
Abstract
Cholesterol homeostasis is among the most intensely regulated processes in biology. Since its isolation from gallstones at the time of the French Revolution, cholesterol has been extensively studied. Insufficient or excessive cellular cholesterol results in pathological processes including atherosclerosis and metabolic syndrome. Mammalian cells obtain cholesterol from the circulation in the form of plasma lipoproteins or intracellularly, through the synthesis of cholesterol from acetyl coenzyme A (acetyl-CoA). This process is tightly regulated at multiple levels. In this review, we provide an overview of the multiple mechanisms by which cellular cholesterol metabolism is regulated. We also discuss the recent advances in the post-transcriptional regulation of cholesterol homeostasis, including the role of small non-coding RNAs (microRNAs). These novel findings may open new avenues for the treatment of dyslipidemias and cardiovascular diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
For over 100 years, cholesterol has been extensively studied. Since its discovery in 1815 by the French chemist, M.E. Chevreul, the structure has been revealed, the biosynthetic pathway determined, and the feedback mechanisms that regulate cholesterol metabolism explained [1]. An important constituent of mammalian cell membranes, cholesterol serves to modulate membrane fluidity and permeability. Cholesterol is also the precursor of all steroid hormones and bile acids and plays a key role in membrane trafficking, transmembrane signaling processes, as well as cell proliferation [2, 3]. Despite its undeniable importance, however, abnormal levels of cholesterol can have serious cellular consequences and may lead to diseases such as atherosclerosis and type II diabetes [4, 5]. Because of this, cells have developed complex mechanisms to regulate the abundance and distribution of sterols within cells.
Vertebrate cells contain cholesterol and although different tissues have characteristic patterns of cholesterol metabolism, the basic pattern is similar in all cells. Animal cells acquire cholesterol endogenously, through a highly regulated process beginning with acetyl coenzyme A (acetyl-CoA) [6, 7], and exogenously, from the circulation in the form of apolipoprotein B-containing lipoproteins, such as low-density lipoprotein (LDL) [8, 9]. LDL particles are delivered to peripheral cells by receptor-mediated endocytosis and hydrolyzed to free cholesterol in the lysosome [8, 9]. Intracellular cholesterol levels are tightly controlled by feedback mechanisms that operate at both transcriptional and post-transcriptional levels [10, 11]. When intracellular levels of cholesterol are low, the ER-bound sterol regulatory element-binding proteins (SREBPs) coordinate the transcriptional activation of 3-hydroxy-3methylglutaryl coenzyme A reductase (HMGCR, the rate limiting enzyme of cholesterol biosynthesis) and almost all down-stream enzymes of the mevalonate (MVA)-pathway [12]. In addition to sterol metabolism, the MVA-pathway is important for the production of dimethylallyl pyrophosphate (DMAPP) and isopentenyl pyrophosphate (IPP), which serves as the basis for the biosynthesis of molecules used in processes as diverse as terpenoid synthesis, protein prenylation, cell membrane maintenance, hormones, protein anchoring, and N-glycosylation [11]. SREBPs also activate the transcription of the LDL receptor (LDLr), which leads to an increase in cellular cholesterol uptake [11].
The liver X receptor (LXR) nuclear hormone receptors also contribute to cholesterol homeostasis. LXRs are important transcriptional regulators of genes involved in the response to cholesterol excess by activating the transcription of genes involved in reverse cholesterol transport (RCT), such as the ABC transporters, ATP-binding cassette transporter A1 (ABCA1) and ATP-binding cassette transporter G1 (ABCG1) [13–15].
In addition to the classical transcriptional regulators of cholesterol metabolism, members of a class of non-coding RNAs, termed microRNAs (miRNAs), have lately been identified as important post-transcriptional regulators of cholesterol homeostasis. In particular, several groups have shown that microRNA-33 (miR-33) regulates cholesterol efflux and high-density lipoprotein (HDL) biogenesis by down-regulating the expression of ABCA1 and ABCG1 [16–20]. In addition, miR-33 also inhibits the translation of several genes involved in fatty acid oxidation, including carnitine palmitoyltransferase 1A (CPT1a), carnitine O-octanoyltransferase (CROT), hydroxyacyl-CoA dehydrogenase/3-ketoacyl-CoA thiolase/enoyl-CoA hydratase (trifunctional protein), beta subunit (HADHB), and AMP-activated protein kinase (AMPK), thereby reducing fatty acid degradation [21, 22]. Other miRNAs such as miR-122, miR-370, miR-378/378*, miR-125a, miR-27 and miR-355 have also been shown to regulate cholesterol homeostasis and fatty acid metabolism [23–29].
While significant advances have been made in understanding the balance between cholesterol synthesis and transport, the mechanisms surrounding cholesterol homeostasis remain incompletely understood. In this review, we summarize the general aspects of cholesterol metabolism, as well as the current evidence surrounding the critical role of miRNAs in regulating cholesterol balance. In particular, we will focus on the contribution of miR-33a and miR-33b in the epigenetic regulation of cholesterol homeostasis.
Cholesterol synthesis and trafficking
Cholesterol synthesis
Mammalian cells acquire cholesterol mainly from the diet and endogenous biosynthesis. The dietary intake of cholesterol is limited, and therefore the physiological requirements for cholesterol are supplied mostly through de novo synthesis. Almost all cells are involved in the synthesis of cholesterol, with the liver accounting for as much cholesterol as the extrahepatic tissues combined [30]. Cholesterol is synthesized in the endoplasmic reticulum (ER) and cytoplasm from acetyl-CoA through the MVA-pathway [31]. The rate-limiting enzyme of this pathway is HMGCR, which catalyzes the conversion of HMG-CoA to mevalonic acid and is a common target for cholesterol lowering drugs, such as statins [32]. In the first committed step of cholesterol biosynthesis, squalene is converted to lanosterol. From this precursor, cholesterol is synthesized in a 19-step process involving the activity of nine different enzymes. The last steps of cholesterol synthesis have been divided into two pathways and can proceed via lathosterol and 7-dehydrocholesterol (Kandutsch–Russell pathway) [33] or via desmosterol to cholesterol (Bloch pathway) [6, 7, 31]. These pathways share the same enzymatic steps but differ in the stage at which the C-24 bond is reduced.
