
Abstract
Although contributing to inflammatory responses and to the development of certain autoimmune pathologies, type I interferons (IFNs) are used for the treatment of viral, malignant, and even inflammatory diseases. Interleukin-1 (IL-1) is a strongly pyrogenic cytokine and its importance in the development of several inflammatory diseases is clearly established. While the therapeutic use of IL-1 blocking agents is particularly successful in the treatment of innate-driven inflammatory disorders, IFN treatment has mostly been appreciated in the management of multiple sclerosis. Interestingly, type I IFNs exert multifaceted immunomodulatory effects, including the reduction of IL-1 production, an outcome that could contribute to its efficacy in the treatment of inflammatory diseases. In this review, we summarize the current knowledge on IL-1 and IFN effects in different inflammatory disorders, the influence of IFNs on IL-1 production, and discuss possible therapeutic avenues based on these observations.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In this review, we focus on IFNs and give a background on their biological functions, emphasizing their anti-inflammatory effects in infections as well as in their therapeutic use. We summarize the knowledge on the complex mechanisms leading to IL-1 production and on its role in the development of various inflammatory disorders. Increasing evidence underlines the ability of IFNs to affect IL-1 production, and these findings potentially impact the management of infectious and inflammatory disorders. Here, we concentrate on the interplay between these two cytokines and possible clinical implications.
Inflammation
An inflammatory reaction is initiated whenever the normal equilibrium of the tissue is altered. Inflammation is required to remove the agent that caused the damage and to restore normality [1]. This is mainly achieved by cells of the immune system and by the action of different factors found in blood plasma and in extracellular fluids.
The classical signs of inflammation were already recognized by Celsus at the time of Ancient Greece and are described as the characteristic redness (rubor), swelling (tumor), increased temperature (calor), and pain (dolor) in the inflamed area. In fact, the primary goal of inflammatory responses is to increase blood supply and vessel permeability within the damaged tissues, thus promoting plasma protein and immune cell exudation. This facilitates tissue repair processes and minimizes the risk of infection. Indeed, when tightly controlled and actively terminated, inflammation represents a protective response, which coordinates tissue healing as well as immune cell activation and migration.
However, if not properly held in check, overriding inflammatory reactions can be detrimental to the tissue and lead to important collateral damage. This is the reason why a fifth hallmark, the ‘Functio laesa’ (i.e., loss of fuction), attributed to the Roman physician Galen, has been added to the four cardinal signs of inflammation. Importantly, chronic inflammation-driven diseases are a major health issue, particularly in developed countries, and selected examples are discussed later.
Mechanisms inducing innate-driven inflammation
Classical triggers of inflammatory responses are tissue damage and infectious agents. Innate immune cells, being strategically located at the site of injury, propagate the first alarm signal to recruit immune cells and to coordinate local tissue repair. Cells of the innate immune system have the ability to immediately respond to pathogen- and danger-associated molecular patterns (PAMPs and DAMPs, respectively) through dedicated germ-line encoded receptors, the so-called pattern recognition receptors (PRRs). Whilst PRRs lack the specificity and plasticity of their more sophisticated adaptive counterparts (T and B cell receptors), they mediate rapid and robust innate immune responses. Upon engagement, these receptors activate multiple signaling pathways that orchestrate the overall immune response, including the adaptive system.
To date, several classes of innate receptors have been defined [2–5]. We mainly focus on Toll-, Retinoic acid-inducible gene (RIG)-, and Nucleotide-binding oligomerization domain (NOD)-like receptors (TLRs, RLRs, and NLRs, respectively), that are particularly relevant to the induction of IFNs and IL-1.
Interleukin-1
The IL-1 family consists of 11 related ligands. Although a few members have been extensively investigated, others are neglected and await more in depth analysis. The most studied family members are the functionally related IL-1α and IL-1β (IL-1α and IL-1β are referred here as to ‘IL-1’), IL-1 receptor antagonist (IL-1Ra), and the IFN- γ-inducing cytokine IL-18.
Biological effects of IL-1
IL-1α and IL-1β, which share 26 % sequence homology, exert pleiotropic activities. Interleukin-1 represents an important mediator linking the immune, the endocrine, and the central nervous system, therefore affecting several physiological functions [6–8]. Yet, IL-1 is best known for its ability to induce fever and for this reason was formerly called ‘endogenous pyrogen’ [7].
In fact, this cytokine is instrumental in inducing inflammation through activation of the nuclear factor-κB (NF-κB) pathway. Both IL-1α and IL-1β signal via a common IL-1 receptor (IL-1R) that is formed by the association of IL-1RI and the IL-1R accessory protein [9]. Interleukin-1R represents the prototypical NF-κB activating Toll/Interleukin-1 receptor (TIR) domain-containing receptor and engages the NF-κB signaling cascade through the adapter myeloid differentiation factor 88 (MyD88) and the IL-1 receptor-associated kinases (IRAKs) [10–13].
Interleukin-1 induces a variety of inflammatory mediators, such as the neutrophil chemoattractant CXCL1, and promotes growth/activation of hematopoietic cells by inducing cytokines such as IL-6 or the granulocyte–macrophage colony-stimulating factor [14–18]. In the early 1990s, the properties of IL-1 in helping hematopoietic reconstitution in bone marrow-transplanted patients have been evaluated, as summarized by Dinarello [15]. Unfortunately, side effects such as inflammation and hypotension were too severe to pursue the use of IL-1 as a therapeutic agent. However, IL-1α and IL-1β were efficacious in reducing thrombocytopenia and leucopenia, in particular with regard to neutrophil counts. Another name that has been assigned to IL-1 in the past was ‘lymphocyte activating factor’, due to its ability to promote lymphocyte proliferation [19]. The interest in this field was recently revitalized, as it was shown that IL-1 plays a crucial role in promoting CD4+ T cell polarization towards a T helper type 17-phenotype (Th17) [20, 21]. These are helper T cells secreting IL-17 (also known as IL-17A), which are mainly involved in autoimmune diseases and antifungal responses. Furthermore, another report demonstrated that IL-1 had the property of opposing the function of regulatory T cells, thus restoring the response of conventional T lymphocytes [22]. Altogether, these studies highlight the contribution of IL-1 in both innate and adaptive immune responses.
Such important inflammatory effects require tight regulation of IL-1 production and signaling. As discussed in more detail in the next paragraph, the generation of bioactive IL-1 is a complex process controlled at multiple levels. In addition, IL-1 signaling is held in check by its antagonist IL-1Ra, which is a protein sharing 26 % homology with IL-1β and 18 % with IL-1α. This naturally occurring inhibitor strongly binds IL-1RI without activating it and defects in its production lead to early onset life-threatening inflammatory diseases [23–26]. The crucial role of IL-1 in the development of inflammatory pathologies has been increasingly studied and recognized over the past few years (reviewed in [14, 27]).
Multilevel control of bioactive IL-1 production
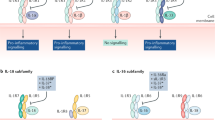
Interleukin-1 is mainly, but not exclusively, produced by myeloid cells. Given its strong and complex effects, its production is regulated at the transcriptional, translational, and posttranslational levels (Fig. 1). In fact, IL-1α and the inactive precursor of IL-1β (called pro-IL-1β) are virtually undetectable in blood cells under normal conditions, and their induction needs the presence of stress signals [28]. In well-studied myeloid immune cells, IL-1 transcription relies on NF-κB activating signals, such as tumor necrosis factor (TNF) or lipopolysaccharide (LPS) [29]. This ‘priming’ step is thus essential to raise intracellular levels of IL-1α and pro-IL-1β, along with enhancing the competence of the cell to proteolytically activate and secrete IL-1β [30–32].

The production of bioactive IL-1. Pattern recognition receptor engagement enhances NLRP3 expression and induces IL-1α and pro-IL-1β. Upon sensing specific inflammasome-activating stimuli, NLRP3 assembles into and recruits ASC and procaspase-1 to the inflammasome complex. This multi-protein platform leads to the activation of caspase-1, which proteolytically converts pro-IL-1β into bioactive IL-1β. In contrast, pro-IL-1α does not require cleavage for its activity. The release of both cytokines triggers NF-κB signaling by binding to IL-1R
Interleukin-1β mRNA is short-lived, giving rise to a tightly controlled burst of protein [33, 34]. Moreover, dissociation between the transcriptional and the translational regulation of this gene has been observed. Whereas, the stimulation of peripheral blood mononuclear cells with LPS increased both IL-1β mRNA and protein, the adhesion of these cells to plastic surface or exposure to the complement system factor C5a augmented transcript abundance, leading, however, to abortive translation [35–37]. Under such conditions, IL-1β transcripts assemble into large polyribosomes without giving rise to substantial protein levels.
Whereas IL-1α is biologically active as it is synthesized, IL-1β is produced as a precursor protein, which is per se inactive; proteolytic processing is required to convert the precursor protein into its active counterpart [38]. Several years ago, caspase-1 was identified as the crucial protease responsible for pro-IL-1β maturation and was called ‘IL-1β-converting enzyme’ thereafter [39–41]. However, the molecular mechanism leading to caspase-1 activation has only been described recently [42]. Its activation is accomplished upon formation of multiprotein complexes, called inflammasomes, which are able to bring caspases in close proximity to promote their autoactivation (discussed in more detail in the next section). A second member of the IL-1 family, IL-18, also requires caspase-1-dependent processing in order to be bioactive. Notably, although IL-1α activity is independent of its cleavage, its secretion has often been observed in association with inflammasome function [17, 40, 43, 44].
NLRs and inflammasomes
NOD-like receptors are mainly recognized for their important role in the activation of inflammatory cascades in response to different PAMPs and DAMPs [4, 45, 46]. This family of proteins is characterized by a tripartite structure. The N-terminus, which is also called ‘effector domain’, is responsible for the recruitment of downstream executioner proteins and consists in most cases of a caspase recruitment or a pyrin domain (CARD or PYD, respectively). In addition, NLRs have a central NOD domain and a C-terminal tail of leucine-rich repeats (LRRs) [45].
Amongst the best-characterized NLRs, we find NLRP3 (previously called NALP3 or cryopyrin), NLRP1, and NLR family CARD-containing (NLRC) 4 (previously known as IPAF). These three family members share the ability to form inflammasome platforms, which have been studied in detail by means of biochemical and genetic approaches. Upon activation, NLRP3 recruits caspase-1 into the inflammasome complex via the adaptor protein ASC (apoptosis-associated speck-like protein containing a CARD), which is a bipartite protein formed by a CARD and a PYD domain. ASC joins the oligomerized NLRP3 proteins through PYD–PYD interactions, while its CARD is available for homotypic interaction with the N-terminal CARD of caspase-1. In contrast to NLRP3, NLRC4 bears an N-terminal CARD, whereby caspase-1 can be directly recruited into the complex via homotypic CARD–CARD interactions. Nonetheless, the presence of ASC has been shown to favor NLRC4 inflammasome activity [47].
NOD-like receptor family CARD-containing protein 4 (NLRC4) with the assistance of neuronal apoptosis inhibitory proteins (NAIPs), another subgroup of NLR proteins, senses distinct Gram-negative bacteria. In particular, the role of NLRC4 in the response to flagellin has been investigated (Table 1) [46, 48–50]. The NLRP1 inflammasome was historically the first to be described, and the NLRP1b murine paralog activates caspase-1 upon exposure to lethal toxin of Bacillus anthracis [42, 51] (Table 1). To date, the activation of the NLRP3 inflammasome remains the most enigmatic. Indeed, this cytoplasmic protein leads to inflammasome assembly upon an ever-growing number of triggers, which belong to completely different chemical and physical categories (as summarized in Table 1, and reviewed in several articles [46, 52–54]). For example, NLRP3 can respond to stimulation with extracellular ATP, with pathogens and PAMPs, or to particulate stimuli such as monosodium urate crystals and asbestos fibers. How such diverse stimuli activate inflammasome formation specifically through NLRP3 is subject of intense research, and proposed models suggest the activation or induction of a common secondary effector molecule [31, 53]. Recent reports on Nlrp6-knockout animals and on human NLRP12 variants support the idea that these NLRs may be similarly involved in inflammasome complex formation [55–57].
Finally, also absent in melanoma 2 (AIM2), not belonging to the NLR family, has been shown to form an inflammasome. This protein of the IFI200 family of proteins recruits and activates caspase-1 upon binding to double-stranded (ds) DNA in the cytoplasm (Table 1) [58].
Interferons
IFNs were discovered over half a century ago as endogenous antiviral effector molecules. These cytokines were named after their ability to ‘interfere’ with viral replication in the host cell. However, IFNs mediate a variety of biological functions not limited to the defense against viral infections, extending to antitumor and immunomodulatory effects [59]. According to their amino acid sequence, chromosomal location, and receptor specificity, IFNs are further subdivided into three groups, which we describe below.
IFN subtypes
Introduction to type I IFNs
Interferon-α and -β are the most studied and therefore best-characterized members of this class. Interferon-β is encoded by a single gene in human and mouse, and more than 20 different genes code for IFN-α, thereof 13 give rise to a functional protein in humans and 14 in mice. Whereas IFN-α and -β regulate a widely overlapping set of genes, these two cytokines are described as slightly differing in their downstream effects and in their expression pattern, which varies depending on the stimulation, on the cell type, and among individuals [60–63].
Other type I subtypes are IFN-ε, -κ, -ω, -δ, and -τ. Interferon-δ and -τ are only found in pigs and cattle, respectively, and have no human homologs. IFN-ε, -κ, and -ω exist in humans but are less well described and show restricted tissue distribution [60]. Induction, signaling, and downstream effects of IFN-α and -β are discussed in detail in the following sections.
Type II IFN
The type II IFN subtype is constituted by a single gene product, IFN-γ. It is structurally different from type I IFNs, but was classified in the IFN family due to its antiviral effects [64, 65]. Interferon-γ binds to the nearly ubiquitously expressed IFN-γ receptor (IFNGR), and signals through Janus kinase 1 (JAK1) and JAK2 to phosphorylate signal transducer and activator of transcription 1 (STAT1), thereby allowing STAT1 homodimer formation and nuclear translocation.
Interferon-γ is involved in the modulation of immune and inflammatory responses and is predominantly produced by NK, NKT, and activated T cells. In the latter, IFN-γ leads to the upregulation of the transcription factor T-bet, which is crucial for controlling commitment to the Th1 phenotype [66]. T helper 1 cells are characterized by IFN-γ production and suited to fight viral infections. In fact, T-bet upregulation drives IFN-γ expression and creates a positive feedback loop, which forces undifferentiated CD4+ cells to Th1 polarization. Furthermore, IFN-γ favors Th1 commitment indirectly by suppressing polarization to Th17 and Th2 [66, 67], the latter being a subtype of helper T cells mainly involved in allergic and helminth responses, distinguished by the production of IL-4 and IL-13.
