paus S, Adam D (2005) Ceramide mediates caspase-independent programmed cell death. FASEB J 19(14):1945–1956. doi:10.1096/fj.05-3726com
Saddoughi SA, Gencer S, Peterson YK, Ward KE, Mukhopadhyay A, Oaks J, Bielawski J, Szulc ZM, Thomas RJ, Selvam SP, Senkal CE, Garrett-Mayer E, De Palma RM, Fedarovich D, Liu A, Habib AA, Stahelin RV, Perrotti D, Ogretmen B (2013) Sphingosine analogue drug FTY720 targets I2PP2A/SET and mediates lung tumour suppression via activation of PP2A-RIPK1-dependent necroptosis. EMBO Mol Med 5(1):105–121. doi:10.1002/emmm.201201283
Yamanaka K, Urano Y, Takabe W, Saito Y, Noguchi N (2014) Induction of apoptosis and necroptosis by 24(S)-hydroxycholesterol is dependent on activity of acyl-CoA:cholesterol acyltransferase 1. Cell Death Dis 5:e990. doi:10.1038/cddis.2013.524
Dai X, Zhang J, Arfuso F, Chinnathambi A, Zayed ME, Alharbi SA, Kumar AP, Ahn KS, Sethi G (2015) Targeting TNF-related apoptosis-inducing ligand (TRAIL) receptor by natural products as a potential therapeutic approach for cancer therapy. Exp Biol Med 240(6):760–773. doi:10.1177/1535370215579167
Philipp S, Sosna J, Plenge J, Kalthoff H, Adam D (2015) Homoharringtonine, a clinically approved anti-leukemia drug, sensitizes tumor cells for TRAIL-induced necroptosis. Cell Commun Signal 13:25. doi:10.1186/s12964-015-0103-0
Jouan-Lanhouet S, Arshad MI, Piquet-Pellorce C, Martin-Chouly C, Le Moigne-Muller G, Van Herreweghe F, Takahashi N, Sergent O, Lagadic-Gossmann D, Vandenabeele P, Samson M, Dimanche-Boitrel MT (2012) TRAIL induces necroptosis involving RIPK1/RIPK3-dependent PARP-1 activation. Cell Death Differ 19(12):2003–2014. doi:10.1038/cdd.2012.90
Omoto S, Guo H, Talekar GR, Roback L, Kaiser WJ, Mocarski ES (2015) Suppression of RIP3-dependent necroptosis by human cytomegalovirus. J Biol Chem 290(18):11635–11648. doi:10.1074/jbc.M115.646042
Irrinki KM, Mallilankaraman K, Thapa RJ, Chandramoorthy HC, Smith FJ, Jog NR, Gandhirajan RK, Kelsen SG, Houser SR, May MJ, Balachandran S, Madesh M (2011) Requirement of FADD, NEMO, and BAX/BAK for aberrant mitochondrial function in tumor necrosis factor alpha-induced necrosis. Mol Cell Biol 31(18):3745–3758. doi:10.1128/MCB.05303-11
Lamothe B, Lai Y, Xie M, Schneider MD, Darnay BG (2013) TAK1 is essential for osteoclast differentiation and is an important modulator of cell death by apoptosis and necroptosis. Mol Cell Biol 33(3):582–595. doi:10.1128/MCB.01225-12
O’Donnell MA, Hase H, Legarda D, Ting AT (2012) NEMO inhibits programmed necrosis in an NFkappaB-independent manner by restraining RIP1. PLoS ONE 7(7):e41238. doi:10.1371/journal.pone.0041238
Liu M, Gu XH, Zhang K, Ding Y, Wei XB, Zhang XM, Zhao YX (2013) Gold nanoparticles trigger apoptosis and necrosis in lung cancer cells with low intracellular glutathione. J Nanopart Res. doi:10.1007/S11051-013-1745-8
Akhtar MJ, Alhadlaq HA, Kumar S, Alrokayan SA, Ahamed M (2015) Selective cancer-killing ability of metal-based nanoparticles: implications for cancer therapy. Arch Toxicol. doi:10.1007/s00204-015-1570-1
van Marion DM, Domanska UM, Timmer-Bosscha H, Walenkamp AM (2016) Studying cancer metastasis: existing models, challenges and future perspectives. Crit Rev Oncol Hematol 97:107–117. doi:10.1016/j.critrevonc.2015.08.009
Steeg PS (2006) Tumor metastasis: mechanistic insights and clinical challenges. Nat Med 12(8):895–904. doi:10.1038/nm1469
Chambers AF, Groom AC, MacDonald IC (2002) Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer 2(8):563–572. doi:10.1038/nrc865
Luzzi KJ, MacDonald IC, Schmidt EE, Kerkvliet N, Morris VL, Chambers AF, Groom AC (1998) Multistep nature of metastatic inefficiency—dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am J Pathol 153(3):865–873. doi:10.1016/S0002-9440(10)65628-3