Following synthesis, cholesterol and a considerable fraction of its precursors, including lathosterol and desmosterol, leave the ER in order to maintain low sterol levels. The newly synthesized cholesterol is targeted to the plasma membrane via non-vesicular mechanisms to become available to extracellular acceptors; additionally, the newly synthesized cholesterol may access other sites, such as the endosomes [34]. In order to prevent over-accumulation of free cholesterol in the plasma and intracellular membranes, cholesterol is converted to cholesterol esters (CEs) primarily by the enzyme, acyl-CoA acyltransferase (ACAT) [35]. Cholesterol esters are stored as cytosolic lipid droplets and can be liberated by a different enzyme, cholesterol ester hydrolase (CEH). This cholesterol/cholesterol ester cycle occurs rapidly, and releases free cholesterol to be trafficked to other intracellular compartments [36].
De novo cholesterol and its precursors play essential roles in many cellular and developmental pathways. Both isoprenoids and post-squalene sterol intermediates serve as precursor molecules for the biosynthesis of many end-products, such as dolichol, coenzyme Q, isopentenyl-tRNA, and heme a. Cholesterol is also a major component of the plasma membrane and is needed for the formation of caveolae and lipid rafts [37]. In addition, cholesterol serves as the precursor for metabolites, such as bile acids, steroids, and fat-soluble vitamins. These important functions of cholesterol are underscored by an increasing number of genetic disorders that are attributed to mutations in cholesterol biosynthetic enzymes [38]. The prototype of these disorders, RSH/Smith–Lemli–Opitz syndrome (RSH/SLOS), is caused by the deficiency of 7-dehydrocholesterol reductase, and results in decreased cholesterol levels [39]. More recently, other multiple malformation syndromes, including desmosterolosis and lathosterolosis, have been characterized [40].
Unlike the liver and peripheral tissues, almost 95% of cholesterol is provided by de novo synthesis in the brain [41]. Only small amounts of cholesterol can be delivered to the brain from the periphery through high-density lipoproteins (HDLs). These lipoproteins can cross the blood–brain barrier (BBB), whereas larger lipoproteins, such us low-density lipoproteins and very low density lipoproteins (LDL/VLDL), are unable to do so. Approximately 70% of the brain’s cholesterol is associated with the myelin sheath [42, 43]. Studies performed in culture cells have demonstrated that oligodendrocytes have the highest capacity of cholesterol synthesis, followed by astrocytes, which synthesize at least three times more cholesterol than neurons. Despite this, neurons have a particularly high demand for cholesterol to form and maintain the axons, dendrites, and synaptic connections [44, 45]. In neurons, LDL receptor-related protein (LRP) serves as a neuronal receptor for astrocyte-produced ApoE-containing lipid particles [46]. Many neurological diseases, including Alzheimer disease (AD) and Huntington’s disease, have been associated with defective cholesterol metabolism in the brain [47]. Indeed, the e4 allele of the ApoE gene was identified as the strongest genetic risk factor for AD [48, 49].
Cholesterol influx and endosomal trafficking
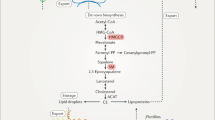
Aside from de novo synthesis, cells obtain cholesterol from the uptake of circulating plasma lipoproteins through the LDL receptor pathway [50]. Cholesterol obtained from food is initially transported from the small intestine to the liver from which it is delivered to the rest of the body. For long-distance transfer between cells in the circulatory system, enterocytes and hepatocytes package cholesterol and cholesteryl esters into lipoproteins, complex spherical particles that range in size and composition (for review see [51]). Briefly, dietary cholesterol is absorbed by enterocytes of the small intestine where it is packaged, along with triglycerides, into chylomicrons. As the chylomicrons reach the circulation via the lymph some of the triglycerides are hydrolyzed by lipoprotein lipase and the chylomicron remnants are taken up by hepatocytes. In turn, hepatocytes secrete lipids in VLDL particles that are processed in the circulation into LDL, the main lipoprotein that delivers cholesterol to the peripheral cells. The uptake of LDL and other ApoE/ApoB containing lipoproteins occurs through the LDLr and is a classic example of receptor-mediated endocytosis [8] (Fig. 1). The LDL that is bound to the cell surface LDLr is endocytosed by clathrin-coated vesicles and transported to acidic endocytic compartments, where cholesterol esters are hydrolyzed by acid lipase [9]. The LDLr dissociates from LDL in the early endosome (due to the lower pH) and is recycled back to the plasma membrane by vesicular mechanisms that depend on small Rab GTPases, such as Rab8 [52] and Rab11 [53]. The non-recycled contents of the early endosome, namely cholesterol, proceed to the late endosome/lysosome and upon release, are delivered to other membranes, such as the plasma membrane, ER, and mitochondria. Two proteins, Niemann–Pick disease, type C1 and C2 (NPC1 and NPC2, respectively), are crucial for moving cholesterol out of the late endosomal system and into the cytosol. A deficiency of either protein leads to Niemann–Pick type C (NPC) disease, which is characterized by an accumulation of free cholesterol in late endocytic organelles [54]. Although the exact roles of these proteins are not clear, it is predicted that NPC2, a small soluble protein, accepts cholesterol in the late endosomal lumen and transports it to membrane-bound NPC1, which helps transfer cholesterol out of the endosomal system [55]. The LDL-cholesterol can also be transported out of the late endosomal system by vesicular mechanisms mediated by Rab7 [56] and Rab9 [57].