Interferon-γ also exerts immunomodulatory effects on innate immune cells, primarily by increasing lysosomal enzymatic activity and bactericidal oxidative burst [68]. Moreover, type II IFN directly enhances antigen presentation by promoting antigen processing and by inducing the expression of major histocompatibility complex (MHC) molecules [65, 68].
Type III IFNs
The third class of IFNs is composed of IFN-λ1, -λ2, and -λ3, or IL-28A, IL-28B, and IL-29, respectively. They are produced by most cell types, but particularly by plasmacytoid dendritic cells (DCs) in response to viral or bacterial infection [69, 70]. λ-Interferons form a separate group as they signal through a distinct receptor complex, consisting of IL-10R2 and IL-28R, which is expressed on a limited number of cells like hepatocytes and epithelial cells [70, 71]. However, type III IFNs activate similar signaling pathways and partly induce the same genes as type I IFNs, resulting in a potent antiviral response [72, 73].
Type I IFN production
Interferon-α and -β can be produced by almost all cell types upon stimulation of PRRs. Constitutive expression of low type I IFN levels is maintained by macrophages, skin DCs, and thymic epithelial cells and is also found in organs like liver, spleen, and kidney [74–77]. Basal IFN signaling is important to keep immune cells in a ‘primed’ state to rapidly and effectively mount an antiviral immune response [78].
Cytoplasmic receptors
Most cell types can induce type I IFN in response to the activation of cytoplasmic RLRs (Fig. 2a), which are nearly ubiquitously expressed [5]. Importantly, the strategic intracellular location of these PRRs allows infected cells to activate the antiviral response. Retinoic acid-inducible gene-like receptors detect microbial ribonucleic acids, which can derive from the genome of RNA viruses or from replication intermediates of DNA viruses [79]. These receptors therefore enable infected cells to locally produce antiviral type I IFNs, which are essential in opposing a number of viral infections [5, 80].

Induction and effects of type I IFNs. a The best characterized mechanisms leading to type I IFN induction are initiated by cytoplasmic RLRs and transmembrane TLRs. Signaling pathways activated by these receptors converge in the phosphorylation of IRFs, which translocate into the nucleus and activate the transcription of type I IFN genes. b Type I IFNs signal through a dimeric receptor and activate JAK1 and TYK2. In turn, these kinases phosphorylate STAT1 and STAT2, enabling recruitment of IRF9 to the so-called ISGF3 complex. This trimeric complex binds to ISREs, thereby modulating the expression of ISGs exerting antiviral, immunomodulatory, proapoptotic, and anti-inflammatory functions
Retinoic acid-inducible gene-like receptors include the two DExD/H-box helicases RIG-I and melanoma differentiation-associated gene 5 (MDA5), which are engaged by distinct ribonucleic acid species. Retinoic acid-inducible gene-I detects 5′ triphosphate single-stranded (ss) RNA with pairing at the 5′ end and rather short dsRNA, while MDA5 is activated by long dsRNA [5].
Retinoic acid-inducible gene-I and MDA5 have a common downstream adaptor, called mitochondrial antiviral signaling protein (MAVS) (Fig. 2a) [5]. Engagement of MAVS is followed by signaling through TBK1 [TNF receptor-associated factor (TRAF) family member-associated NF-κB activator (TANK)-binding kinase 1] and the IκB kinase ε (IKKε), leading to the activation of the transcription factors interferon-regulatory factor (IRF) 3, IRF7, and NF-κB [5, 80, 81]. Phosphorylation of IRFs allows their homo- or heterodimerization and promotes their subsequent localization to the nucleus, where they cooperate with NF-κB to stimulate the transcription of type I IFN genes [82] (Fig. 2a). In most cells, IRF3 is the only constitutively expressed IRF. The phosphorylation of IRF3 results in the induction of IFN-β and low levels of IFN-α4 [83]. IRF3-triggered type I IFN increases IRF7 levels, which then lead to the proficient expression of IFN-α [84].
In addition to RNA, sensing of cytoplasmic DNA also results in the induction of type I IFNs. Proposed DNA receptors are the DNA-dependent activator of IRFs (DAI) and the DNA-dependent RNA polymerase III [85]. The latter transcribes dsDNA into a 5′triphosphate dsRNA, which is recognized by RIG-I. More recently, the AIM2-like molecule IFI16 and other members of the DExD/H-box helicase superfamily have been described as contributing to the sensing of cytoplasmic DNA [85]. Downstream of these DNA sensors, the ER-localized stimulator of IFN genes (STING) serves as a recruitment platform to activate TBK1, which in turn phosphorylates IRF3/7, resulting in the induction of type I IFNs [86].
Transmembrane receptors
Interferon production in immune cells can also be induced by the activation of transmembrane TLRs (Fig. 2a). The expression of TLRs is mainly restricted to tissues and cell types involved in innate immunity, such as macrophages and dendritic cells (DCs) [87, 88]. Toll-like receptors recognize structurally conserved molecules derived from microbes through their LRRs. While TLR3 senses viral dsRNA, TLR7 and TLR8 recognize ssRNA. Toll-like receptor 9 detects unmethylated CpG-containing oligonucleotides, typical of microbial DNA, and TLR4 responds to LPS, a cell wall component of Gram-negative bacteria (a detailed description of TLR ligands can be found in [2, 88]). These receptors are localized either in endosomal compartments (TLR3, 7, 8, 9,) or at the cell surface (TLR3, 4). They are thus devoted to the recognition of PAMPs upon phagocytosis or after lysis and release of material by a pathogen or an infected cell.
All TLRs except TLR3 trigger signaling cascades via MyD88. TLR3 associates instead with TRIF (TIR-domain-containing adapter inducing IFN-β), whereas TLR4 uses both TRIF and MyD88 [2]. Similarly to RLRs, engagement of the TRIF-dependent pathway downstream of TLR3 and TLR4 activates TBK1 and IKKε, leading to the phosphorylation of IRFs. Type I IFN induction by endoplasmic TLR7, TLR8, and TLR9 is instead mediated by an MyD88-dependent complex including IRAK1 and IKKα, which phosphorylate and thereby activate IRF7 [89].
The main producers of type I (and III) IFNs are plasmacytoid DCs, a population of circulating DCs [90]. These ‘specialized IFN-producing cells’ are characterized by rapid and potent IFN production, which is dependent on the activation of TLR7 and TLR9. Indeed, high constitutive expression of these TLRs and of the downstream signaling molecule IRF7 leads to a very efficient coupling of ligand detection to cytokine production [91]. Thus, plasmacytoid DC are particularly important to produce systemic type I IFN, which is crucial to alert the entire organism.
Type I IFN signaling
Interferon-α and -β share a common receptor, the IFN-α receptor (IFNAR), composed of a IFNAR1 and a IFNAR2 subunit (Fig. 2b) [64]. These cytokines can signal in virtually every cell, as the receptor is ubiquitously expressed. Ligand-induced receptor dimerization leads to the auto- and trans-phosphorylation of receptor-associated tyrosine kinases JAK1 (on IFNAR2) and TYK2 (on IFNAR1). Janus kinase 1 and Tyrosine kinase 2 then phosphorylate the intracellular domain of IFNAR, which creates docking sites for STAT1 and STAT2. These two transcription factors heterodimerize, bind to IRF9, and translocate to the nucleus (Fig. 2b) [92]. This heterotrimer [also known as IFN-stimulated gene factor 3 (ISGF3)] binds to genomic IFN-stimulated response elements (ISREs), thereby modulating the expression of numerous IFN-stimulated genes (ISGs). Interferon-stimulated genes encode factors involved in the antiviral and anti-inflammatory response, in immunomodulation, and factors endowed with pro-apoptotic and anti-proliferative activities (Fig. 2b) [61, 93].
Importantly, ISGs also code for proteins that augment IFN signaling by a positive feedback loop. Firstly, the expression of the nucleic acid receptors RIG-I, MDA5, DAI, and of TLRs is positively regulated by type I IFNs [94]. Secondly, ISGs encode signaling molecules involved in the IFN pathway, such as IRF7 and STAT1. Furthermore, all genes encoding type I IFNs contain ISREs in their promoters, resulting in a self-amplifying loop (Fig. 2b) [61, 95].
Although ISGF3 mediates the most studied and probably the major effects of type I IFNs, other STAT family members can participate in type I IFN signaling. Homo- and heterodimers of STAT1, STAT3, STAT4, STAT5, and STAT6 can play a role downstream of IFNAR [64]. Moreover, type I IFNs have also been described to activate other signaling pathways such as mitogen-activated protein and phosphatidylinositol-3 kinases [96].
Effects of type I IFNs
The following sections focus on the antiviral, pro-apoptotic, immunomodulatory, and in particular anti-inflammatory activities of type I IFNs. Emerging evidence indicates that IFN-β and the different α-subtypes can vary in these effects [97, 98]. This may be explained by the different binding properties of IFN-α and -β to IFNAR, influencing downstream signaling [97, 99]. Therefore, in some cases, we discuss the diverging activities of β- and α-IFNs.
Antiviral effects
Type I IFNs were initially discovered and best described as effector molecules in the protection against viral infections. They have the ability to induce an antiviral state in infected, but also in neighboring cells by autocrine and paracrine effects. Interferon-stimulated genes code for proteins that target virtually all steps of the virus life cycle, including entry, transcription, and translation, and lead to viral RNA degradation.
A prime example of an antiviral effector molecule is the dsRNA-dependent protein kinase R (PKR). Upon PKR activation, the α-subunit of the eukaryotic translational initiation factor 2 (eIF2α) is phosphorylated, which results in the inhibition of cellular as well as viral mRNA translation [100]. Further, the translational suppression triggered by PKR is also linked to IFN-induced cell-cycle arrest and apoptosis [101–103]. Not being central to this work, we refer the reader to excellent reviews in which the antiviral effects of IFNs have been summarized [103, 104].
Immunomodulation
In addition to their direct antiviral effects, type I IFNs play a major role in modulating innate and adaptive immunity. Notably, they are involved in regulating homeostasis, survival, differentiation, and trafficking of immune cells [105].
To start with, these cytokines play an important role in promoting the maturation of DCs, which are required for efficient activation of T cells [106, 107]. In fact, type I IFNs enhance antigen presentation, in particular by increasing the expression of costimulatory and MHC class I molecules [108, 109].
Type I IFN signaling has also been described to directly act on T cells as it can induce the production of IFN-γ through activation of STAT4, thereby favoring induction and maintenance of Th1 cells [110, 111]. Some reports claim that type I IFNs are, however, not sufficient to promote Th1 differentiation as the phosphorylation of STAT4 occurs only transiently. In both human and mouse cells, IL-12 has been proven to be necessary to induce adequate levels of the transcription factor T-bet, thereby leading to Th1 polarization [112, 113]. Nevertheless, IL-18 has been described to act with type I IFNs to activate STAT4 in the absence of IL-12 [114]. Type I IFNs are likely to also indirectly contribute to Th1 differentiation. They do so by inhibiting the ability of IL-4 to promote Th2 commitment and to antagonize Th1 development [115, 116]. Similarly, IFNs are involved in the negative regulation of Th17 development [117, 118]. Interferon-β has been described to induce the expression of IL-27 via STAT1. Interleukin-27 in turn downregulates IL-17 and IL-23, the latter being required for Th17 maintenance [119, 120].
In addition, type I IFNs seem to play a role in the survival of activated CD4+ T cells. Despite the fact that IFNs have been described as promoting cell growth arrest and even apoptosis [121, 122], these cytokines seem to protect T cells from apoptosis induction upon antigen encounter and enhance the development of central memory-like CD4+ T cells [123, 124].
Type I IFNs are also able to induce efficient CD8+ T cell responses by promoting DC cross-presentation. This process defines the ability of DCs to take up extracellular antigens by endocytosis and present them via MHCI to CD8+ T cells [107]. Furthermore, the augmented MHCI levels increase the presentation of peptides to cytotoxic T cells, thus leading to augmented recognition and killing of infected cells. Type I IFNs also support CD8+ T cell responses by directly enhancing effector functions, including IFN-γ secretion and expression of perforin and granzymes [125, 126]. An increase in the cytotoxic capacity has also been observed for NK cells and macrophages [127], highlighting the immunomodulatory effects of type I IFNs on these cell types as well. Notably, high levels of IL-15 induced in DCs by type I IFNs during their maturation lead to a selective stimulation and maintenance of memory CD8+ T cells [128, 129] and also favor NK cell development and differentiation [130]. Besides modulating T cell activation, type I IFNs also influence their migration by the induction of chemoattractants, such as CXCL10 and CXCL11 [131].
Direct and indirect effects of type I IFNs have also been described for B cells. These cytokines can enhance antibody production by promoting isotype switch and by inducing two TNF-family members important for B cell survival and homeostasis, namely B cell-activating factor (BAFF) and a proliferation-inducing ligand (APRIL) [132, 133]. In addition, treatment with type I IFNs has been shown to protect B cells from apoptosis [134]. Therefore, these cytokines seem to also contribute to the induction and maintenance of a potent humoral immune response [132]. Altogether, type I IFNs play a crucial role in the regulation of immune responses, by directly affecting functions of lymphocytes and antigen presentation.
Apoptosis
In contrast to the aforementioned anti-apoptotic effects in lymphocytes, type I IFNs have often been described to have anti-proliferative activity and to sensitize infected or transformed cells to apoptosis [122, 135]. These anti-proliferative effects were correlated with an inhibition of the NF-κB pathway [136], which, however, requires higher doses of IFNs than the induction of an antiviral state.
Although type I IFNs are insufficient per se to induce death in most cell types, they can sensitize cells to apoptosis via upregulation of caspases, pro-apoptotic, and tumor suppressor genes [137–139]. Furthermore, the induction of multiple antiviral proteins affects viral replication by significantly interfering with essential cellular processes. As an example, several studies have assigned a pro-apoptotic role to the IFN-inducible PKR, which is in part linked to the translation blockade induced by this kinase [140–142]. Type I IFNs have also been shown to have in vivo antitumor effects by promoting cell cycle arrest and apoptosis in transformed cells [135, 143, 144].
Moreover, engagement of RIG-I and MDA5 is known to induce pro-apoptotic signaling in addition to transcription of IFNs themselves [145, 146]. Similarly, apoptosis induction has been described downstream of TLR3 and 4 [142, 144, 147]. Interferons positively regulate the expression of these receptors, thereby rendering cells further susceptible to death. Altogether, certain effects of type I IFNs might cumulate, thereby sensitizing cells to apoptosis.
Autoimmunity and pro-inflammatory effects
Although beneficial for host immune responses, type I IFNs are also involved in the pathogenesis of certain diseases. Type I IFNs marry immuno-stimulatory to pro-apoptotic effects, which together make these cytokines particularly suited to favor autoimmune manifestations. Indeed, elevated production of inflammatory cytokines, such as type I IFNs, activates DCs. Moreover, these cytokines can promote tissue damage, therefore inducing the release of apoptotic material, which can be further phagocytosed and presented by activated antigen-presenting cells. These effects might be detrimental in individuals prone to develop autoimmunity due to an increased risk of activating autoreactive lymphocytes [148–150].