Su Z, Yang Z, Xu Y, Chen Y, Yu Q (2015) Apoptosis, autophagy, necroptosis, and cancer metastasis. Mol Cancer 14(1):48. doi:10.1186/s12943-015-0321-5
Trauzold A, Siegmund D, Schniewind B, Sipos B, Egberts J, Zorenkov D, Emme D, Röder C, Kalthoff H, Wajant H (2006) TRAIL promotes metastasis of human pancreatic ductal adenocarcinoma. Oncogene 25(56):7434–7439. doi:10.1038/sj.onc.1209719
Röder C, Trauzold A, Kalthoff H (2011) Impact of death receptor signaling on the malignancy of pancreatic ductal adenocarcinoma. Eur J Cell Biol 90(6–7):450–455. doi:10.1016/j.ejcb.2010.10.008
Lemke J, von Karstedt S, Zinngrebe J, Walczak H (2014) Getting TRAIL back on track for cancer therapy. Cell Death Differ 21(9):1350–1364. doi:10.1038/cdd.2014.81
Haselmann V, Kurz A, Bertsch U, Hübner S, Olempska-Müller M, Fritsch J, Häsler R, Pickl A, Fritsche H, Annewanter F, Engler C, Fleig B, Bernt A, Röder C, Schmidt H, Gelhaus C, Hauser C, Egberts JH, Heneweer C, Rohde AM, Böger C, Knippschild U, Röcken C, Adam D, Walczak H, Schütze S, Janssen O, Wulczyn FG, Wajant H, Kalthoff H, Trauzold A (2014) Nuclear death receptor TRAIL-R2 inhibits maturation of let-7 and promotes proliferation of pancreatic and other tumor cells. Gastroenterology 146(1):278–290. doi:10.1053/j.gastro.2013.10.009
Prasad S, Kim JH, Gupta SC, Aggarwal BB (2014) Targeting death receptors for TRAIL by agents designed by Mother Nature. Trends Pharmacol Sci 35(10):520–536. doi:10.1016/j.tips.2014.07.004
Fu ZZ, Deng BY, Liao YX, Shan LC, Yin F, Wang ZY, Zeng H, Zuo DQ, Hua YQ, Cai ZD (2013) The anti-tumor effect of shikonin on osteosarcoma by inducing RIP1 and RIP3 dependent necroptosis. BMC Cancer 13:580. doi:10.1186/1471-2407-13-580
Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G (2010) Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol 11(10):700–714. doi:10.1038/nrm2970
Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, Dong MQ, Han J (2009) RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 325(5938):332–336. doi:10.1126/science.1172308
Cho YS, Challa S, Moquin D, Genga R, Ray TD, Guildford M, Chan FK (2009) Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137(6):1112–1123. doi:10.1016/j.cell.2009.05.037
Ryter SW, Mizumura K, Choi AM (2014) The impact of autophagy on cell death modalities. Int J Cell Biol 2014:502676. doi:10.1155/2014/502676
Parikh SA, Kantarjian H, Schimmer A, Walsh W, Asatiani E, El-Shami K, Winton E, Verstovsek S (2010) Phase II study of obatoclax mesylate (GX15-070), a small-molecule BCL-2 family antagonist, for patients with myelofibrosis. Clin Lymphoma Myeloma Leuk 10(4):285–289. doi:10.3816/CLML.2010.n.059
Basit F, Cristofanon S, Fulda S (2013) Obatoclax (GX15-070) triggers necroptosis by promoting the assembly of the necrosome on autophagosomal membranes. Cell Death Differ 20(9):1161–1173. doi:10.1038/cdd.2013.45
Urtishak KA, Edwards AY, Wang LS, Hudome A, Robinson BW, Barrett JS, Cao K, Cory L, Moore JS, Bantly AD, Yu QC, Chen IM, Atlas SR, Willman CL, Kundu M, Carroll AJ, Heerema NA, Devidas M, Hilden JM, Dreyer ZE, Hunger SP, Reaman GH, Felix CA (2013) Potent obatoclax cytotoxicity and activation of triple death mode killing across infant acute lymphoblastic leukemia. Blood 121(14):2689–2703. doi:10.1182/blood-2012-04-425033
Bonapace L, Bornhauser BC, Schmitz M, Cario G, Ziegler U, Niggli FK, Schäfer BW, Schrappe M, Stanulla M, Bourquin JP (2010) Induction of autophagy-dependent necroptosis is required for childhood acute lymphoblastic leukemia cells to overcome glucocorticoid resistance. J Clin Invest 120(4):1310–1323. doi:10.1172/JCI39987