Hepatic cellular cholesterol trafficking and compartmentalization. Mammalian cells synthesize cholesterol from acetyl-coA through the mevalonate pathway in the endoplasmic reticulum (ER). The newly synthesized cholesterol can be transported to subcellular membranes by the biosynthetic secretory pathway via the Golgi or by non-vesicular pathways with the help of cholesterol transfer proteins, such as SCP-2, StAR, and caveolae. Excess cholesterol in the ER is esterified by acetyl-coA cholesterol acetyltransferase (ACAT) to be stored in lipid droplets. In addition, cells acquire cholesterol from the circulation in the form of apolipoprotein B-containing lipoproteins, particularly low-density lipoprotein (LDL). The circulating LDL particles are internalized through the LDL receptor (LDLr) and are transported to the early endosomes. The cholesterol is subsequently transported to the late endosome (LE) and the lysosome (LY) where cholesterol esters are hydrolyzed by acid lipase (AL). The LDL receptors are recycled to the plasma membrane. Cholesterol exit from the late endosome is mediated by Niemann–Pick disease, type C1 and 2 (NPC1 and NPC2) proteins. Other sterol-binding proteins, such as MLN64 and OSBP-related protein 1 (ORP1), and the Rab GTPases are also involved in late endosomal trafficking, however, the mechanism by which these proteins transport cholesterol is not well characterized. EE early endosome, MVB multivesicular body, CEH cholesterol ester hydrolase
Intracellular cholesterol transport
Cholesterol is transported throughout the cell by moving as part of the membrane in vesicular transport processes or in a non-vesicular manner [58]. While vesicular transport plays an important role in some cholesterol transport processes, the major mode of intracellular cholesterol transport is non-vesicular [59]. Because cholesterol is insoluble, non-vesicular transport is mediated by diffusible carrier proteins, which have hydrophobic cavities to bind and protect cholesterol from the cytosol. Alternatively, non-vesicular transport may also involve the diffusion of cholesterol from one membrane to another [60]. Although the identity of sterol carriers remains to be determined, several proteins have been implicated in transporting cholesterol throughout the cell, including sterol carrier protein 2 (SCP-2), caveolin, the ORP (oxysterol-binding protein-related protein) family, and the START [StAR (steroidogenic acute regulatory protein)-related lipid transport] domain proteins [61].
SCP-2 is a small, soluble lipid transfer protein that is capable of transporting sterols and a variety of other lipids between membranes in vitro [62]. Many studies demonstrate that SCP-2 can transfer cholesterol between biological membranes [63], and a number of papers show that cellular cholesterol distribution and trafficking are altered when SCP-2 expression is increased or decreased [64]. Several studies have also shown a role for caveolins, membrane proteins that associate with cholesterol and spingolipids, in cholesterol transport [65, 66]. It is thought that caveolins form a chaperone complex with cholesterol and that these intracellular lipid particles transport cholesterol throughout the cell [67, 68]; however, some studies have failed to find evidence that this complex delivers newly synthesized cholesterol [69]. The ORP family members were first identified as cytosolic receptors for 25-hydroxycholesterol [70] and several members have been shown to mediate cholesterol trafficking in the late endosomal system, including ORP-9, ORP4-S, ORP-1L, and ORP5. OSBP, the founding member, was originally isolated because of its ability to bind oxysterols. It has subsequently been found that it also binds cholesterol [71]. ORP9 binds and transports cholesterol between membranes and may play a role in cholesterol transport between the ER and the Golgi [72]. ORP4-S associates with intermediate filaments and its overexpression decreases esterification of LDL-derived cholesterol [71]. Other studies have shown that ORP-1L localizes to late endosomal surfaces, and enhances LXR reporter activity when overexpressed [73]. Recently, it was also found that ORP5 plays a role in the non-vesicular transport of cholesterol from the late endosome to the ER [74].
Lastly, a large family of proteins containing START domains has been shown to bind lipids and play an important role in intracellular trafficking. The START domain proteins are located in the cytoplasm, bound to membranes or in the nucleus, and to date, 16 mammalian START domain proteins have been identified [61, 75]. StAR, the prototypic member of this family, is required for the transfer of cholesterol from the outer to the inner mitochondria membrane. Various other START domain proteins have also been identified in the intracellular transport of cholesterol, yet their exact roles remain to be determined [76]. Markedly, the ceramide transfer protein (CERT), which contains a START domain, plays a key role in the non-vesicular transport of ceramide from the ER to the Golgi [77]. Furthermore, MLN64/StarD3 has been implicated in transferring cholesterol from the endolysosomal compartment to the mitochondria, due to its localization and its topology [78]. Indeed, Charman et al. [79] recently showed that MLN64 mediates the transport of cholesterol to the mitochondria independently of NPC1.
Cholesterol efflux
Cholesterol removal is imperative for cholesterol homeostasis and serves to prevent cholesterol over-accumulation in cells. Because cells cannot degrade cholesterol, excess cholesterol must be removed and transported to the liver for reutilization and excretion in a process that is traditionally referred to as reverse cholesterol transport (RCT) [80]. HDL serves as the major acceptor for cellular cholesterol released from extrahepatic tissues, and is inversely correlated with the risk of atherosclerotic cardiovascular disease [81].