First indications that type I IFNs might be key players in autoimmunity came from the observation that the administration of these cytokines as antiviral or anti-proliferative therapy was associated with autoimmune manifestations [149, 151]. However, most of the time, symptoms resolved when the therapy was stopped, indicating that type I IFNs may initiate the development of autoimmunity but other factors are required to sustain the disease [97, 149]. Administration of IFN-α more than IFN-β was found to be associated with the onset of such autoimmune manifestations, which supports the hypothesis that there are differences in the action of type I IFN subtypes. Accordingly, reports suggest a more pronounced pathologic and driving role for systemic IFN-α in the development of autoimmunity [97, 98].
Indeed, an ‘IFN signature’, characterized by elevated IFN-α levels and expression of type I IFN-regulated genes, is found in systemic lupus erythematosus (SLE) patients and correlates with immune cell activation and clinical disease manifestations (Table 2). Systemic lupus erythematosus is a systemic autoimmune disorder that can affect any part of the body. This disease is characterized by the deposition of immune complexes leading to inflammation, and one of the pathologic features and diagnostic criteria of SLE is the presence of anti-nuclear antibodies.
Dying cells release cellular material, which is then recognized by PRRs, inducing an inflammatory response. Released intracellular components are also antigenic determinants for B cells, triggering antibody production against self-DNA and -RNA. This leads to the formation of immune complexes, which may even promote delivery of nucleic acids to immune cells, therefore enhancing IFN production [152–154]. The overproduction of type I IFNs results in a chronic activation of DCs, which further sustains the response of autoreactive lymphocytes in predisposed individuals, thus promoting the vicious circle [133].
The specific nature of the autoantigens in SLE may be instrumental in dictating the deleterious role of IFN-α in this disease. Cytoplasmic delivery of nucleic acids leads to activation of innate immune sensors, including the AIM2 inflammasome, whose components are further induced by type I IFNs, as discussed below. Under these circumstances, AIM2 inflammasome engagement may lead to enhanced IL-18 production, which has been involved in disease progression [155]. Interestingly, AIM2 belongs to the Ifi200 gene family, which in the mouse clusters within the Nba2 lupus susceptibility locus [156].
Although the strongest genetic factor associated to SLE is the human leukocyte antigen (HLA) region on chromosome 6 [157], many polymorphisms map to genes of the type I IFN pathway, further suggesting that these cytokines might play a detrimental role in this disease. In fact, IRF5 and TYK2 polymorphisms have been linked to increased risk of SLE [97, 158]. Moreover, a pathological role of type I IFNs is supported by studies from mouse lupus models showing that genetic ablation of Ifnar1 leads to less severe autoimmune manifestations [159, 160]. Against this commonly accepted view, few recent reports claim a protective role for type I IFNs in SLE, a surprising finding that requires more in-depth analysis [161, 162].
An IFN signature could also be detected in other autoimmune diseases like Sjögren’s syndrome and in a subgroup of rheumatoid arthritis patients [97, 163, 164]. Rheumatoid arthritis is mainly characterized by inflammation of the joints and autoantibody production, while Sjögren’s syndrome is a lymphoproliferative disease characterized by xerostomia (dry mouth) and xerophtalmia (dry eyes). Whereas, in SLE, a pathologic role for type I IFNs is established, a causative role for these cytokines in rheumatoid arthritis and Sjögren’s syndrome has not been determined, leaving open the possibility that they are produced in the inflammatory context as anti-inflammatory mediators [97, 163, 164]. In the case of Sjögren’s syndrome, this is supported by the fact that elevated levels of IL-1Ra have been detected in the cerebrospinal fluid of patients, and that oromucosal administration of IFN-α significantly increased unstimulated whole saliva flow, thus having a modest therapeutic effect [165, 166]. In the case of rheumatoid arthritis, a possible anti-inflammatory function of type I IFN is suggested by the protective role of IFN-β in animal models of collagen-induced as well as antigen-induced arthritis [167–169]. Moreover, an open phase I study, in which 12 patients with rheumatoid arthritis were treated with IFN-β subcutaneously, demonstrated modest clinical improvement [170]. However, a double-blinded placebo-controlled trial of IFN-β-1a (recombinant IFN-β produced in mammalian cells) in rheumatoid arthritis patients did not show any significant difference in radiological scores or clinical outcomes [171].
Anti-inflammatory effects of type I IFNs
Experimental evidence
Anti-inflammatory effects in sterile and infectious models
Multiple lines of evidence indicate that type I IFNs also exert anti-inflammatory functions. It has been known for a long time that type I IFN administration can be beneficial in a number of sterile inflammatory models, such as skin reactivity test, collagen-induced arthritis, or allotransplant rejection [59, 172–174]. Interferon-β also leads to a reduced reaction upon neutrophil-induced blood–brain barrier rupture [175] and in experimental autoimmune encephalomyelitis (EAE) [176], both models mimicking inflammatory aspects of the central nervous system found in MS. Interestingly, IFN has a direct stabilizing effect on cerebral endothelial cells lining the blood–brain barrier in in vitro and EAE models [176, 177]. These data suggest that the decreased passage of autoreactive lymphocytes through the endothelium might be one of the modes of action of IFN-β in MS, as discussed later.
In addition, while in virtually all viral and most bacterial infections type I IFNs are advantageous or even required for resistance [178], recent evidence shows that in some instances IFN induction can also have unfavorable outcomes. On the one hand, these effects can be due to a disproportionate production of type I IFN leading to an excessive inflammation, such as for cerebral malaria [179]. On the other hand, anti-inflammatory effects can dampen the immune response, which results in an exacerbated infection. This is the case for Francisella tularensis [180] and Listeria monocytogenes, as well as for the protozoan Leishmania amazonensis [181–183]. These infections were found to be less detrimental and also have reduced infectious burden in Ifnar −/− mice, showing that IFNs can indirectly favor microbial outgrowth.
Mycobacterium tuberculosis infection was also shown to be less severe in Ifnar −/− mice, with similar or reduced bacterial burdens as compared to control mice [184, 185]. Of note, patients infected with M. tuberculosis show an IFN signature, which might therefore have a pathogenic role during infection [186].
Finally, another line of evidence for the anti-inflammatory effects of IFNs comes from superinfections. In most cases, influenza-related deaths do not derive from the viral infection per se but from bacterial superinfections. A recent study revealed that secondary Streptococcus pneumoniae infections are worsened by the anti-inflammatory effects of influenza-induced type I IFNs [187]. Similarly, fungal infections can be exacerbated by type I IFNs. Mice pre-treated with type I IFN-inducing RNAs are more susceptible to candidiasis than untreated controls, an effect abolished in Ifnar −/− mice [188–190].
Type I IFN-dependent inhibition of IL-1 production
The first molecular mechanisms for these anti-inflammatory phenomena was proposed in 1990 when Schindler et al. showed that IFN-α has the ability to reduce IL-1 production [191, 192]. As discussed above, production of active IL-1 has to fulfill several conditions, and its activity is further regulated by the existence of a natural antagonist and a decoy receptor, IL-1RII. Additionally, IL-1 synthesis and inflammasome function are targeted by several negative regulatory mechanisms (reviewed in [46, 193]). Interestingly, type I IFNs have the ability to suppress pro-IL-1 levels and decrease the activity of NLRP1 and NLRP3 inflammasomes [188]. The effects of type I IFNs on IL-1 production have been known for decades, and several studies contributed to the understanding of the underlying mechanisms, as schematically illustrated in Fig. 3 [59, 188, 192, 194–196]. Of note, both IFN-α and -β are able to exert this anti-inflammatory effect.

Type I IFNs inhibit IL-1 production. Type I IFN signaling suppresses caspase-1-dependent IL-1β maturation by a STAT1-dependent mechanism, which might involve de novo transcription of a target protein. In addition, type I IFNs induce the expression of IL-1Ra, the natural IL-1R antagonist, and the anti-inflammatory cytokine IL-10. In murine macrophages, enhancement of IL-10 production by type I IFNs also requires the transcription factor STAT1. Interleukin-10, in turn, contributes to the induction of IL-1Ra and to the decrease of pro-IL-1 levels in a STAT3-dependent manner
In addition to reducing the expression of a number of inflammatory genes, including TNF and the IL-12/IL-23 subunit p40, type I IFNs reduce the levels of IL-1α and pro-IL-1β [120, 188, 192, 194–198]. Suppression of IL-1α and -β has been reported to occur both at the transcriptional as well as at the translational level [199, 200].
It recently became clear that this mechanism is relevant to infectious diseases. In fact, IL-1 is instrumental for resistance to both M. tuberculosis infection and systemic candidiasis, suggesting that IFN-dependent suppression of IL-1 production might be one mechanism explaining the detrimental effects of type I IFN in these models [184, 199, 201–204]. Interestingly, in the context of M. tuberculosis infection, adaptive immune cell-derived type II IFN has also been found to suppress IL-1 production [184]. Although the anti-inflammatory contribution by IFN-γ is controversial, emerging evidence highlights an inhibitory role of type II IFN on IL-1 levels, which seems to be dependent on the context and on the target cell type [184, 188, 192, 205].
Additionally, type I IFNs induce the expression of anti-inflammatory genes, such as IL-1Ra and IL-10 (Fig. 3) [194, 195, 206]. In fact, type I IFNs synergize with LPS to induce IL-10 [188, 207–210]. In the human system, this effect is thought to occur through the transcription factor STAT3 or the PI3K pathway [211, 212]. However, the use of macrophages derived from different knockout mice indicated that IL-10 induction downstream of type I IFNs depends on STAT1, most likely as part of the canonical signaling complex comprising STAT2 and IRF9 [188].
Interleukin-10 is one of the major anti-inflammatory cytokines, which increases IL-1Ra transcription and negatively regulates IL-1α and pro-IL-1β levels (Fig. 3) [188, 208, 209, 213, 214]. We found that the inhibitory effects of type I IFNs on pro-IL-1 levels in macrophages are in part achieved through autocrine IL-10 signaling [188]. Interleukin-10-dependent regulation of pro-IL-1 has been shown to mainly occur at the transcriptional level [208, 213, 214]. Furthermore, as IL-10 induces IL-1Ra [214], the question arises as to which extent the IFN-dependent IL-1Ra induction could also be mediated by autocrine IL-10 signaling.
Emphasizing their highly complex role in the balance between inflammatory and anti-inflammatory effects, it has been shown that type I IFNs have the ability to both positively and negatively regulate inflammasome activity. In fact, IFNs play an important pro-inflammatory role, for instance in sensitizing cells to certain bacterial inflammasome activators such as F. tularensis, and in the maintenance of AIM2 expression [215, 216]. Interferons are thus favoring the activity of the DNA-sensing AIM2 inflammasome, which is important upon infection by cytosolic bacteria and DNA viruses [215, 216]. In addition, caspase-1 itself, although being constitutively expressed, is a transcriptional target of type I IFN [155, 217]. Interestingly, IFNs not only seem to alter the activity of different inflammasomes but also the abundance of their substrates; while IL-1α and pro-IL-1β levels are decreased, the amount of pro-IL-18 is augmented [155, 198].
In contrast, exogenous type I IFNs are clearly anti-inflammatory, suppressing the activity of the NLRP3 inflammasome, which is a sensor for a broad range of stimuli [188]. This effect is transient and requires higher doses of IFNs than the one on pro-IL-1, at least in macrophages [188]. Although experimental data indicate that this inhibitory effect requires the transcription factor STAT1, the underlying mechanisms currently remain unclear. Interestingly, it was recently suggested that IFI16, which is strongly induced by type I IFN treatment, might be involved in this process [218]. In fact, knockdown of this DNA-binding protein in THP1 cells, a monocytic myeloid cell line, clearly increased the basal caspase-1 processing, indicating an augmented inflammasome activity.
Altogether, the complex effects of type I IFNs on inflammasome activity suggest that these cytokines favor the activity of the AIM2 inflammasome, possibly in concert with the production of IL-18, while impairing the activity of the NLRP3 inflammasome along with the production of IL-1. In the case of a viral infection, this might hinder development of a Th17 and favor a tailored Th1 type of response.
Established type I IFN-based clinical treatments
Therapeutic use of type I IFNs in viral and neoplastic diseases
Type I interferons are classical antiviral effector molecules. It is therefore not surprising that they have been studied as potential therapies for a number of viral diseases. Interferon-α is currently used in combination with other medications as a standard treatment for hepatitis B and C infections and may also reduce the occurrence of associated hepatocellular carcinoma [219–221]. Moreover, IFN-α was shown to be effective against human herpesvirus 8-driven Kaposi sarcoma and genital warts caused by papilloma viruses [222, 223]. Although its antiviral properties could per se explain the beneficial effects of IFN-α in these virus-associated neoplasms, the synergy with its ability to promote apoptosis, modulate immune functions, and alter tumor microenvironment is likely to contribute to this outcome [224]. On the one hand, IFN-dependent reduction of viral load could alone decrease inflammation by removing viral-derived PAMPs. On the other hand, IFN-α has also been successfully used in the therapy of malignancies, which are not considered to be associated with viral infections, where its effects are not mediated by the control of viral load. In particular, beneficial effects were observed in hairy cell leukemia, chronic myeloid leukemia, and melanoma [224–226].
Therapeutic use of type I IFNs in multiple sclerosis
Furthermore, for almost 20 years, IFN has been known to reduce inflammation in MS (Table 2) [97]. Multiple sclerosis is an inflammatory autoimmune disorder of the brain and the spinal cord and represents the second most common cause of neurological disability in young adults, exceeded only by trauma [227]. Affected patients suffer from relapses of neurological deficits that affect the white matter of the brain and the spinal cord. These are caused by autoreactive T cells that attack the myelin sheath, resulting in defective neural transmission. At least in the early stage of the disease, there is a more or less full recovery after each relapse, a form named relapsing-remitting phase (RR-MS). However, more than 80 % of patients with RR-MS, after 10–15 years, see their neurological symptoms progress unremittingly, without any relapses. At this stage, one speaks of secondary-progressive MS.
Interferon-β is used for the treatment of patients suffering from a first episode suggestive of MS (clinically isolated syndrome) and of patients with RR-MS. Pivotal studies have shown a reduction in the frequency of relapses by about 30 % over 2–3 years of treatment with IFN (reviewed in [228]). In a large clinical trial, administration of IFN-β was shown to marginally, but still significantly, decrease the rate of accumulation of disability over 3 years [229]. However, once the secondary progressive stage of the disease is reached, these therapies lose their efficacy, suggesting that IFN-β acts more on the inflammatory than on the neurodegenerative phase of the disease.
Interferon-α has also been studied as a treatment modality in MS. In one small study, IFN-α-2a (recombinant IFN-α produced in Escherichia coli) has been found to be effective in reducing exacerbations in MS patients as compared to placebo [230]. Additional encouraging findings have been reported in a trial of natural human leukocyte IFN-α in MS patients [231]. However, several reports have appeared on the potential of recombinant IFN-α to exacerbate the course of MS, including in patients with the progressive form of the disease [232, 233]. An MS-like syndrome has also been reported in a patient with chronic myeloid leukemia treated with IFN-α [234]. Thus, currently, IFN-α is not recommended as a treatment for MS.