He W, Wang Q, Srinivasan B, Xu J, Padilla MT, Li Z, Wang X, Liu Y, Gou X, Shen HM, Xing C, Lin Y (2014) A JNK-mediated autophagy pathway that triggers c-IAP degradation and necroptosis for anticancer chemotherapy. Oncogene 33(23):3004–3013. doi:10.1038/onc.2013.256
Colbert LE, Fisher SB, Hardy CW, Hall WA, Saka B, Shelton JW, Petrova AV, Warren MD, Pantazides BG, Gandhi K, Kowalski J, Kooby DA, El-Rayes BF, Staley CA 3rd, Adsay NV, Curran WJ Jr, Landry JC, Maithel SK, Yu DS (2013) Pronecrotic mixed lineage kinase domain-like protein expression is a prognostic biomarker in patients with early-stage resected pancreatic adenocarcinoma. Cancer 119(17):3148–3155. doi:10.1002/cncr.28144
He L, Peng K, Liu Y, Xiong J, Zhu FF (2013) Low expression of mixed lineage kinase domain-like protein is associated with poor prognosis in ovarian cancer patients. OncoTargets Ther 6:1539–1543. doi:10.2147/OTT.S52805
Wang HY, Sun LM, Su LJ, Rizo J, Liu L, Wang LF, Wang FS, Wang XD (2014) Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell 54(1):133–146. doi:10.1016/j.molcel.2014.03.003
Forbes SA, Bhamra G, Bamford S, Dawson E, Kok C, Clements J, Menzies A, Teague JW, Futreal PA, Stratton MR (2008) The Catalogue of Somatic Mutations in Cancer (COSMIC). Curr Protoc Hum Genet Chapter 10:Unit 10 11. doi:10.1002/0471142905.hg1011s57
Linkermann A, Stockwell BR, Krautwald S, Anders HJ (2014) Regulated cell death and inflammation: an auto-amplification loop causes organ failure. Nat Rev Immunol 14(11):759–767. doi:10.1038/nri3743
Brenner D, Blaser H, Mak TW (2015) Regulation of tumour necrosis factor signalling: live or let die. Nat Rev Immunol 15(6):362–374. doi:10.1038/nri3834
Bonnet MC, Preukschat D, Welz PS, van Loo G, Ermolaeva MA, Bloch W, Haase I, Pasparakis M (2011) The adaptor protein FADD protects epidermal keratinocytes from necroptosis in vivo and prevents skin inflammation. Immunity 35(4):572–582. doi:10.1016/j.immuni.2011.08.014
Welz PS, Wullaert A, Vlantis K, Kondylis V, Fernandez-Majada V, Ermolaeva M, Kirsch P, Sterner-Kock A, van Loo G, Pasparakis M (2011) FADD prevents RIP3-mediated epithelial cell necrosis and chronic intestinal inflammation. Nature 477(7364):330–334. doi:10.1038/nature10273
Günther C, Martini E, Wittkopf N, Amann K, Weigmann B, Neumann H, Waldner MJ, Hedrick SM, Tenzer S, Neurath MF, Becker C (2011) Caspase-8 regulates TNF-alpha-induced epithelial necroptosis and terminal ileitis. Nature 477(7364):335–339. doi:10.1038/nature10400
Pierdomenico M, Negroni A, Stronati L, Vitali R, Prete E, Bertin J, Gough PJ, Aloi M, Cucchiara S (2014) Necroptosis is active in children with inflammatory bowel disease and contributes to heighten intestinal inflammation. Am J Gastroenterol 109(2):279–287. doi:10.1038/ajg.2013.403
Huang CY, Kuo WT, Huang YC, Lee TC, Yu LC (2013) Resistance to hypoxia-induced necroptosis is conferred by glycolytic pyruvate scavenging of mitochondrial superoxide in colorectal cancer cells. Cell Death Dis 4:e622. doi:10.1038/cddis.2013.149
Cerhan JR, Ansell SM, Fredericksen ZS, Kay NE, Liebow M, Call TG, Dogan A, Cunningham JM, Wang AH, Liu-Mares W, Macon WR, Jelinek D, Witzig TE, Habermann TM, Slager SL (2007) Genetic variation in 1253 immune and inflammation genes and risk of non-Hodgkin lymphoma. Blood 110(13):4455–4463. doi:10.1182/blood-2007-05-088682
Nugues AL, El Bouazzati H, Hetuin D, Berthon C, Loyens A, Bertrand E, Jouy N, Idziorek T, Quesnel B (2014) RIP3 is downregulated in human myeloid leukemia cells and modulates apoptosis and caspase-mediated p65/RelA cleavage. Cell Death Dis 5:e1384. doi:10.1038/cddis.2014.347