Excess cellular cholesterol is eliminated by diffusion-mediated or apolipoprotein-mediated mechanisms [82–84]. During passive diffusion, the removal of cholesterol is mediated by the exchange of cholesterol between the cell membrane and HDL. Many factors are likely involved in this process, such as cholesterol compartments in the plasma membrane and HDL structure. The enzyme lecithin-cholesterol acyltransferase (LCAT) has been suggested to also participate in this process [84]. LCAT is found on the surface of lipoproteins (namely, HDL) and converts free cholesterol into cholesterol esters by transferring fatty acids from phosphatidylcholine (PC) to unesterified cholesterol [85]. The cholesterol esters are then sequestered into the core of the lipoprotein and transported to the liver and steroidogenic tissues where they are selectively removed by the scavenger receptor class B type 1 (SRB1) [86]. The role of LCAT, however, is still controversial since overexpression of LCAT in mice does not increase RCT [87] and cholesterol efflux can still occur in LCAT deficient conditions [88]. A large amount of CEs that are formed within HDL are also transferred to the triglyceride rich lipoprotein (TRL) by the cholesteryl ester transfer protein (CETP). The remnants of this protein are converted to LDL and removed by the LDL receptor pathway or directly removed in the liver for excretion into bile [89]. Two ATP-binding cassette transporters, ATP-binding cassette, subfamily G, member 5 (ABCG5) and ATP-binding cassette, subfamily G, member 8 (ABCG8), are half-transporters that function as heterodimers to mediate the excretion of cholesterol into bile [90]. Mutations in either gene lead to sitosterolemia, a recessive disease that is characterized by increased cholesterol absorption and impaired biliary secretion [91, 92].
Another mechanism for cholesterol removal is mediated mostly by ApoA-I (the major apoprotein of HDL) and leads to the assembly of discoidal HDL along with phospholipids and cholesterol [93]. ABCA1 initiates HDL formation in the liver and is the first step in RCT [94]. This finding was brought to light when the mutation in Tangier disease, a condition characterized by low plasma HDL, was mapped to the Abca1 gene [95]. Several mechanisms have been proposed to facilitate ABCA1-mediated cholesterol efflux to lipid-poor ApoA-I. In one mechanism, ApoA-I forms complexes with phospholipids and cholesterol at the cell surface, and is subsequently internalized and targeted to intracellular compartments, while lipidation of ApoA-I occurs as part of the retroendocytosis pathway [96–98]. Although several studies have suggested an important role for late endosomes and lysosomes in the ABCA1-mediated lipid efflux pathway [99, 100], the mechanism by which this occurs remains unclear. In another mechanism, ApoA-I forms complexes with phospholipids and cholesterol at the plasma membrane. This process is also mediated by ABCA1 and is supported by multiple lines of evidence [101–103]. Vedhachalam et al. have recently proposed a scheme in which a small pool of ApoA-I binds to ABCA1, enhancing net phospholipid translocation and thus, membrane strain [103]. The membrane strain is relieved by the bending and creation of exovesiculated lipid domains, which promotes the binding of ApoA-I. In addition to ApoA-I, this lipid transport pathway has broad specificity for multiple exchangeable apolipoproteins including ApoA-II, ApoE, ApoC and ApoA-IV [104]. While ABCA1 is the predominant factor needed for cholesterol efflux to ApoA-I and the formation of pre-β HDL, other factors, such as LCAT, and the ABC transporters, ABCG1 and ATP-binding cassette, subfamily 8, member 4 (ABCG4), are needed for the maturation of HDL [105]. After pre-β HDL is formed it must undergo further lipidation. Studies have shown that ABCA1 and ABCG1 and ABCG1/ABCG4 heterodimers synergistically mediate cholesterol efflux to HDL [106, 107], however, the mechanism by which these transporters promote cholesterol efflux remains to be determined.
ABCA1 also plays an important role in regulating cholesterol metabolism in the brain. Several studies have demonstrated that ABCA1 facilitates cholesterol efflux of central nervous system (CNS) cholesterol to ApoE [108]. Changes in ABCA1 expression also modulate the amount of Aβ peptide, most probably in a cholesterol dependent manner. ABCA1 deficiency exacerbates amyloidogenesis, whereas ABCA1 overexpression ameliorates amyloid plaque formation [109, 110].
Transcriptional regulation of cholesterol metabolism
Cholesterol homeostasis is regulated in a tightly controlled manner by a family of membrane-bound transcription factors called SREBPs. SREBPs are members of the basic helix-loop-helix leucine zipper (bHLH-Zip) family and activate the expression of a variety of genes required for cholesterol, fatty acid, triglyceride, and phospholipid uptake and synthesis [10, 111, 112]. In mammals, there are three SREBP isoforms: SREBP-1a and SREBP-1c, encoded by Srebp-1, and SREBP-2, encoded by Srebp-2 [10, 113]. The SREBP transcription factors differ in their tissue-specific expression, their target gene selectivity, and the relative potencies of their trans-activation domains [10, 111, 112]. While SREBP-1a is a potent activator of all SREBP-responsive genes, SREBP-1c preferentially enhances the transcription of genes involved in fatty acid synthesis. Conversely, SREBP-2 activates genes involved in cholesterol biosynthesis [10, 111, 112].
SREBPs are synthesized as inactive precursors that are attached to the ER membrane in a hairpin fashion [114, 115]. Following translation, SREBPs bind to the SREBP cleavage-activating protein (SCAP), a transmembrane protein that serves as an escort protein and sterol sensor [116]. In mammals, ER exit is controlled by the regulated binding of SCAP to the insulin-induced gene, INSIG [117, 118]. This ER anchor protein binds to the sterol-sensing domain of SCAP and prevents the SCAP/SREBP complex from entering the COPII-dependent ER-Golgi trafficking pathway [118]. Similar to SREBP-1a and -1c, SREBP-2 is controlled in a highly regulated negative feedback mechanism by downstream products of the cholesterol biosynthetic pathway [10, 119]. When intracellular cholesterol levels are low, the SCAP–INSIG interaction is disrupted, allowing SCAP to interact with the COPII trafficking complex and deliver SREBP to the Golgi where it is cleaved by two membrane-bound proteases, site-1 protease (S1P) and site-2 protease (S2P) [120]. These sequential cleavages release the active, N-terminal fragment from the membrane so that it can translocate to the nucleus and activate the expression of cholesterol related genes, such as HMGCR and the LDLr [121]. During sterol-rich conditions, SREBP is retained in the ER by the SCAP/INSIG complex and can no longer be processed by the proteases in the Golgi. Because of this, the active SREBPs cannot enter the nucleus and cholesterol uptake and synthesis decline [122, 123].