Autoreactive lymphocytes are traditionally considered to be the primary immuno-pathogenic mechanism in MS for several reasons: (1) the adoptive transfer of myelin-specific autoreactive T cells induces EAE in mice [235], (2) perivascular cuffs of T lymphocytes are found in the center of demyelinating lesions in the white matter in human MS [236], (3) there is an oligo-clonal expansion of CD8+ T cells in brain lesions of MS patients, suggesting that these cells recognize an antigen in the central nervous system [237], (4) the central role of T cells is supported by genetic association of specific HLA alleles with MS [228], and (5) B cell infiltration and immunoglobulins are detected in MS lesions [238].
However, despite a sustained effort in research, the precise etiology(ies) of MS remain(s) to be established. Comprehensive studies of the adaptive immunity have not provided the whole picture of its immuno pathogenesis, which points to the fact that MS is a complex and/or heterogeneous disease. In particular, no consistent auto-antigen has been found. Therefore, an alternative model proposes a pivotal role of a dysregulated innate immune response that may impact the adaptive immune system by the generation of autoreactive T and B cells [239, 240]. Interestingly, experimental evidence highlights a role for inflammasome components and IL-1 in EAE progression, as mice deficient for caspase-1, ASC, NLRP3, IL-1RI, or IL-1α/β were significantly protected, while EAE in animals deficient for IL-1Ra showed exacerbated progression [20, 241–244]. The fact that IFN-β targets the innate at least as much as the adaptive arm of the immune system supports the aforementioned hypothesis [245].
Proposed mechanisms of IFN-β efficacy in multiple sclerosis
Originally, the efficacy of IFN-β in MS was attributed to its ability to decrease inflammatory cytokine production while increasing the expression of anti-inflammatory mediators [191, 192, 194–198, 206]. Of note, monocytes derived from IFN-β-treated MS patients produced significantly lower amounts of IL-1β upon ex vivo challenge with inflammasome activators, as compared to monocytes from healthy donors [188]. This suggests that the efficacy of IFN-β treatment might in part be caused by this anti-inflammatory mechanism. Moreover, the pro-apoptotic effects on activated autoreactive T cells, which cause damage in the central nervous system, were believed to contribute to IFN-β effectiveness in MS treatment [192, 194, 195, 246]. Later studies added IFN-β-activated STAT3 signaling as a main player in the anti-inflammatory effects. It does so by promoting the expression of Src homology phosphatase-1, an inhibitor of cytokine signaling, and blocking the activation of inflammatory mediators, such as NF-κB and STAT6 [247–249].
Moreover, IFN-β has been proposed to negatively influence trafficking of immune cells to the inflamed central nervous system. One mechanism involves the inhibition of lymphocyte egress from lymph nodes. Sphingosine 1-phosphate receptor-1 is required for this process and is negatively regulated by type I IFNs, thus reducing the number of circulating effector T cells [250]. In addition, IFN-β also decreases the ability of T cells to migrate to the central nervous system by stabilizing the blood–brain barrier, downregulating integrin expression on T cells, and affecting the function of matrix metalloproteases, which are important for cell migration and adhesion [177, 251–254].
More recently, the Th17-opposing effects of type I IFNs have gained attention. In fact, studies in MS patients and EAE have shown that Th17 cells are involved in these autoimmune manifestations [255]. Interferons could therefore also exert their beneficial effects in the treatment of MS by hindering the development of Th17, a possible mechanism being the reduction of IL-1 levels [120].
Only about two-thirds of MS patients respond well to treatment with IFN-β in terms of decreased relapse rates and fewer new brain and spinal cord lesions [256]. Identifying so-called ‘non-responders’ prior to the initiation of therapy is of obvious clinical interest, and multiple studies have looked at ways to predict a patient’s ‘responder’ status. One study of MS patients treated with IFN-β demonstrated that non‐responder and responder phenotypes differ in their ex vivo gene expression profiles as assessed by magnetic resonance imaging scanning and clinical disease activity. In particular, IL-8, an important chemotactic mediator recruiting neutrophils to sites of inflammation, was found to be significantly downregulated ex vivo and in vitro in IFN-β treated responders but not in non-responders. In addition, expression of a number of genes involved in either regulation of proliferation or apoptosis were modified in responders towards an anti-proliferative pro-apoptotic state. In non-responders, however, this shift was either absent or observed to a significantly lesser degree [257]. Another study has suggested that high concentration of IL-17F, another member of the IL-17 family, in the serum of patients with RR-MS is associated with non-responsiveness to IFN-β therapy [258]. Furthermore, it has been proposed that, in certain subpopulations of MS patients, IFN-β treatment could worsen the disease [259]. This differential response to therapy highlights the fact that MS is a heterogeneous disease and that other factors, such as genetic ones, are important players in the susceptibility to the treatment [238].
Adverse effects and drawbacks of IFN treatment
Just as any treatment, type I IFNs can have side effects. Up to 75 % of patients treated with IFN-β experience flu-like symptoms such as fever, headache, muscle pain, fatigue, and chills [260]. The most common observed laboratory abnormalities are elevation of liver enzymes and leukopenia. These changes are seldom serious, generally reversible, and rarely warrant the discontinuation of the treatment [261–267]. However, reports on fulminant liver failure as well as on unmasking of pre-existing autoimmune hepatitis or psoriasis do exist [268–270]. Hence, the regular monitoring of liver enzymes is recommended.
Reports of depression associated with IFN therapy (both IFN-α and -β) are well known. However, controversy still exists on whether it is caused by IFN. For instance, depression is quite common in MS patients irrespective of the treatment they are undergoing. A recent review has described 11 cases of severe depression with suicide attempts among patients treated with IFN-β who had no prior psychiatric history [271]. Yet, other studies found no evidence to support the claim that IFN-β can cause or exacerbate depression [272].
Occasionally, IFN therapy can lead to the development of autoimmune manifestations in the form of SLE, arthritis, and diabetes [97, 149, 151]. However, most of these complications resolve upon discontinuation of treatment. Rarely, a syndrome resembling thrombotic thrombocytopenic purpura with fever, thrombocytopenia, and renal failure has been reported in MS patients treated with IFN-β-1a [260].
In addition, chronic therapy with IFN-β can be associated with development of antibodies neutralizing IFN. In initial trials of different IFN-β preparations in MS patients, frequencies of neutralizing antibodies varied from 7 to 42 % [262, 273–275]. However, these percentages often depend on the method used to detect antibodies. It has previously been reported that up to 80 % of serum samples from patients treated with IFN-β for more than 1 year contained measurable amounts of neutralizing antibodies when assessed with an optimized assay [276]. It has been demonstrated that the presence of antibodies against IFN-β in the sera of MS patients may reduce the clinical efficacy of this medication as manifested by increased relapse rates [277]. Switching the therapy is recommended for neutralizing antibody-positive patients that have experienced worsening of their disease course. However, provided that the patient is without exacerbations on IFN-β treatment, it is generally not necessary to check the neutralizing antibody status.
Potential clinical treatments based on type I IFN
Excessive inflammatory reactions can originate from a disproportioned inflammatory response or from a malfunctioning control mechanism, and can have an acquired or a genetic origin. In fact, we define a wide range of pathological manifestations with the term ‘inflammatory disorders’, all sharing inflammatory features, but resulting from profoundly different causes [278]. Thus, the spectrum of inflammatory diseases spans from pure ‘auto-inflammatory’ ones, which are disorders without any involvement of the adaptive immune system, to classical autoimmune pathologies.
The origin of several auto-inflammatory diseases can be solely attributed to deregulated IL-1 signaling [27]. This is the case for diseases such as cryopyrin-associated periodic syndromes (CAPS; which include: familial cold auto-inflammatory syndrome, Muckle–Wells syndrome, and neonatal-onset multisystem inflammatory disease), where activating mutations within the NLRP3 gene lead to constitutive inflammasome activity [279]. The pathological role of excessive inflammasome activity and IL-1 signaling in this disorder has been irrevocably proven by the therapeutic efficacy of agents blocking IL-1 [280–285]. Given the clear molecular basis and the straightforward therapeutic approaches, we do not discuss such auto-inflammatory disorders in this review. Instead, we focus on diseases with more complex origin, with IL-1 being involved but not being the unique cause of the pathogenesis, and where novel therapeutic approaches are needed.
In the following paragraphs, we therefore discuss representative disorders with regard to the role of type I IFNs, inflammasomes and IL-1 (as summarized in Table 2).
Familial Mediterranean fever
Familial Mediterranean fever (FMF) is manifested by fever episodes along with local inflammatory reactions such as peritonitis, arthritis, skin rashes, and in some cases amyloidosis. Patients bear mutations in the Mediterranean fever (MEFV) gene, also known as pyrin, and such variants are encountered with higher frequency in populations from the Mediterranean basin and the Middle East [286, 287]. Although to date an excessive IL-1 production in FMF patients has not been consistently measured and the nature of mutations affecting pyrin is not firmly established, it is clear that these patients respond to IL-1 blocking biologics [27, 288–295]. Interestingly, IFN-α treatment was also found to be promising in the management of colchicine-resistant FMF patients [296]. This suggests that the ability of type I IFN to interfere with IL-1 production may be relevant in this therapeutic context.
Inflammatory bowel disease
Inflammatory bowel disease (IBD) is a heterogeneous group of inflammatory disorders of the intestinal tract, including Crohn’s disease and ulcerative colitis. Crohn’s disease is characterized by discontinuous and transmural inflammatory manifestations. In 2001, two independent studies showed a clear association of this disease with variants of NOD2, a member of the NLR family [297, 298]. Later studies identified a number of other genes as hotspots for mutations, including IL-23R, STAT3, immunity-related GTPase family M (IRGM), and autophagy-related 16-like 1 (ATG16L1) [299–303]. Nucleotide-binding oligomerization domain-containing protein 2 (NOD2), immunity-related GTPase family M protein (IRGM) and autophagy-related protein 16-1 (ATG16L1) control autophagy, which interestingly seems to be an important negative regulator of inflammasome activity as shown in IBD models [304–306]. Furthermore, Atg16L1-deficient mice show increased intestinal inflammation and IL-1 secretion [304, 306].
Whereas in Crohn’s disease the inflammation can be found anywhere in the digestive tract, ulcerative colitis is characterized by an inflamed large intestine and rectum, only in some cases spreading to the small intestine. A direct cause of ulcerative colitis is so far not known, but environmental factors, immune dysfunction, and a presumed genetic predisposition were proposed to contribute to disease pathology. Interestingly, IL-23R, and STAT3 are common risk loci associated with both ulcerative colitis and Crohn’s disease. Interleukin-23R also signals through the transcription factor STAT3, clearly identifying the IL-23 pathway as a key player in the development of IBD. Interleukin-23 plays a very important role in Th17 responses and has been associated with autoimmune disorders, thus highlighting the contribution of the adaptive immune branch to this disease. However, STAT3 is also implicated in signaling downstream of the anti-inflammatory IL-10, and several reports strongly link IBD to mutations affecting IL-10 signaling [301, 307, 308].
Besides the classical anti-inflammatory treatments, anti-TNF biologics have been successfully used in the management of IBD, and shown to prevent deleterious tissue damage [309]. Interestingly, type I IFNs have been reported to be beneficial in the treatment of ulcerative colitis [310, 311]. A proposed mechanism is the shift towards a Th1 polarization mediated by type I IFNs in a Th2-predominant disease like ulcerative colitis [312].
Behçet disease
Behçet disease is a chronic multifocal inflammatory disorder with mucocutaneous, ocular, vascular, skeletal, and central nervous system manifestations. Strong associations with specific HLA alleles have been identified, and also, albeit less pronounced, with IL-10 and IL-23 signaling pathways [313, 314]. Despite the strong link to certain HLAs and the important presence of CD4+ T cells in inflamed areas, direct contribution by these HLAs to a potential autoreactive response has not been determined [315].
For patients refractory to traditional immunosuppressive and anti-inflammatory therapies, encouraging results have been observed using TNF and IL-1 antagonist, and, interestingly, IFN-α [295, 316–319]. Therefore, both IL-1 inhibition and type I IFN treatment are helpful for the management of Behçet disease, suggesting that part of type I IFN efficacy in this context might be due to its suppression of IL-1 production.
Atopic disorders
Allergies are frequent inflammatory disorders originating from both genetic and environmental factors. One of the hypotheses to explain the widespread prevalence of these disorders in developed countries relies on the hygienic habits adopted by our society. Under these circumstances, the immune system would compensate its inactivity towards pathogens by attacking otherwise inert antigenic determinants, such as pollen-derived ones [320]. Nonetheless, several allergens are endowed with enzymatic functions, thus having immuno-stimulatory properties [321].
Atopic responses can be found in different tissues, including skin, airway mucosa, and conjunctiva, and are associated with Th2 polarization. In fact, Th2 inflammatory cytokines promote all the typical features of allergic reactions, such as excessive production of immunoglobulin E, mast cell activation, and eosinophilia. Antibody-allergen complexes are crucial to crosslink Fc-receptors on mast cells, thus leading to the release of pre-stored granules containing, among other substances, histamines.
Multiple links between IL-1 and allergy have been reported. Firstly, IL-1 supports the production of several chemokines and cytokines, which are involved in atopic reactions, and can synergize with other Th2 cytokines to activate mast cells [321–323]. Secondly, IL-1 is detected upon allergen challenge, and mast cells are capable of producing bioactive IL-1 [324, 325]. Besides their role in Th17 differentiation, inflammasome activators have also been suggested to promote Th2-biased polarization, although a specific role for inflammasome or IL-1 in this process has not been unambiguously shown [326–335]. Interestingly, IFN treatment alleviates asthma; this could rely on a type I IFN-induced Th2 to Th1 shift, and the suppression of IL-1 production could also contribute to this effect [336].
Cancer
Our understanding of the interplay between inflammation and cancer has profoundly changed. A decade ago, an increased inflammation within tumor environment was perceived as a potentially beneficial feature, able to attract and activate immune cells. However, there is now increasing evidence that inflammation can also exert detrimental effects, favoring tumor growth, angiogenic switch, and metastasis [337]. This makes IL-1 a double-edged sword, which in some settings favors tumor regression, but in others helps its progression (for a more detailed discussion, see [46, 338]).
Interferon-α therapy has proven efficacious for treating several virus-associated tumors, myeloproliferative disorders, hairy cell leukemia, and melanomas, particularly ulcerated ones, as discussed in the section “Therapeutic use of type I IFNs in viral and neoplastic diseases” [224, 226]. Besides its pro-apoptotic features, the anti-angiogeneic properties of type I IFNs could play an important role in the case of solid tumors [339].