Koo GB, Morgan MJ, Lee DG, Kim WJ, Yoon JH, Koo JS, Kim SI, Kim SJ, Son MK, Hong SS, Levy JM, Pollyea DA, Jordan CT, Yan P, Frankhouser D, Nicolet D, Maharry K, Marcucci G, Choi KS, Cho H, Thorburn A, Kim YS (2015) Methylation-dependent loss of RIP3 expression in cancer represses programmed necrosis in response to chemotherapeutics. Cell Res 25(6):707–725. doi:10.1038/cr.2015.56
Chen W, Wu J, Li L, Zhang Z, Ren J, Liang Y, Chen F, Yang C, Zhou Z, Su SS, Zheng X, Zhang Z, Zhong CQ, Wan H, Xiao M, Lin X, Feng XH, Han J (2015) Ppm1b negatively regulates necroptosis through dephosphorylating Rip3. Nat Cell Biol 17(4):434–444. doi:10.1038/ncb3120
Lu X, An H, Jin R, Zou M, Guo Y, Su PF, Liu D, Shyr Y, Yarbrough WG (2014) PPM1A is a RelA phosphatase with tumor suppressor-like activity. Oncogene 33(22):2918–2927. doi:10.1038/onc.2013.246
Li D, Xu T, Cao Y, Wang H, Li L, Chen S, Wang X, Shen Z (2015) A cytosolic heat shock protein 90 and cochaperone CDC37 complex is required for RIP3 activation during necroptosis. Proc Natl Acad Sci USA 112(16):5017–5022. doi:10.1073/pnas.1505244112
Gray PJ Jr, Prince T, Cheng J, Stevenson MA, Calderwood SK (2008) Targeting the oncogene and kinome chaperone CDC37. Nat Rev Cancer 8(7):491–495. doi:10.1038/nrc2420
Lee EW, Kim JH, Ahn YH, Seo J, Ko A, Jeong M, Kim SJ, Ro JY, Park KM, Lee HW, Park EJ, Chun KH, Song J (2012) Ubiquitination and degradation of the FADD adaptor protein regulate death receptor-mediated apoptosis and necroptosis. Nat Commun 3:978. doi:10.1038/ncomms1981
Moquin DM, McQuade T, Chan FK (2013) CYLD deubiquitinates RIP1 in the TNFalpha-induced necrosome to facilitate kinase activation and programmed necrosis. PLoS One 8(10):e76841. doi:10.1371/journal.pone.0076841
Alameda JP, Moreno-Maldonado R, Navarro M, Bravo A, Ramirez A, Page A, Jorcano JL, Fernandez-Acenero MJ, Casanova ML (2010) An inactivating CYLD mutation promotes skin tumor progression by conferring enhanced proliferative, survival and angiogenic properties to epidermal cancer cells. Oncogene 29(50):6522–6532. doi:10.1038/onc.2010.378
Schworer SA, Smirnova II, Kurbatova I, Bagina U, Churova M, Fowler T, Roy AL, Degterev A, Poltorak A (2014) Toll-like receptor-mediated down-regulation of the deubiquitinase cylindromatosis (CYLD) protects macrophages from necroptosis in wild-derived mice. J Biol Chem 289(20):14422–14433. doi:10.1074/jbc.M114.547547
Mohammad RM, Muqbil I, Lowe L, Yedjou C, Hsu HY, Lin LT, Siegelin MD, Fimognari C, Kumar NB, Dou QP, Yang H, Samadi AK, Russo GL, Spagnuolo C, Ray SK, Chakrabarti M, Morre JD, Coley HM, Honoki K, Fujii H, Georgakilas AG, Amedei A, Niccolai E, Amin A, Ashraf SS, Helferich WG, Yang X, Boosani CS, Guha G, Bhakta D, Ciriolo MR, Aquilano K, Chen S, Mohammed SI, Keith WN, Bilsland A, Halicka D, Nowsheen S, Azmi AS (2015) Broad targeting of resistance to apoptosis in cancer. Semin Cancer Biol. doi:10.1016/j.semcancer.2015.03.001
Acknowledgments
This work was supported by grants from the Deutsche Krebshilfe (110055) and the Deutsche Forschungsgemeinschaft (SFB 877, project B2 and Cluster of Excellence “Inflammation at Interfaces”, EXC306-PMTP1 and EXC306-PWTP2) to D. A. and a junior grant from the Medical Faculty of the Christian-Albrechts-University to S. P.
Author information
Authors and Affiliations
Corresponding author
Additional information
S. Philipp and J. Sosna contributed equally to this work.
Rights and permissions
About this article

Cite this article
Philipp, S., Sosna, J. & Adam, D. Cancer and necroptosis: friend or foe?. Cell. Mol. Life Sci. 73, 2183–2193 (2016). https://doi.org/10.1007/s00018-016-2193-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-016-2193-2