Aside from being regulated at the processing level, SREBPs are also regulated transcriptionally and post-transcriptionally in an isoform specific manner. SREBP-2 and SREBP-1c are subject to distinct forms of transcriptional regulation, while SREBP-1a is constitutively expressed at low levels in the liver and most mammalian tissues [124]. Both SREBP-2 and SREBP-1c are regulated in a feed-forward manner by the sterol response elements (SRE) in their promoter regions. In the presence of sterols, these nSREBPs activate the expression of their own genes. Contrastingly, there is evidence that the SREBP-1c promoter (which activates fatty acid synthesis) is regulated by several other factors, including insulin and oxysterols [125, 126]. Additionally, studies have shown that post-transcriptional modifications also affect the activity of SREBPs. Nuclear SREBPs are modified by ubiquitination and are subject to degradation by the 26 S proteasome [127]. SREBP-1 and SREBP-2 can also be modified by SUMO-1, which has been reported to repress transcriptional activity [128]. Alternatively, the acetylation of SREBP-1a and SREBP-1c at lysine residues by the transcriptional activator, p300, can increase the stability of these transcription factors [124]. More recently, it has been shown that sirtuin 1 (SIRT1), a NAD+-dependent deacetylase, plays a critical role in controlling SREBP activity. SIRT1 down regulates SREBP during fasting by deacetylating SREBP, resulting in ubiquitination and protein instability, and therefore decreases in target gene expression [129]. Furthermore, SRT2183, a chemical activator of SIRT, inhibits SREBP-dependent gene expression in vitro and in vivo, decreasing hepatic lipid and cholesterol levels and reducing liver steatosis in obese mice [129].
In addition to SREBPs, the LXRs also contribute to cholesterol homeostasis. The LXRs, LXRα and LXRβ, are nuclear receptors that form heterodimers with retinoid X receptors (RXRs) and are activated by a variety of sterol metabolites [130]. Whereas LXRβ is expressed ubiquitously, LXRα is primarily expressed in the liver, adipose tissue, and macrophages and thus plays an important role in lipid metabolism [131]. LXRs activate the transcription of genes involved in cholesterol efflux, including ABCA1, ABCG1, and ABCG5/8. When cholesterol levels surpass the biosynthetic rate, a feed-forward pathway is initiated that leads to the clearance of cholesterol. The binding of oxysterols to LXRs triggers a conformational change in the receptor that enhances interaction with co-activator proteins, thereby facilitating transcription of the aforementioned target genes and RCT [15]. Specifically, ABCA1 and ABCG1 promote cellular cholesterol efflux to HDL and ApoA-I [132], while ABCG5/8 promote cholesterol excretion into bile [133]. ABCA1 also plays a crucial role in HDL formation, and thus atherosclerosis susceptibility. In vivo studies show that LXR-deficient mice accumulate sterols in their tissues and develop accelerated atherosclerosis [134], whereas synthetic LXR agonists stimulate ABCA1 expression and reverse cholesterol transport [135, 136].
Post-transcriptional regulation of cholesterol metabolism
Cholesterol metabolism is also regulated at the post-transcriptional level by various mechanisms including the degradation and phosphorylation of HMGCR, the proprotein convertase subtilisin/kexin type 9 (PCSK9)-dependent degradation of the LDLr, the IDOL-dependent ubiquitination of the LDL receptor and the ABCA1, ABCG1 and NPC1 post-transcriptional repression by the miR-33.
HMGCR degradation/phosphorylation and squalene monooxygenase degradation
The HMGCR protein, a rate-limiting enzyme in cholesterol biosynthesis, displays a long half-life, thereby maximizing mevalonate production for sterol and isoprenoid synthesis [11]. Products from both pathways (sterols and isoprenoids) feed back to control HMGCR activity including the degradation of the enzyme [120, 137]. Although it has been known for some time that products of the MVA-pathway control the stability of HMGCR activity, DeBose-Boyd et al. have recently uncovered the molecular details behind this post-transcriptional regulation [120, 137–139]. A key discovery was the finding that the sterol intermediates, lanosterol and 24,25-dihydrolanosterol, rather than the end product cholesterol, stimulate HMGCR degradation [137]. Lanosterol is the first sterol synthesized from mevalonate flux into the sterol pathway [31]. Accumulation of lanosterol stimulates binding of the HMGCR to INSIG, resulting in the ubiquitination of HMGCR on two cytosolic lysine residues and subsequent degradation by the 26S proteasome [137]. In addition to lanosterol, oxysterols promote the ubiquitination and degradation of the HMGCR [138]. Although it remains to be determined, oxysterols are presumably acting through their ability to bind INSIG and to stimulate INGIG binding to HMGCR [138].
High concentrations of mevalonate are required for the rapid degradation of HMGCR. In addition to lanosterol, the complete and rapid degradation of HMGCR requires additional non-sterol isoprenoid signaling [140]. The requirement for this signal can be filled by addition of geranylgeraniol, a 20-carbon isoprenyl alcohol, but not the 15-carbon alcohol farnesol [140]. It is not known, however, which geranylgeraniol-derived metabolite is acting to control the HMGCR degradation.