Interestingly, the properties of IL-1 in promoting myeloproliferation through positive regulation of granulocyte–macrophage colony-stimulating factor, and in induction of vascular endothelial growth factor have been consistently reported [14, 340–343]. This suggests that the action of IFN-α on IL-1 production might also contribute to the suppressive effects of IFNs on myeloproliferative disorders and angiogenesis.
Concluding remarks
As discussed, IFN therapies are effective in the management of viral, malignant, and autoimmune disorders. It goes without saying that in diseases of viral origin, such as hepatitis, the contribution by IFNs in controlling viral load is crucial. Nonetheless, suppression of certain inflammatory components can be beneficial in all these diseases. Indeed, the anti-inflammatory effects of IFN-α and -β have been recognized in sterile and infectious experimental models as well as in the clinic. The strongest evidence for the surprising anti-inflammatory role of type I IFNs comes from the treatment of MS, where pro-apoptotic, immunomodulatory, and anti-inflammatory effects of IFN-β converge to a beneficial outcome. Whereas IFN-β is virtually uniquely used in the treatment of MS, the therapeutic administration of IFN-α ranges from viral infections to tumors. However, a comprehensive analysis of the possible molecular mechanisms behind this divergent use of type I IFNs is so far missing.
One important feature of inflammatory pathologies resides in the nature of the inflammation; at one extreme, we can place disorders of pure innate origin. In many of these pathologies, IL-1 plays an important role, as witnessed by the efficacy of treatments targeting this cytokine. It is, however, important to mention that in some auto-inflammatory syndromes, a possible contribution of autoreactive T and B cells has so far been disregarded. At the opposite end of the inflammatory disease spectrum are rare monogenic autoimmune disorders affecting the adaptive immune system, such as autoimmune polyendocrinopathy syndrome. Inbetween these extremes, there are disorders that involve both innate and adaptive immune cells. Recently, the effectiveness of anti-IL-1 treatment is becoming appreciated in disorders with such complex etiology as Behçet or Type 2 diabetes. These observations encourage extending the use of anti-IL-1 agents, which are specific, safe, and well tolerated [283–285].
In addition, experimental results point to a crucial contribution of IL-1 in a number of inflammatory syndromes, including those currently treated with IFNs. Given the strong ability of type I IFNs to dampen IL-1 production, it is conceivable that IFNs might be considered as anti-inflammatory agents in a broader range of complex inflammatory diseases where a role for IL-1 has been reported. The combined administration of type I IFNs together with IL-1 neutralizing agents might enforce the anti-inflammatory properties of IFNs, thus increasing treatment efficacy. Possibly, this may also allow the reduction of the administered dose of IFNs, thereby improving tolerability.
However, the effects of IFNs are pleiotropic and their use warrants caution because of their potential side effects. Instead of broadening the use of type I IFNs in the clinic, we might take advantage of the growing knowledge on the multiple anti-inflammatory effects of type I IFNs and use agents that specifically target selected pathways downstream of these cytokines. Such approaches might represent valuable alternatives to the use of IFNs. We have highlighted how therapies targeting IL-1 signaling can mimic one of the important anti-inflammatory effects of IFNs. Another example are sphingosine analogs, which have been recently approved for MS treatment and that act by sequestering lymphocytes within lymph nodes. Finally, it is possible that IL-1-blocking agents could be well complemented by or even synergize with treatments that target other specific aspects of inflammatory diseases and could therefore provide an even more efficient therapy.
References
Medzhitov R (2008) Origin and physiological roles of inflammation. Nature 454(7203):428–435
Kawai T, Akira S (2011) Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 34(5):637–650
Osorio F, Reis e Sousa C (2011) Myeloid C-type lectin receptors in pathogen recognition and host defense. Immunity 34(5):651–664
Elinav E et al (2011) Regulation of the antimicrobial response by NLR proteins. Immunity 34(5):665–679
Loo YM, Gale M Jr (2011) Immune signaling by RIG-I-like receptors. Immunity 34(5):680–692
Goshen I, Yirmiya R (2009) Interleukin-1 (IL-1): a central regulator of stress responses. Front Neuroendocrinol 30(1):30–45
Dinarello CA (2004) Infection, fever, and exogenous and endogenous pyrogens: some concepts have changed. J Endotoxin Res 10(4):201–222
Rasmussen AK, Bendtzen K, Feldt-Rasmussen U (2000) Thyrocyte-interleukin-1 interactions. Exp Clin Endocrinol Diabetes 108(2):67–71
Dower SK et al (1986) The cell surface receptors for interleukin-1 alpha and interleukin-1 beta are identical. Nature 324(6094):266–268
Stylianou E et al (1992) Interleukin 1 induces NF-kappa B through its type I but not its type II receptor in lymphocytes. J Biol Chem 267(22):15836–15841
Leung K et al (1994) The cytoplasmic domain of the interleukin-1 receptor is required for nuclear factor-kappa B signal transduction. J Biol Chem 269(3):1579–1582
Croston GE, Cao Z, Goeddel DV (1995) NF-kappa B activation by interleukin-1 (IL-1) requires an IL-1 receptor-associated protein kinase activity. J Biol Chem 270(28):16514–16517
O’Neill LA (2008) The interleukin-1 receptor/Toll-like receptor superfamily: 10 years of progress. Immunol Rev 226:10–18
Dinarello CA (2011) A clinical perspective of IL-1 beta as the gatekeeper of inflammation. Eur J Immunol 41(5):1203–1217
Dinarello CA (1996) Biologic basis for interleukin-1 in disease. Blood 87(6):2095–2147
Fantuzzi G, Dinarello CA (1996) The inflammatory response in interleukin-1 beta-deficient mice: comparison with other cytokine-related knock-out mice. J Leukoc Biol 59(4):489–493
Yazdi AS et al (2010) Nanoparticles activate the NLR pyrin domain containing 3 (Nlrp3) inflammasome and cause pulmonary inflammation through release of IL-1 alpha and IL-1 beta. Proc Natl Acad Sci USA 107(45):19449–19454
Chen CJ et al (2006) MyD88-dependent IL-1 receptor signaling is essential for gouty inflammation stimulated by monosodium urate crystals. J Clin Invest 116(8):2262–2271
Smith KA, Gilbride KJ, Favata MF (1980) Lymphocyte activating factor promotes T-cell growth factor production by cloned murine lymphoma cells. Nature 287(5785):853–855
Sutton C et al (2006) A crucial role for interleukin (IL)-1 in the induction of IL-17-producing T cells that mediate autoimmune encephalomyelitis. J Exp Med 203(7):1685–1691
Acosta-Rodriguez EV et al (2007) Interleukins 1 beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol 8(9):942–949
O’Sullivan BJ et al (2006) IL-1 beta breaks tolerance through expansion of CD25+ effector T cells. J Immunol 176(12):7278–7287
Hannum CH et al (1990) Interleukin-1 receptor antagonist activity of a human interleukin-1 inhibitor. Nature 343(6256):336–340
Carter DB et al (1990) Purification, cloning, expression and biological characterization of an interleukin-1 receptor antagonist protein. Nature 344(6267):633–638
Aksentijevich I et al (2009) An auto inflammatory disease with deficiency of the interleukin-1-receptor antagonist. N Engl J Med 360(23):2426–2437
Reddy S et al (2009) An auto inflammatory disease due to homozygous deletion of the IL1RN locus. N Engl J Med 360(23):2438–2444
Masters SL et al (2009) Horror autoinflammaticus: the molecular pathophysiology of auto inflammatory disease. Annu Rev Immunol 27:621–668
Netea MG et al (2009) Differential requirement for the activation of the inflammasome for processing and release of IL-1 beta in monocytes and macrophages. Blood 113(10):2324–2335
Hiscott J et al (1993) Characterization of a functional NF-kappa B site in the human interleukin 1 beta promoter: evidence for a positive autoregulatory loop. Mol Cell Biol 13(10):6231–6240
Bauernfeind FG et al (2009) Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol 183(2):787–791
Bauernfeind F et al (2011) Inflammasomes: current understanding and open questions. Cell Mol Life Sci 68(5):765–783
Guarda G et al (2011) Differential expression of NLRP3 among hematopoietic cells. J Immunol 186(4):2529–2534
Lu JY, Sadri N, Schneider RJ (2006) Endotoxic shock in AUF1 knockout mice mediated by failure to degrade proinflammatory cytokine mRNAs. Genes Dev 20(22):3174–3184
Fenton MJ et al (1987) Transcriptional regulation of the human prointerleukin 1 beta gene. J Immunol 138(11):3972–3979
Schindler R, Clark BD, Dinarello CA (1990) Dissociation between interleukin-1 beta mRNA and protein synthesis in human peripheral blood mononuclear cells. J Biol Chem 265(18):10232–10237
Schindler R, Gelfand JA, Dinarello CA (1990) Recombinant C5a stimulates transcription rather than translation of interleukin-1 (IL-1) and tumor necrosis factor: translational signal provided by lipopolysaccharide or IL-1 itself. Blood 76(8):1631–1638
Kaspar RL, Gehrke L (1994) Peripheral blood mononuclear cells stimulated with C5a or lipopolysaccharide to synthesize equivalent levels of IL-1 beta mRNA show unequal IL-1 beta protein accumulation but similar polyribosome profiles. J Immunol 153(1):277–286
Mosley B et al (1987) The interleukin-1 receptor binds the human interleukin-1 alpha precursor but not the interleukin-1 beta precursor. J Biol Chem 262(7):2941–2944
Thornberry NA et al (1992) A novel hetero dimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature 356(6372):768–774
Kuida K et al (1995) Altered cytokine export and apoptosis in mice deficient in interleukin-1 beta converting enzyme. Science 267(5206):2000–2003
Li P et al (1995) Mice deficient in IL-1 beta-converting enzyme are defective in production of mature IL-1 beta and resistant to endotoxic shock. Cell 80(3):401–411
Martinon F, Burns K, Tschopp J (2002) The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell 10(2):417–426
Narayan S et al (2011) Octacalcium phosphate crystals induce inflammation in vivo through interleukin-1 but independent of the NLRP3 inflammasome in mice. Arthritis Rheum 63(2):422–433
Fettelschoss A et al (2011) Inflammasome activation and IL-1beta target IL-1alpha for secretion as opposed to surface expression. Proc Natl Acad Sci USA 108(44):18055–18060
Martinon F, Mayor A, Tschopp J (2009) The inflammasomes: guardians of the body. Annu Rev Immunol 27:229–265
Davis BK, Wen H, Ting JP (2011) The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol 29:707–735
Suzuki T et al (2007) Differential regulation of caspase-1 activation, pyroptosis, and autophagy via Ipaf and ASC in Shigella-infected macrophages. PLoS Pathog 3(8):e111
Lightfield KL et al (2008) Critical function for Naip5 in inflammasome activation by a conserved carboxy-terminal domain of flagellin. Nat Immunol 9(10):1171–1178
Kofoed EM, Vance RE (2011) Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature 477(7366):592–595
Mariathasan S et al (2004) Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 430(6996):213–218
Boyden ED, Dietrich WF (2006) Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat Genet 38(2):240–244
Menu P, Vince JE (2011) The NLRP3 inflammasome in health and disease: the good, the bad and the ugly. Clin Exp Immunol 166(1):1–15
Tschopp J, Schroder K (2010) NLRP3 inflammasome activation: the convergence of multiple signalling pathways on ROS production? Nat Rev Immunol 10(3):210–215
Chen G et al (2009) NOD-like receptors: role in innate immunity and inflammatory disease. Annu Rev Pathol 4:365–398
Elinav E et al (2011) NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 145(5):745–757
Jeru I et al (2011) Identification and functional consequences of a recurrent NLRP12 missense mutation in periodic fever syndromes. Arthritis Rheum 63(5):1459–1464
Borghini S et al (2011) Clinical presentation and pathogenesis of cold-induced auto inflammatory disease in a family with recurrence of an NLRP12 mutation. Arthritis Rheum 63(3):830–839
Schattgen SA, Fitzgerald KA (2011) The PYHIN protein family as mediators of host defenses. Immunol Rev 243(1):109–118
Billiau A (2006) Anti-inflammatory properties of type I interferons. Antivir Res 71(2–3):108–116
Schlaak JF et al (2002) Cell-type and donor-specific transcriptional responses to interferon-alpha. Use of customized gene arrays. J Biol Chem 277(51):49428–49437
Honda K, Taniguchi T (2006) IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat Rev Immunol 6(9):644–658
Goodbourn S, Didcock L, Randall RE (2000) Interferons: cell signalling, immune modulation, antiviral response and virus countermeasures. J Gen Virol 81(Pt 10):2341–2364
Basler CF, Garcia-Sastre A (2002) Viruses and the type I interferon antiviral system: induction and evasion. Int Rev Immunol 21(4–5):305–337
Platanias LC (2005) Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol 5(5):375–386
Young HA, Bream JH (2007) IFN-gamma: recent advances in understanding regulation of expression, biological functions, and clinical applications. Curr Top Microbiol Immunol 316:97–117
Zhu J, Yamane H, Paul WE (2010) Differentiation of effector CD4 T cell populations. Annu Rev Immunol 28:445–489
Hwang ES (2010) Transcriptional regulation of T helper 17 cell differentiation. Yonsei Med J 51(4):484–491
Schroder K et al (2004) Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol 75(2):163–189
Coccia EM et al (2004) Viral infection and Toll-like receptor agonists induce a differential expression of type I and lambda interferons in human plasmacytoid and monocyte-derived dendritic cells. Eur J Immunol 34(3):796–805
Witte K et al (2010) IL-28A, IL-28B, and IL-29: promising cytokines with type I interferon-like properties. Cytokine Growth Factor Rev 21(4):237–251
Sheppard P et al (2003) IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat Immunol 4(1):63–68
Kotenko SV et al (2003) IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat Immunol 4(1):69–77
Uze G, Monneron D (2007) IL-28 and IL-29: newcomers to the interferon family. Biochimie 89(6–7):729–734
Nickolaus P, Zawatzky R (1994) Inhibition by interleukin-4 of constitutive beta interferon synthesis in mouse macrophages. J Virol 68(10):6763–6766