In addition to the degradation of HMGCR, it has recently been shown that cholesterol accelerates the proteosomal degradation of squalene monooxygenase (SM), the enzyme that catalyzes the first oxygenation step in cholesterol synthesis [141]. Unlike HMGCR, SM degradation is not mediated by INSIG, 24,25-didydrolanosterol, or side-chain oxysterols, but rather by cholesterol itself [141]. This finding suggests an additional control point in cholesterol metabolism beyond HMGCR.
In 1973, David Gibson’s group reported that a soluble fraction from rat liver caused a time-dependent inactivation of microsomal HMGCR activity in the presence of ATP [142]. Years later AMPK was characterized and identified as a kinase that was able to phosphorylate the HMGR at Ser871 and inhibit its activity [143, 144]. Elegant confirmation that AMPK had a role in regulation of HMGCR in intact cells was obtained by the Brown and Goldstein laboratory [145]. They expressed recombinant Chinese hamster HMGCR by stable transfection of UT-2 cells, a mutant of CHO cells that lacks endogenous reductase. In cells expressing the wild type enzyme, activation of AMPK resulted in total cessation of sterol synthesis, whereas this effect was completely abolished in cells expressing a Ser871-Ala mutant. Rodwell’s group confirmed Ser871 as the site of phosphorylation and inactivation of HMGCR by AMPK [146].
PCSK9
PCSK9 enhances the degradation of the LDL receptor [147, 148]. Recent advances have revealed a large number of genetic variants of PCSK9 that may modulate plasma cholesterol levels either positively or negatively, therefore influencing the risk of atherosclerosis. The PCSK9 gene is 25-kb long and comprises 12 exons and 11 introns. It encodes a 692-amino acid serine protease formerly called neural apoptosis regulated convertase-1 (NARC1), which is a member of the proprotein convertase family of enzymes [147, 148]. These convertases act as molecular scissors for tissue-specific processing of multiple precursor proteins. The catalytic domain of PCSK9 contains the main binding structure of PCSK9 to the epidermal growth factor-like repeat domain (EGF-A) of the LDLr [149]. The major function of the heterodimeric pro-segment PCSK9 is to degrade the LDLr intracellularly and extracellularly, thereby acting as a chaperone that binds to the LDLr to promote its lysosomal degradation. Mice lacking PCSK9 have LDL levels more than 50% lower than wild-type mice [149]. The deletion of PCSK9 protein results in an increase in LDL receptors and a significant lowering of LDL cholesterol in mice. Notably, recent studies have reported that PCSK9 inhibition using antibodies reduces LDL cholesterol over 60% [150].
The E3 ubiquitin ligase IDOL
While investigating the mechanism by which LXR regulates LDLr activity, Tontonoz et al. discovered myosin regulatory light chain interacting protein (MYLIP), later renamed IDOL (for Inducible Degrader of the LDL receptor) to reflect its biological function [151]. IDOL is an E3 ubiquitin ligase (transcriptionally activated by LXR agonists) that triggers ubiquitination of the LDLr on its cytoplasmatic domain, thereby targeting it for degradation. Unlike the LDLr and Pcsk9 genes, Idol is not regulated by SREBPs. IDOL knockdown in hepatocytes increases LDLr protein levels and promotes LDL uptake [151]. Conversely, adenovirus-mediated expression of IDOL in mouse liver promotes LDLr degradation and elevates plasma LDL levels. Interestingly, IDOL targets two closely related LDLr family members, VLDLr and ApoE receptor 2 (ApoER2), two proteins implicated in both neuronal development and lipid metabolism [152]. IDOL triggers ubiquitination of the VLDLr and ApoER2 on their cytoplasmic tails, leading to their degradation [152].
MicroRNAs and cholesterol metabolism
MiRNA biogenesis
In addition to classic regulatory mechanisms, miRNAs have also been shown to regulate the expression of key genes in cholesterol metabolism. miRNAs are small (~22 nt), single-stranded, non-coding RNAs that regulate gene expression post-transcriptionally [153–156]. This novel class of RNAs was first discovered in the nematode Caenorhabditis elegans, and since then has been identified in the genomes of most plants, animals, and viruses [153–156]. In animals, miRNAs control the expression of their target genes by primarily acting as sequence specific inhibitors of messenger RNA (mRNA). miRNAs are transcribed in the nucleus by RNA polymerase II from individual miRNA genes, introns of protein coding genes, or from polycistronic transcripts that often encode multiple related miRNAs [153–156]. The primary transcripts (pri-miRNA) are usually thousands of nucleotides long and contain local hairpin structures. Following transcription, they are processed sequentially in the nucleus and cytoplasm by a complex of RNase-III endonucleases, Drosha and Dicer. Specifically, Drosha processes the pri-miRNA transcript to a 70–100 nucleotide stem-loop precursor (pre-miRNA), which is then delivered to the cytoplasm by Exportin 5, where it is subsequently cleaved by Dicer to produce a ~22 nt miRNA:miRNA* duplex [154–156]. Once in the cytoplasm, one of the duplex strands is preferentially incorporated into the RNA-induced silencing complex (RISC) in association with an Ago family member [154–156]. Within the RISC, the miRNA guides the complex to its RNA target, thereby mediating its repression. In animals, miRNAs control gene expression by binding to the 3′UTRs of their targets through Watson–Crick base pairing between the target and the 5′-end of the miRNAs, the ‘seed’ sequence (nt 2–8) [154–156]. This interaction typically leads to the translational repression of target mRNAs by either transcript destabilization, translational inhibition, or both [154–156].
MiRNAs and cholesterol
Several microRNAs have been described to regulate lipid metabolism, including miR-122, miR-370, miR-378/378*, miR-143, miR-27, miR-335, and miR-33 [25–27, 29, 157–159]. miR-122 was initially identified as the most highly expressed miRNA in the liver and since, has been associated with the regulation of liver metabolism, as well as hepatitis C infection [158]. Silencing of miR-122 down-regulates genes implicated in cholesterol biosynthesis and triglyceride metabolism leading to the reduction of total cholesterol levels and making miR-122 a viable candidate for therapeutic inhibitors to lower cholesterol in humans. miR-122 targets genes involved in the regulation of glucose metabolism such us aldolase A (ALDO A) and pyruvate kinase (PKM2). In addition, several genes involved in fatty acid synthesis and oxidation, including fatty acid synthase (FAS), acetyl-coA carboxylase 1 (ACC1), and acetyl-coA carboxylase 2 (ACC2), were altered in mice treated with anti-miR-122. Similarly to miR-122, miR-370 has been recently shown to control fatty acid metabolism by regulating miR-122 expression levels and direct targeting of CPT1a, a mitochondrial enzyme that promotes fatty acid oxidation [27]. miR-378/378*, an intronic miRNA located within the peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PGC1α) genomic sequence, also plays important roles in regulating lipid metabolism. Overexpression of miR-378/378* during adipogenesis increases triacylglycerol accumulation, and an increase in fatty acid metabolism genes, including fatty acid binding protein 4 (FABP4), FAS, and stearoyl-CoA desaturase 1 (SCD1), was observed in ST2 cells that overexpressed miR-378/378* [26]. More recently, miR-143, miR-27 and miR-335 have also emerged as regulators of lipid metabolism and adipocyte differentiation [28, 29, 160]. While miR-143 was shown to play a role in adipocyte differentiation by targeting extracellular signal-related kinase 5 (ERK5), miR-27 was shown to inhibit the expression of peroxisome proliferator-activated receptor gamma (PPARγ) and CCAAT/enhancer binding protein (C/EBP), alpha. miR-335, a miRNA that is highly expressed in the liver and adipose tissue of obese mice, is up-regulated in response to lipid loading, while miR-335’s role in the regulation of lipid metabolism and adipogenesis remains to be discovered [28, 29, 160].
MiR-33 is a key regulator of cholesterol metabolism
Most recently, several independent groups have identified miR-33a and miR-33b, intronic miRNAs located within the Srebp-2 and Srebp-1 genes, respectively [18–20]. Metabolic stimuli that activate the expression of Srebp-2 and Srebp-1 lead to an increased expression of miR-33a and miR-33b, respectively, suggesting that both host genes and miRNAs are co-regulated. Moreover, Horie et al. recently cloned fragments of the mouse Srebp-2 gene that included exon 16 through exon 17, including miR-33a, and showed that miR-33 is coordinately expressed when Srebp-2 is activated [17].
miR-33a targets genes involved in cholesterol trafficking, including ABCA1, ABCG1, and NPC1 [16, 18–20]. Interestingly, ABCA1, a transporter responsible for the movement of cholesterol out of the cell, was among the top predicted target genes for miR-33a. miR-33a overexpression strongly represses ABCA1 expression and decreases cellular cholesterol efflux to ApoA-I. Conversely, antagonism of endogenous miR-33 up-regulates ABCA1 expression in vitro and in vivo, and promotes cholesterol efflux to ApoA-I, further confirming the physiological effects of miR-33 [16, 18–20]. Similar results were observed in mouse peritoneal macrophages isolated from miR-33 null mice [17]. miR-33 also targets ABCG1, but only in rodents. Accordingly, miR-33 repressed cholesterol efflux to mature HDL in mouse cells but not in human cells. Together, these findings establish a reciprocal pathway in which, during sterol-poor conditions, miR-33a is coincidentally generated with SREBP-2 and works to increase cellular cholesterol levels by down-regulating ABCA1 and ABCG1 and thus, limit cholesterol efflux (Fig. 2).

Maintenance of intracellular lipid levels by sterol regulatory element binding proteins (SREBPs) and miR-33a and -b in hepatocytes. When intracellular cholesterol levels are low, SREBP-2 is delivered to the Golgi where it is sequentially cleaved by two membrane-bound proteases, site-1 protease (S1P) and site-2 protease (S2P). The N-terminal fragment is released and translocates to the nucleus, where it acts as a transcription factor to regulate genes containing a sterol response element (SRE), including Srebp-2. Conversely, the Srebp-1c promoter contains a liver X receptor (LXR) binding site that activates SREBP-1c transcription in the presence of LXR agonists or insulin. Activation of SREBP-2 or SREBP-1 results in the co-transcription of miR-33a and miR-33b, respectively. These miRNAs simultaneously inhibit the expression of genes involved in cholesterol transport (ABCA1, ABCG1, and NPC1) and fatty acid oxidation (CROT, CPT1a, HADHB, and AMPK), thereby decreasing reverse cholesterol transport and reducing fatty acid β-oxidation. miR-33a and -b also contribute to the regulation of glucose metabolism, by targeting IRS2 and SIRT6. HDL high-density lipoprotein, ABCA1 ATP-binding cassette transporter A1, ABCG1 ATP-binding cassette transporter G1, NPC1 Niemann–Pick disease, type C1, LE late endosome, ER endoplasmic reticulum, SCAP SREBP cleavage activating protein, INSIG insulin induced gene, RXR retinoid X receptor, LXRE liver X receptor element, CPT1a carnitine palmitoyltransferase 1A, CROT carnitine O-octanoyltransferase, HADHB hydroxyacyl-CoA dehydrogenase/3-ketoacyl-CoA thiolase/enoyl-CoA hydratase (trifunctional protein), beta subunit, IRS2 insulin receptor substrate 2, SIRT6 sirtuin 6, PM plasma membrane
As mentioned above, ABCA1 plays a key role in regulating HDL biogenesis in vivo. Remarkably, antagonists of miR-33 in vivo using LNA-modified oligonucleotides, lentivirus, and adenovirus increase significantly the expression of ABCA1 in the liver and plasma HDL levels. Similar results were observed in the miR-33 knockout mice [17]. Because low levels of HDL correlate with increased cardiovascular disease risk, there is an increasing interest in studying the regulation, mechanism of action, and suitability of ABCA1 as a target to increase HDL levels for the treatment and prevention of atherosclerosis. To assess whether or not anti-miR-33 therapy increases reverse cholesterol transport and promotes regression of atherosclerosis, Rayner and colleagues treated Ldlr −/− mice with established plaques with anti-miR-33 oligonucleotides. Interestingly, mice treated with anti-miR-33 oligonucleotides presented smaller plaques with increased fibrous caps and reduced necrotic cores, phenotypic characteristics of stable plaques [161].