Bautista EM et al (2005) Constitutive expression of alpha interferon by skin dendritic cells confers resistance to infection by foot-and-mouth disease virus. J Virol 79(8):4838–4847
Lienenklaus S et al (2009) Novel reporter mouse reveals constitutive and inflammatory expression of IFN-beta in vivo. J Immunol 183(5):3229–3236
Pulverer JE et al (2010) Temporal and spatial resolution of type I and III interferon responses in vivo. J Virol 84(17):8626–8638
Taniguchi T, Takaoka A (2001) A weak signal for strong responses: interferon-alpha/beta revisited. Nat Rev Mol Cell Biol 2(5):378–386
Weber F et al (2006) Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J Virol 80(10):5059–5064
McCartney SA, Colonna M (2009) Viral sensors: diversity in pathogen recognition. Immunol Rev 227(1):87–94
Meylan E et al (2005) Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 437(7062):1167–1172
Merika M, Thanos D (2001) Enhanceosomes. Curr Opin Genet Dev 11(2):205–208
Schafer SL et al (1998) Regulation of type I interferon gene expression by interferon regulatory factor-3. J Biol Chem 273(5):2714–2720
Honda K et al (2005) IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 434(7034):772–777
Rathinam VA, Fitzgerald KA (2011) Innate immune sensing of DNA viruses. Virology 411(2):153–162
Ishikawa H, Barber GN (2011) The STING pathway and regulation of innate immune signaling in response to DNA pathogens. Cell Mol Life Sci 68(7):1157–1165
Hornung V et al (2002) Quantitative expression of toll-like receptor 1–10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. J Immunol 168(9):4531–4537
Takeda K, Kaisho T, Akira S (2003) Toll-like receptors. Annu Rev Immunol 21:335–376
Takeuchi O, Akira S (2009) Innate immunity to virus infection. Immunol Rev 227(1):75–86
Colonna M, Trinchieri G, Liu YJ (2004) Plasmacytoid dendritic cells in immunity. Nat Immunol 5(12):1219–1226
Colonna M, Krug A, Cella M (2002) Interferon-producing cells: on the front line in immune responses against pathogens. Curr Opin Immunol 14(3):373–379
Silvennoinen O et al (1993) Interferon-induced nuclear signalling by Jak protein tyrosine kinases. Nature 366(6455):583–585
Borden EC et al (2007) Interferons at age 50: past, current and future impact on biomedicine. Nat Rev Drug Discov 6(12):975–990
de Veer MJ et al (2001) Functional classification of interferon-stimulated genes identified using microarrays. J Leukoc Biol 69(6):912–920
Harada H, Taniguchi T, Tanaka N (1998) The role of interferon regulatory factors in the interferon system and cell growth control. Biochimie 80(8–9):641–650
Joshi S et al (2010) Mechanisms of mRNA translation of interferon stimulated genes. Cytokine 52(1–2):123–127
Crow MK (2010) Type I interferon in organ-targeted autoimmune and inflammatory diseases. Arthritis Res Ther 12(Suppl 1):S5
Choubey D, Moudgil KD (2011) Interferons in autoimmune and inflammatory diseases: regulation and roles. J Interferon Cytokine Res 31(12):857–865
Kalie E et al (2008) The stability of the ternary interferon-receptor complex rather than the affinity to the individual subunits dictates differential biological activities. J Biol Chem 283(47):32925–32936
Bowie AG, Unterholzner L (2008) Viral evasion and subversion of pattern-recognition receptor signalling. Nat Rev Immunol 8(12):911–922
Garcia MA, Meurs EF, Esteban M (2007) The ds RNA protein kinase PKR: virus and cell control. Biochimie 89(6–7):799–811
Hovanessian AG (2007) On the discovery of interferon-inducible, double-stranded RNA activated enzymes: the 2′-5′oligoadenylate synthetases and the protein kinase PKR. Cytokine Growth Factor Rev 18(5–6):351–361
Randall RE, Goodbourn S (2008) Interferons and viruses: an inter play between induction, signalling, antiviral responses and virus countermeasures. J Gen Virol 89(Pt 1):1–47
Sadler AJ, Williams BR (2008) Interferon-inducible antiviral effectors. Nat Rev Immunol 8(7):559–568
Mattei F, Schiavoni G, Tough DF (2010) Regulation of immune cell homeostasis by type I interferons. Cytokine Growth Factor Rev 21(4):227–236
Longhi MP et al (2009) Dendritic cells require a systemic type I interferon response to mature and induce CD4+ Th1 immunity with poly IC as adjuvant. J Exp Med 206(7):1589–1602
Le Bon A et al (2003) Cross-priming of CD8+ T cells stimulated by virus-induced type I interferon. Nat Immunol 4(10):1009–1015
Theofilopoulos AN et al (2005) Type I interferons (alpha/beta) in immunity and autoimmunity. Annu Rev Immunol 23:307–336
Gallucci S, Lolkema M, Matzinger P (1999) Natural adjuvants: endogenous activators of dendritic cells. Nat Med 5(11):1249–1255
Brinkmann V et al (1993) Interferon alpha increases the frequency of interferon gamma-producing human CD4+ T cells. J Exp Med 178(5):1655–1663
Krug A et al (2003) CpG-A oligonucleotides induce a monocyte-derived dendritic cell-like phenotype that preferentially activates CD8 T cells. J Immunol 170(7):3468–3477
Persky ME, Murphy KM, Farrar JD (2005) IL-12, but not IFN-alpha, promotes STAT4 activation and Th1 development in murine CD4+ T cells expressing a chimeric murine/human Stat2 gene. J Immunol 174(1):294–301
Berenson LS et al (2006) Distinct characteristics of murine STAT4 activation in response to IL-12 and IFN-alpha. J Immunol 177(8):5195–5203
Matikainen S et al (2001) IFN-alpha and IL-18 synergistically enhance IFN-gamma production in human NK cells: differential regulation of Stat4 activation and IFN-gamma gene expression by IFN-alpha and IL-12. Eur J Immunol 31(7):2236–2245
Rogge L et al (1997) Selective expression of an interleukin-12 receptor component by human T helper 1 cells. J Exp Med 185(5):825–831
Huber JP et al (2010) Cutting edge: Type I IFN reverses human Th2 commitment and stability by suppressing GATA3. J Immunol 185(2):813–817
Harrington LE et al (2005) Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol 6(11):1123–1132
Moschen AR et al (2008) Interferon-alpha controls IL-17 expression in vitro and in vivo. Immunobiology 213(9–10):779–787
Sweeney CM et al (2011) IL-27 mediates the response to IFN-beta therapy in multiple sclerosis patients by inhibiting Th17 cells. Brain Behav Immun 25(6):1170–1181
Ramgolam VS et al (2009) IFN-beta inhibits human Th17 cell differentiation. J Immunol 183(8):5418–5427
Geng Y et al (1995) Tumor suppressor activity of the human consensus type I interferon gene. Cytokines Mol Ther 1(4):289–300
Tanaka N et al (1998) Type I interferons are essential mediators of apoptotic death in virally infected cells. Genes Cells 3(1):29–37
Marrack P, Kappler J, Mitchell T (1999) Type I interferons keep activated T cells alive. J Exp Med 189(3):521–530
Davis AM et al (2008) Cutting edge: a T-bet-independent role for IFN-alpha/beta in regulating IL-2 secretion in human CD4+ central memory T cells. J Immunol 181(12):8204–8208
Curtsinger JM et al (2005) Type I IFNs provide a third signal to CD8 T cells to stimulate clonal expansion and differentiation. J Immunol 174(8):4465–4469
Le Bon A et al (2006) Direct stimulation of T cells by type I IFN enhances the CD8+ T cell response during cross-priming. J Immunol 176(8):4682–4689
Ronnblom L, Alm GV, Eloranta ML (2009) Type I interferon and lupus. Curr Opin Rheumatol 21(5):471–477
Zhang X et al (1998) Potent and selective stimulation of memory-phenotype CD8+ T cells in vivo by IL-15. Immunity 8(5):591–599
Mattei F et al (2001) IL-15 is expressed by dendritic cells in response to type I IFN, double-stranded RNA, or lipopolysaccharide and promotes dendritic cell activation. J Immunol 167(3):1179–1187
Di Sabatino A et al (2011) Role of IL-15 in immune-mediated and infectious diseases. Cytokine Growth Factor Rev 22(1):19–33
Groom JR, Luster AD (2011) CXCR3 ligands: redundant, collaborative and antagonistic functions. Immunol Cell Biol 89(2):207–215
Le Bon A et al (2001) Type I interferons potently enhance humoral immunity and can promote isotype switching by stimulating dendritic cells in vivo. Immunity 14(4):461–470
Jego G et al (2003) Plasmacytoid dendritic cells induce plasma cell differentiation through type I interferon and interleukin 6. Immunity 19(2):225–234
Badr G et al (2010) Type I interferon (IFN-alpha/beta) rescues B-lymphocytes from apoptosis via PI3Kdelta/Akt, Rho-A, NFkappaB and Bcl-2/Bcl(XL). Cell Immunol 263(1):31–40
Bekisz J et al (2010) Antiproliferative properties of type I and type II interferon. Pharmaceuticals 3(4):994–1015
Rath PC, Aggarwal BB (2001) Antiproliferative effects of IFN-alpha correlate with the downregulation of nuclear factor-kappa B in human Burkitt lymphoma Daudi cells. J Interferon Cytokine Res 21(7):523–528
Takaoka A et al (2003) Integration of interferon-alpha/beta signalling to p53 responses in tumour suppression and antiviral defence. Nature 424(6948):516–523
Fuertes Marraco SA et al (2011) Type I interferon drives dendritic cell apoptosis via multiple BH3-only proteins following activation by Poly IC in vivo. PLoS ONE 6(6):e20189
Eitz Ferrer P et al (2011) Induction of Noxa-mediated apoptosis by modified vaccinia virus Ankara depends on viral recognition by cytosolic helicases, leading to IRF-3/IFN-beta-dependent induction of pro-apoptotic Noxa. PLoS Pathog 7(6):e1002083
Lee SB, Esteban M (1994) The interferon-induced double-stranded RNA-activated protein kinase induces apoptosis. Virology 199(2):491–496
Gil J, Esteban M (2000) The interferon-induced protein kinase (PKR), triggers apoptosis through FADD-mediated activation of caspase 8 in a manner independent of Fas and TNF-alpha receptors. Oncogene 19(32):3665–3674
Hsu LC et al (2004) The protein kinase PKR is required for macrophage apoptosis after activation of Toll-like receptor 4. Nature 428(6980):341–345
Chawla-Sarkar M et al (2003) Apoptosis and interferons: role of interferon-stimulated genes as mediators of apoptosis. Apoptosis 8(3):237–249
Salaun B, Romero P, Lebecque S (2007) Toll-like receptors’ two-edged sword: when immunity meets apoptosis. Eur J Immunol 37(12):3311–3318
Haller O, Kochs G, Weber F (2006) The interferon response circuit: induction and suppression by pathogenic viruses. Virology 344(1):119–130
Besch R et al (2009) Proapoptotic signaling induced by RIG-I and MDA-5 results in type I interferon-independent apoptosis in human melanoma cells. J Clin Invest 119(8):2399–2411
Hasan UA et al (2007) Cell proliferation and survival induced by Toll-like receptors is antagonized by type I IFNs. Proc Natl Acad Sci USA 104(19):8047–8052
Colonna M (2006) Toll-like receptors and IFN-alpha: partners in autoimmunity. J Clin Invest 116(9):2319–2322
Hall JC, Rosen A (2010) Type I interferons: crucial participants in disease amplification in autoimmunity. Nat Rev Rheumatol 6(1):40–49
Akeno N et al (2011) IFN-alpha mediates the development of autoimmunity both by direct tissue toxicity and through immune cell recruitment mechanisms. J Immunol 186(8):4693–4706
Borg FA, Isenberg DA (2007) Syndromes and complications of interferon therapy. Curr Opin Rheumatol 19(1):61–66
Vallin H et al (1999) Anti-double-stranded DNA antibodies and immuno stimulatory plasmid DNA in combination mimic the endogenous IFN-alpha inducer in systemic lupus erythematosus. J Immunol 163(11):6306–6313
Bave U et al (2003) Fc gamma RIIa is expressed on natural IFN-alpha-producing cells (plasmacytoid dendritic cells) and is required for the IFN-alpha production induced by apoptotic cells combined with lupus IgG. J Immunol 171(6):3296–3302
Means TK et al (2005) Human lupus autoantibody-DNA complexes activate DCs through cooperation of CD32 and TLR9. J Clin Invest 115(2):407–417
Kahlenberg JM et al (2011) Inflammasome activation of IL-18 results in endothelial progenitor cell dysfunction in systemic lupus erythematosus. J Immunol 187(11):6143–6156
Panchanathan R et al (2010) Aim2 deficiency stimulates the expression of IFN-inducible Ifi202, a lupus susceptibility murine gene within the Nba2 autoimmune susceptibility locus. J Immunol 185(12):7385–7393
Martens HA et al (2009) An extensive screen of the HLA region reveals an independent association of HLA class I and class II with susceptibility for systemic lupus erythematosus. Scand J Rheumatol 38(4):256–262
Sigurdsson S et al (2005) Polymorphisms in the tyrosine kinase 2 and interferon regulatory factor 5 genes are associated with systemic lupus erythematosus. Am J Hum Genet 76(3):528–537
Santiago-Raber ML et al (2003) Type-I interferon receptor deficiency reduces lupus-like disease in NZB mice. J Exp Med 197(6):777–788
Braun D, Geraldes P, Demengeot J (2003) Type I Interferon controls the onset and severity of autoimmune manifestations in lpr mice. J Autoimmun 20(1):15–25
Hron JD, Peng SL (2004) Type I IFN protects against murine lupus. J Immunol 173(3):2134–2142
Wu X, Peng SL (2006) Toll-like receptor 9 signaling protects against murine lupus. Arthritis Rheum 54(1):336–342
Vakaloglou KM, Mavragani CP (2011) Activation of the type I interferon pathway in primary Sjogren’s syndrome: an update. Curr Opin Rheumatol 23(5):459–464
van der Pouw Kraan TC et al (2007) Rheumatoid arthritis subtypes identified by genomic profiling of peripheral blood cells: assignment of a type I interferon signature in a subpopulation of patients. Ann Rheum Dis 66(8):1008–1014
Harboe E et al (2009) Fatigue in primary Sjogren’s syndrome—a link to sickness behaviour in animals? Brain Behav Immun 23(8):1104–1108
Meijer JM et al (2007) The future of biologic agents in the treatment of Sjogren’s syndrome. Clin Rev Allergy Immunol 32(3):292–297
Tak PP (2004) IFN-beta in rheumatoid arthritis. Front Biosci 9:3242–3247
Ying F et al (2011) Type I IFN protects against antigen-induced arthritis. Eur J Immunol 41(6):1687–1695
van Holten J et al (2004) Treatment with recombinant interferon-beta reduces inflammation and slows cartilage destruction in the collagen-induced arthritis model of rheumatoid arthritis. Arthritis Res Ther 6(3):R239–R249
Tak PP et al (1999) The effects of interferon beta treatment on arthritis. Rheumatology (Oxford) 38(4):362–369
Vervoordeldonk MJ, Aalbers CJ, Tak PP (2009) Interferon beta for rheumatoid arthritis: new clothes for an old kid on the block. Ann Rheum Dis 68(2):157–158
Koltai M, Meos E (1973) Inhibition of the acute inflammatory response by interferon inducers. Nature 242(5399):525–526
Triantaphyllopoulos KA et al (1999) Amelioration of collagen-induced arthritis and suppression of interferon-gamma, interleukin-12, and tumor necrosis factor alpha production by interferon-beta gene therapy. Arthritis Rheum 42(1):90–99
Hirsch MS et al (1974) Immunosuppressive effects of an interferon preparation in vivo. Transplantation 17(2):234–236
Veldhuis WB et al (2003) Interferon-beta prevents cytokine-induced neutrophil infiltration and attenuates blood-brain barrier disruption. J Cereb Blood Flow Metab 23(9):1060–1069
Yu M et al (1996) Interferon-beta inhibits progression of relapsing-remitting experimental autoimmune encephalomyelitis. J Neuroimmunol 64(1):91–100
Kraus J et al (2004) Interferon-beta stabilizes barrier characteristics of brain endothelial cells in vitro. Ann Neurol 56(2):192–205
Trinchieri G (2010) Type I interferon: friend or foe? J Exp Med 207(10):2053–2063
Sharma S et al (2011) Innate immune recognition of an AT-rich stem-loop DNA motif in the Plasmodium falciparum genome. Immunity 35(2):194–207
Henry T et al (2010) Type I IFN signaling constrains IL-17A/F secretion by gammadelta T cells during bacterial infections. J Immunol 184(7):3755–3767
Auerbuch V et al (2004) Mice lacking the type I interferon receptor are resistant to Listeria monocytogenes. J Exp Med 200(4):527–533
O’Connell RM et al (2004) Type I interferon production enhances susceptibility to Listeria monocytogenes infection. J Exp Med 200(4):437–445
Xin L et al (2010) Type I IFN receptor regulates neutrophil functions and innate immunity to Leishmania parasites. J Immunol 184(12):7047–7056
Mayer-Barber KD et al (2011) Innate and adaptive interferons suppress IL-1 alpha and IL-1 beta production by distinct pulmonary myeloid subsets during Mycobacterium tuberculosis infection. Immunity 35(6):1023–1034
Manca C et al (2005) Hypervirulent M. tuberculosis W/Beijing strains upregulate type I IFNs and increase expression of negative regulators of the Jak-Stat pathway. J Interferon Cytokine Res 25(11):694–701
Berry MP et al (2010) An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature 466(7309):973–977
Shahangian A et al (2009) Type I IFNs mediate development of postinfluenza bacterial pneumonia in mice. J Clin Invest 119(7):1910–1920
Guarda G et al (2011) Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity 34(2):213–223
Worthington M, Hasenclever HF (1972) Effect of an interferon stimulator, polyinosinic: polycytidylic acid, on experimental fungus infections. Infect Immun 5(2):199–202
Jensen J, Vazquez-Torres A, Balish E (1992) Poly (I. C)-induced interferons enhance susceptibility of SCID mice to systemic candidiasis. Infect Immun 60(11):4549–4557
Reznikov LL et al (1998) Spontaneous and inducible cytokine responses in healthy humans receiving a single dose of IFN-alpha2b: increased production of interleukin-1 receptor antagonist and suppression of IL-1-induced IL-8. J Interferon Cytokine Res 18(10):897–903
Schindler R, Ghezzi P, Dinarello CA (1990) IL-1 induces IL-1. IV. IFN-gamma suppresses IL-1 but not lipopolysaccharide-induced transcription of IL-1. J Immunol 144(6):2216–2222
Guarda G, So A (2010) Regulation of inflammasome activity. Immunology 130(3):329–336
Huang Y, Blatt LM, Taylor MW (1995) Type 1 interferon as an antiinflammatory agent: inhibition of lipopolysaccharide-induced interleukin-1 beta and induction of interleukin-1 receptor antagonist. J Interferon Cytokine Res 15(4):317–321
Coclet-Ninin J, Dayer JM, Burger D (1997) Interferon-beta not only inhibits interleukin-1 beta and tumor necrosis factor-alpha but stimulates interleukin-1 receptor antagonist production in human peripheral blood mononuclear cells. Eur Cytokine Netw 8(4):345–349
Zang YC et al (2004) Regulation of differentiation and functional properties of monocytes and monocyte-derived dendritic cells by interferon beta in multiple sclerosis. Mult Scler 10(5):499–506
Byrnes AA et al (2001) Type I interferons and IL-12: convergence and cross-regulation among mediators of cellular immunity. Eur J Immunol 31(7):2026–2034
Nagai T et al (2003) Timing of IFN-beta exposure during human dendritic cell maturation and naive Th cell stimulation has contrasting effects on Th1 subset generation: a role for IFN-beta-mediated regulation of IL-12 family cytokines and IL-18 in naive Th cell differentiation. J Immunol 171(10):5233–5243
Novikov A et al (2011) Mycobacterium tuberculosis triggers host type I IFN signaling to regulate IL-1{beta} production in human macrophages. J Immunol 187(5):2540–2547
Radwan M et al (2010) Tyrosine kinase 2 controls IL-1 beta production at the translational level. J Immunol 185(6):3544–3553
Bellocchio S et al (2004) The contribution of the Toll-like/IL-1 receptor superfamily to innate and adaptive immunity to fungal pathogens in vivo. J Immunol 172(5):3059–3069
Vonk AG et al (2006) Endogenous interleukin (IL)-1 alpha and IL-1 beta are crucial for host defense against disseminated candidiasis. J Infect Dis 193(10):1419–1426
Fremond CM et al (2007) IL-1 receptor-mediated signal is an essential component of MyD88-dependent innate response to Mycobacterium tuberculosis infection. J Immunol 179(2):1178–1189
Mayer-Barber KD et al (2010) Caspase-1 independent IL-1 beta production is critical for host resistance to mycobacterium tuberculosis and does not require TLR signaling in vivo. J Immunol 184(7):3326–3330
Masters SL et al (2010) Regulation of interleukin-1 beta by interferon-gamma is species specific, limited by suppressor of cytokine signalling 1 and influences interleukin-17 production. EMBO Rep 11(8):640–646
Aman MJ et al (1994) Regulation of cytokine expression by interferon-alpha in human bone marrow stromal cells: inhibition of hematopoietic growth factors and induction of interleukin-1 receptor antagonist. Blood 84(12):4142–4150
Chang EY et al (2007) Cutting edge: involvement of the type I IFN production and signaling pathway in lipopolysaccharide-induced IL-10 production. J Immunol 178(11):6705–6709
de Waal Malefyt R et al (1991) Interleukin 10(IL-10) inhibits cytokine synthesis by human monocytes: an autoregulatory role of IL-10 produced by monocytes. J Exp Med 174(5):1209–1220
Fiorentino DF et al (1991) IL-10 inhibits cytokine production by activated macrophages. J Immunol 147(11):3815–3822
Berg DJ et al (1996) Enterocolitis and colon cancer in interleukin-10-deficient mice are associated with aberrant cytokine production and CD4(+) Th1-like responses. J Clin Invest 98(4):1010–1020
Wang H et al (2011) The role of glycogen synthase kinase 3 in regulating IFN-beta-mediated IL-10 production. J Immunol 186(2):675–684
Ziegler-Heitbrock L et al (2003) IFN-alpha induces the human IL-10 gene by recruiting both IFN regulatory factor 1 and Stat3. J Immunol 171(1):285–290
Wang P et al (1994) IL-10 inhibits transcription of cytokine genes in human peripheral blood mononuclear cells. J Immunol 153(2):811–816
Jenkins JK, Malyak M, Arend WP (1994) The effects of interleukin-10 on interleukin-1 receptor antagonist and interleukin-1 beta production in human monocytes and neutrophils. Lymphokine Cytokine Res 13(1):47–54
Fernandes-Alnemri T et al (2010) The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat Immunol 11(5):385–393
Rathinam VA et al (2010) The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat Immunol 11(5):395–402
Zwaferink H et al (2008) IFN-beta increases listeriolysin O-induced membrane permeabilization and death of macrophages. J Immunol 180(6):4116–4123
Veeranki S et al (2011) IFI16 protein mediates the anti-inflammatory actions of the type-I interferons through suppression of activation of caspase-1 by inflammasomes. PLoS ONE 6(10):e27040
Dienstag JL (2008) Hepatitis B virus infection. N Engl J Med 359(14):1486–1500
Foster GR (2010) Pegylated interferons for the treatment of chronic hepatitis C: pharmacological and clinical differences between peginterferon-alpha-2a and peginterferon-alpha-2b. Drugs 70(2):147–165
Vezali E et al (2011) Does interferon therapy prevent hepatocellular carcinoma in patients with chronic viral hepatitis? Clin Res Hepatol Gastroenterol 35(6–7):455–464
Yang J et al (2009) Interferon for the treatment of genital warts: a systematic review. BMC Infect Dis 9:156
Krown SE (2007) AIDS-associated Kaposi’s sarcoma: is there still a role for interferon alfa? Cytokine Growth Factor Rev 18(5–6):395–402
Kiladjian JJ, Mesa RA, Hoffman R (2011) The renaissance of interferon therapy for the treatment of myeloid malignancies. Blood 117(18):4706–4715
Habermann TM, Rai K (2011) Historical treatments of in hairy cell leukemia, splenectomy and interferon: past and current uses. Leuk Lymphoma 52(Suppl 2):18–20
Eggermont AM et al (2012) Ulceration and stage are predictive of interferon efficacy in melanoma: results of the phase III adjuvant trials EORTC 18952 and EORTC 18991. Eur J Cancer 48(2):218–25
Anderson DW et al (1992) Revised estimate of the prevalence of multiple sclerosis in the United States. Ann Neurol 31(3):333–336
Compston A, Coles A (2008) Multiple sclerosis. Lancet 372(9648):1502–1517
Kappos L et al (2007) Effect of early versus delayed interferon beta-1b treatment on disability after a first clinical event suggestive of multiple sclerosis: a 3-year follow-up analysis of the BENEFIT study. Lancet 370(9585):389–397
Durelli L et al (1994) Chronic systemic high-dose recombinant interferon alfa-2a reduces exacerbation rate, MRI signs of disease activity, and lymphocyte interferon gamma production in relapsing-remitting multiple sclerosis. Neurology 44(3 Pt 1):406–413
Squillacote D, Martinez M, Sheremata W (1996) Natural alpha interferon in multiple sclerosis: results of three preliminary series. J Int Med Res 24(3):246–257
Kinnunen E et al (1993) Effects of recombinant alpha-2b-interferon therapy in patients with progressive MS. Acta Neurol Scand 87(6):457–460
Larrey D et al (1989) Exacerbation of multiple sclerosis after the administration of recombinant human interferon alfa. JAMA 261(14):2065
Kataoka I et al (2002) Multiple sclerosis associated with interferon-alpha therapy for chronic myelogenous leukemia. Am J Hematol 70(2):149–153
Ben-Nun A, Wekerle H, Cohen IR (1981) The rapid isolation of clonable antigen-specific T lymphocyte lines capable of mediating autoimmune encephalomyelitis. Eur J Immunol 11(3):195–199
Lassmann H, Wekerle H (2006) The pathology of multiple sclerosis. In: Compston A (ed) Mc Alpine’s multiple sclerosis. Elsevier, London, pp 557–599
Babbe H et al (2000) Clonal expansions of CD8(+) T cells dominate the T cell infiltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. J Exp Med 192(3):393–404
Lucchinetti C et al (2000) Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol 47(6):707–717
Glass CK et al (2010) Mechanisms underlying inflammation in neurodegeneration. Cell 140(6):918–934
Fernandez M, Montalban X, Comabella M (2010) Orchestrating innate immune responses in multiple sclerosis: molecular players. J Neuroimmunol 225(1–2):5–12
Furlan R et al (1999) Caspase-1 regulates the inflammatory process leading to autoimmune demyelination. J Immunol 163(5):2403–2409
Shaw PJ et al (2010) Cutting edge: critical role for PYCARD/ASC in the development of experimental autoimmune encephalomyelitis. J Immunol 184(9):4610–4614
Gris D et al (2010) NLRP3 plays a critical role in the development of experimental autoimmune encephalomyelitis by mediating Th1 and Th17 responses. J Immunol 185(2):974–981
Matsuki T et al (2006) Abnormal T cell activation caused by the imbalance of the IL-1/IL-1R antagonist system is responsible for the development of experimental autoimmune encephalomyelitis. Int Immunol 18(2):399–407
Bhat R, Steinman L (2009) Innate and adaptive autoimmunity directed to the central nervous system. Neuron 64(1):123–132
Kaser A et al (1999) Interferon-beta 1b augments activation-induced T-cell death in multiple sclerosis patients. Lancet 353(9162):1413–1414
Christophi GP et al (2008) SHP-1 deficiency and increased inflammatory gene expression in PBMCs of multiple sclerosis patients. Lab Invest 88(3):243–255
Christophi GP et al (2009) Interferon-beta treatment in multiple sclerosis attenuates inflammatory gene expression through inducible activity of the phosphatase SHP-1. Clin Immunol 133(1):27–44
Dickensheets HL et al (1999) Interferons inhibit activation of STAT6 by interleukin 4 in human monocytes by inducing SOCS-1 gene expression. Proc Natl Acad Sci USA 96(19):10800–10805
Shiow LR et al (2006) CD69 acts downstream of interferon-alpha/beta to inhibit S1P1 and lymphocyte egress from lymphoid organs. Nature 440(7083):540–544
Kieseier BC, Archelos JJ, Hartung HP (2004) Different effects of simvastatin and interferon beta on the proteolytic activity of matrix metalloproteinases. Arch Neurol 61(6):929–932
Gilli F et al (2004) Neutralizing antibodies against IFN-beta in multiple sclerosis: antagonization of IFN-beta mediated suppression of MMPs. Brain 127(Pt 2):259–268
Muraro PA et al (2000) VLA-4/CD49d downregulated on primed T lymphocytes during interferon-beta therapy in multiple sclerosis. J Neuroimmunol 111(1–2):186–194
Muraro PA et al (2004) Decreased integrin gene expression in patients with MS responding to interferon-beta treatment. J Neuroimmunol 150(1–2):123–131
Becher B, Segal BM (2011) Th17 cytokines in autoimmune neuro-inflammation. Curr Opin Immunol 26:707–712
Arnason BG (1999) Immunologic therapy of multiple sclerosis. Annu Rev Med 50:291–302
Sturzebecher S et al (2003) Expression profiling identifies responder and non-responder phenotypes to interferon-beta in multiple sclerosis. Brain 126(Pt 6):1419–1429
Axtell RC et al (2010) T helper type 1 and 17 cells determine efficacy of interferon-beta in multiple sclerosis and experimental encephalomyelitis. Nat Med 16(4):406–412
Wang AG et al (2006) Early relapse in multiple sclerosis-associated optic neuritis following the use of interferon beta-1a in Chinese patients. Jpn J Ophthalmol 50(6):537–542
Walther EU, Hohlfeld R (1999) Multiple sclerosis: side effects of interferon beta therapy and their management. Neurology 53(8):1622–1627
The IFNB Multiple Sclerosis Study Group (1993) Interferon beta-1b is effective in relapsing-remitting multiple sclerosis. I. Clinical results of a multicenter, randomized, double-blind, placebo-controlled trial. Neurology 43(4):655–661
The IFNB Multiple Sclerosis Study Group, The University of British Columbia MS/MRI Analysis Group (1995) Interferon beta-1b in the treatment of multiple sclerosis: final outcome of the randomized controlled trial. Neurology 45(7):1277–1285
Paty DW, Li DK, UBC MS/MRI Study Group, The IFNB Multiple Sclerosis Study Group (1993) Interferon beta-1b is effective in relapsing-remitting multiple sclerosis. II. MRI analysis results of a multicenter, randomized, double-blind, placebo-controlled trial. Neurology 43(4):662–667
Jacobs LD, The Multiple Sclerosis Collaborative Research Group (MSCRG) et al (1996) Intramuscular interferon beta-1a for disease progression in relapsing multiple sclerosis. Ann Neurol 39(3):285–294
PRISMS (Prevention of Relapses and Disability by Interferon beta-1a Subcutaneously in Multiple Sclerosis) Study Group (1998) Randomised double-blind placebo-controlled study of interferon beta-1a in relapsing/remitting multiple sclerosis. Lancet 352(9139):1498–1504
European Study Group (1998) Placebo-controlled multicentre randomised trial of interferon beta-1b in treatment of secondary progressive multiple sclerosis. Lancet 352(9139):1491–1497
Reder AT et al (2010) Cross-sectional study assessing long-term safety of interferon-beta-1b for relapsing-remitting MS. Neurology 74(23):1877–1885
Tremlett HL, Oger J (2004) Elevated aminotransferases during treatment with interferon-beta for multiple sclerosis: actions and outcomes. Mult Scler 10(3):298–301
Yoshida EM et al (2001) Fulminant liver failure during interferon beta treatment of multiple sclerosis. Neurology 56(10):1416
Pulicken M et al (2006) Unmasking of autoimmune hepatitis in a patient with MS following interferon beta therapy. Neurology 66(12):1954–1955
Fragoso YD et al (2010) Severe depression, suicide attempts, and ideation during the use of interferon beta by patients with multiple sclerosis. Clin Neuropharmacol 33(6):312–316
Borras C et al (1999) Emotional state of patients with relapsing-remitting MS treated with interferon beta-1b. Neurology 52(8):1636–1639
Abdul-Ahad AK et al (1997) Incidence of antibodies to interferon-beta in patients treated with recombinant human interferon-beta 1a from mammalian cells. Cytokines Cell Mol Ther 3(1):27–32
Rudick RA, Multiple Sclerosis Collaborative Research Group (MSCRG) et al (1998) Incidence and significance of neutralizing antibodies to interferon beta-1a in multiple sclerosis. Neurology 50(5):1266–1272
The IFNB Multiple Sclerosis Study Group, The University of British Columbia MS/MRI Analysis Group (1996) Neutralizing antibodies during treatment of multiple sclerosis with interferon beta-1b: experience during the first three years. Neurology 47(4):889–894
Ross C, Danish Multiple Sclerosis Study Group et al (2000) Immunogenicity of interferon-beta in multiple sclerosis patients: influence of preparation, dosage, dose frequency, and route of administration. Ann Neurol 48(5):706–712
Sorensen PS et al (2003) Clinical importance of neutralising antibodies against interferon beta in patients with relapsing-remitting multiple sclerosis. Lancet 362(9391):1184–1191
McGonagle D, McDermott MF (2006) A proposed classification of the immunological diseases. PLoS Med 3(8):e297
Hoffman HM et al (2001) Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet 29(3):301–305
Hawkins PN et al (2004) Spectrum of clinical features in Muckle-Wells syndrome and response to anakinra. Arthritis Rheum 50(2):607–612
Goldbach-Mansky R et al (2006) Neonatal-onset multisystem inflammatory disease responsive to interleukin-1beta inhibition. N Engl J Med 355(6):581–592
Lachmann HJ et al (2009) Use of canakinumab in the cryopyrin-associated periodic syndrome. N Engl J Med 360(23):2416–2425
Lepore L et al (2010) Follow-up and quality of life of patients with cryopyrin-associated periodic syndromes treated with Anakinra. J Pediatr 157(2):310–315e1
Neven B et al (2010) Long-term efficacy of the interleukin-1 receptor antagonist anakinra in ten patients with neonatal-onset multisystem inflammatory disease/chronic infantile neurologic, cutaneous, articular syndrome. Arthritis Rheum 62(1):258–267
Kuemmerle-Deschner JB et al (2011) Efficacy and safety of anakinra therapy in pediatric and adult patients with the autoinflammatory Muckle-Wells syndrome. Arthritis Rheum 63(3):840–849
The International FMF Consortium (1947) Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. Cell 90(4):797–807
French Familial Mediterranean fever Consortium (1997) A candidate gene for familial Mediterranean fever. Nat Genet 17(1):25–31
Rozenbaum M et al (1992) Decreased interleukin 1 activity released from circulating monocytes of patients with familial Mediterranean fever during in vitro stimulation by lipopolysaccharide. J Rheumatol 19(3):416–418
Papin S et al (2007) The SPRY domain of pyrin, mutated in familial Mediterranean fever patients, interacts with inflammasome components and inhibits proIL-1beta processing. Cell Death Differ 14(8):1457–1466
Chae JJ et al (2011) Gain-of-function pyrin mutations induce NLRP3 protein-independent interleukin-1beta activation and severe autoinflammation in mice. Immunity 34(5):755–768
Mitroulis I et al (2008) Anakinra suppresses familial Mediterranean fever crises in a colchicine-resistant patient. Neth J Med 66(11):489–491
Calligaris L et al (2008) The efficacy of anakinra in an adolescent with colchicine-resistant familial Mediterranean fever. Eur J Pediatr 167(6):695–696
Ozen S et al (2011) Anti-interleukin 1 treatment for patients with familial Mediterranean fever resistant to colchicine. J Rheumatol 38(3):516–518
Meinzer U et al (2011) Interleukin-1 targeting drugs in familial Mediterranean fever: a case series and a review of the literature. Semin Arthritis Rheum 65(2):265–271
Bilginer Y, Ayaz NA, Ozen S (2010) Anti-IL-1 treatment for secondary amyloidosis in an adolescent with FMF and Behcet’s disease. Clin Rheumatol 29(2):209–210
Tweezer-Zaks N et al (2008) Interferon-alpha as a treatment modality for colchicine-resistant familial Mediterranean fever. J Rheumatol 35(7):1362–1365
Hugot JP et al (2001) Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature 411(6837):599–603
Ogura Y et al (2001) A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature 411(6837):603–606
Barrett JC et al (2008) Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat Genet 40(8):955–962
Fisher SA et al (2008) Genetic determinants of ulcerative colitis include the ECM1 locus and five loci implicated in Crohn’s disease. Nat Genet 40(6):710–712
Franke A et al (2008) Replication of signals from recent studies of Crohn’s disease identifies previously unknown disease loci for ulcerative colitis. Nat Genet 40(6):713–715
Cho JH (2008) The genetics and immunopathogenesis of inflammatory bowel disease. Nat Rev Immunol 8(6):458–466
Massey DC, Parkes M (2007) Genome-wide association scanning highlights two autophagy genes, ATG16L1 and IRGM, as being significantly associated with Crohn’s disease. Autophagy 3(6):649–651
Saitoh T et al (2008) Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 456(7219):264–268
Travassos LH et al (2010) Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol 11(1):55–62
Maeda S et al (2005) Nod2 mutation in Crohn’s disease potentiates NF-kappaB activity and IL-1beta processing. Science 307(5710):734–738
Franke A et al (2008) Sequence variants in IL10, ARPC2 and multiple other loci contribute to ulcerative colitis susceptibility. Nat Genet 40(11):1319–1323
Glocker EO et al (2009) Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N Engl J Med 361(21):2033–2045
Oussalah A, Danese S, Peyrin-Biroulet L (2010) Efficacy of TNF antagonists beyond one year in adult and pediatric inflammatory bowel diseases: a systematic review. Curr Drug Targets 11(2):156–175
Sumer N, Palabiyikoglu M (1995) Induction of remission by interferon-alpha in patients with chronic active ulcerative colitis. Eur J Gastroenterol Hepatol 7(7):597–602
Nikolaus S et al (2003) Interferon beta-1a in ulcerative colitis: a placebo controlled, randomised, dose escalating study. Gut 52(9):1286–1290
Tilg H, Kaser A (2004) Type I interferons and their therapeutic role in Th2-regulated inflammatory disorders. Expert Opin Biol Ther 4(4):469–481
de Menthon M et al (2009) HLA-B51/B5 and the risk of Behcet’s disease: a systematic review and meta-analysis of case-control genetic association studies. Arthritis Rheum 61(10):1287–1296
Remmers EF et al (2010) Genome-wide association study identifies variants in the MHC class I, IL10, and IL23R-IL12RB2 regions associated with Behcet’s disease. Nat Genet 42(8):698–702
Yurdakul S, Yazici H (2008) Behcet’s syndrome. Best Pract Res Clin Rheumatol 22(5):793–809
Sfikakis PP et al (2007) Anti-TNF therapy in the management of Behcet’s disease—review and basis for recommendations. Rheumatology (Oxford) 46(5):736–741
Melikoglu M et al (2005) Short-term trial of etanercept in Behcet’s disease: a double blind, placebo controlled study. J Rheumatol 32(1):98–105
Botsios C et al (2008) Resistant Behcet disease responsive to anakinra. Ann Intern Med 149(4):284–286
Kotter I et al (2004) The use of interferon alpha in Behcet disease: review of the literature. Semin Arthritis Rheum 33(5):320–335
Sheikh A, Strachan DP (2004) The hygiene theory: fact or fiction? Curr Opin Otolaryngol Head Neck Surg 12(3):232–236
Hammad H, Lambrecht BN (2008) Dendritic cells and epithelial cells: linking innate and adaptive immunity in asthma. Nat Rev Immunol 8(3):193–204
Lee HC, Ziegler SF (2007) Inducible expression of the proallergic cytokine thymic stromal lymphopoietin in airway epithelial cells is controlled by NFkappaB. Proc Natl Acad Sci USA 104(3):914–919
Allakhverdi Z et al (2007) Thymic stromal lymphopoietin is released by human epithelial cells in response to microbes, trauma, or inflammation and potently activates mast cells. J Exp Med 204(2):253–258
Rusznak C et al (2001) Interaction of cigarette smoke and house dust mite allergens on inflammatory mediator release from primary cultures of human bronchial epithelial cells. Clin Exp Allergy 31(2):226–238
Nakamura Y et al (2009) Mast cells mediate neutrophil recruitment and vascular leakage through the NLRP3 inflammasome in histamine-independent urticaria. J Exp Med 206(5):1037–1046
Okada S et al (1995) Potential role of interleukin-1 in allergen-induced late asthmatic reactions in guinea pigs: suppressive effect of interleukin-1 receptor antagonist on late asthmatic reaction. J Allergy Clin Immunol 95(6):1236–1245
Schmitz N, Kurrer M, Kopf M (2003) The IL-1 receptor 1 is critical for Th2 cell type airway immune responses in a mild but not in a more severe asthma model. Eur J Immunol 33(4):991–1000
Nakae S et al (2003) IL-1 is required for allergen-specific Th2 cell activation and the development of airway hypersensitivity response. Int Immunol 15(4):483–490
Johnson VJ, Yucesoy B, Luster MI (2005) Prevention of IL-1 signaling attenuates airway hyperresponsiveness and inflammation in a murine model of toluene diisocyanate-induced asthma. J Allergy Clin Immunol 116(4):851–858
Li T et al (2006) Pharmacokinetics and anti-asthmatic potential of non-parenterally administered recombinant human interleukin-1 receptor antagonist in animal models. J Pharmacol Sci 102(3):321–330
Wang CC et al (2006) Adenovirus expressing interleukin-1 receptor antagonist alleviates allergic airway inflammation in a murine model of asthma. Gene Ther 13(19):1414–1421
Idzko M et al (2007) Extracellular ATP triggers and maintains asthmatic airway inflammation by activating dendritic cells. Nat Med 13(8):913–919
Eisenbarth SC et al (2008) Crucial role for the Nalp3 inflammasome in the immunostimulatory properties of aluminium adjuvants. Nature 453(7198):1122–1126
Kool M et al (2008) Cutting edge: alum adjuvant stimulates inflammatory dendritic cells through activation of the NALP3 inflammasome. J Immunol 181(6):3755–3759
Kool M et al (2011) An unexpected role for uric acid as an inducer of T helper 2 cell immunity to inhaled antigens and inflammatory mediator of allergic asthma. Immunity 34(4):527–540
Simon HU et al (2003) Clinical and immunological effects of low-dose IFN-alpha treatment in patients with corticosteroid-resistant asthma. Allergy 58(12):1250–1255
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144(5):646–674
Apte RN et al (2006) The involvement of IL-1 in tumorigenesis, tumor invasiveness, metastasis and tumor-host interactions. Cancer Metastasis Rev 25(3):387–408
Sidky YA, Borden EC (1987) Inhibition of angiogenesis by interferons: effects on tumor- and lymphocyte-induced vascular responses. Cancer Res 47(19):5155–5161
Tanaka T et al (2000) Induction of VEGF gene transcription by IL-1 beta is mediated through stress-activated MAP kinases and Sp1 sites in cardiac myocytes. J Mol Cell Cardiol 32(11):1955–1967
El Awad B et al (2000) Hypoxia and interleukin-1beta stimulate vascular endothelial growth factor production in human proximal tubular cells. Kidney Int 58(1):43–50
Jung YD et al (2001) Vascular endothelial growth factor is upregulated by interleukin-1 beta in human vascular smooth muscle cells via the P38 mitogen-activated protein kinase pathway. Angiogenesis 4(2):155–162
Voronov E, Carmi Y, Apte RN (2007) Role of IL-1-mediated inflammation in tumor angiogenesis. Adv Exp Med Biol 601:265–270
Moosig F et al (2004) IL-1RA in refractory systemic lupus erythematosus. Lupus 13(8):605–606
Ostendorf B et al (2005) Preliminary results of safety and efficacy of the interleukin 1 receptor antagonist anakinra in patients with severe lupus arthritis. Ann Rheum Dis 64(4):630–633
Voronov E et al (2006) IL-1 beta-deficient mice are resistant to induction of experimental SLE. Eur Cytokine Netw 17(2):109–116
Davis LS, Hutcheson J, Mohan C (2011) The role of cytokines in the pathogenesis and treatment of systemic lupus erythematosus. J Interferon Cytokine Res 31(10):781–789
Acknowledgments
We thank F. Staehli and K. Maslowski, UNIL, Lausanne, for critical reading of the manuscript. Studies in the laboratory of Jürg Tschopp are funded by grants of the Swiss National Science Foundation, the EU Apo-Sys program, the Institute of Arthritis Research and the Louis-Jeantet Foundation.
Conflict of interest
The authors declare no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ludigs, K., Parfenov, V., Du Pasquier, R.A. et al. Type I IFN-mediated regulation of IL-1 production in inflammatory disorders. Cell. Mol. Life Sci. 69, 3395–3418 (2012). https://doi.org/10.1007/s00018-012-0989-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-012-0989-2