MiR-33 coordinates genes regulating fatty acid and glucose metabolism
In addition to the cholesterol transport genes, ABCA1, ABCG1 and NPC1, two independent studies have recently shown that miR-33a and -b binding sites are highly conserved in the 3′UTR of genes involved in fatty acid oxidation including, Cpt1a, Crot, Hadhb, and Ampk [16]. We and Gerin et al. have shown that miR-33 decreases the expression of CPT1a, CROT, and HADHB at the mRNA and protein level. Furthermore, overexpression of miR-33a and miR-33b reduces fatty acid oxidation and leads to the accumulation of triglycerides in human hepatic cells and in the fat body of miR-33 transgenic flies [21]. Another interesting finding is that miR-33 inhibits the expression of AMPK, a cellular energy sensor that coordinates hepatic lipid metabolism at the transcriptional and post-transcriptional level. In the liver, activation of AMPK promotes fatty acid oxidation, while inhibiting cholesterol and triglyceride synthesis [144]. Taken together, miR-33a and -b appear to be fundamental modulators of lipid metabolism by limiting cellular cholesterol efflux and fatty acid degradation upon SREBP induction (Fig. 2).
Our previous work also reveals an interesting role for miR-33 in glucose metabolism, as miR-33 overexpression inhibits the insulin receptor substrate 2 (IRS2), an essential docking molecule that mediates the effects of insulin. Consistent with these findings, miR-33 overexpression reduces insulin-induced 2-deoxyglucose (2-DOG) uptake in hepatic cells [21]. In addition, miR-33 inhibits sirtuin 6 (SIRT6), which has been involved in regulating fatty acid and glucose homeostasis. Indeed, hepatic-specific disruption of SIRT6 in mice results in fatty liver formation because of enhanced glucolysis and triglyceride synthesis [21].
MiRNAs and cardiovascular diseases
MiRNA expression of different cell types involved in the vascular system has already been widely studied. miR-126, an intronic miRNA located in the epidermal growth factor-like domain 7 (Egfl7), regulates vascular integrity and angiogenesis ([162, 163] and Fish). Interestingly, miR-126 targets VCAM-1, suggesting a role for this miRNA in regulating vascular inflammation and atherosclerosis [164]. miR-126 also may inhibit atherosclerosis by promoting endothelial precursor cell (EPC) recruitment for replacement of damaged endothelium [165]. miR-145 and miR-143 have recently been shown to regulate smooth muscle cell proliferation and migration, thereby contributing to vascular remodeling and preventing restenosis [166–169]. In addition, many other miRNAs has been characterized in human atherosclerotic plaques including miR-21, miR-210, miR-34a and miR-146, however, their role in the progression atherosclerosis remains unknown [170].
Recently, the presence of miRNAs in the plasma has been highlighted [171]. The plasma miRNAs are not cell associated, but packaged in microvesicles that protect them from endogenous RNase activity [172]. Several circulating miRNAs are significantly reduced in patients with coronary artery disease (CAD) including miR-126, miR-17, miR-92a, and miR-155 [173]. In contrast, the cardiac muscle-enriched miRNAs (miR-208a and miR-133a) tend to be higher in patients with CAD [173]. Mayr et al. have also identified plasma miRNAs differentially expressed in patients with diabetes mellitus (DM) [174]. miR-20b, miR-21, miR-24, miR-15a, miR-126, miR-191, miR-197, miR-223, miR-320, and miR-486 expression were reduced in patients with DM. Most differences in miRNA levels were replicated in plasma obtained from hyperglycemic Lepob mice [174].
MiRNAs are transported by plasma lipoproteins
In a series of beautiful experiments, Vickers et al. have demonstrated that HDL transports endogenous miRNAs and delivers them to the recipient cells with functional targeting capabilities [175]. This remarkable finding suggests that some of the biological effects of HDL could be mediated by the miRNAs transported by these lipoproteins. The exact process of how HDL is loaded with miRNAs and what proteins, if any, facilitate this association remains unknown. Interestingly, the miRNAs transferred from HDL to human hepatocytes appears to be SR-BI dependent [175].
miR-223, one of the most abundant miRNAs found in human and mouse HDL, was increased in atherosclerosis mouse models and patients with familial hypercholesterolemia [175]. The role of miR-223 in multiple physiological processes has been extensively investigated [176–178]. Of note, miR-223 has been involved in regulating cell cycle, glucose homeostasis, granulopoiesis and osteoclast differentiation [176–178].
Conclusions
Our understanding of cholesterol homeostasis has advanced significantly over the past years. Much insight has been gained into the transport pathways that regulate sterol trafficking and distribution within cells and advances have been made in understanding the regulatory mechanisms that control cholesterol uptake and efflux. Furthermore, recent studies indicate that miRNAs (particularly miR-33a and -b) play a significant role in regulating cholesterol and fatty acid metabolism.
miRNAs represent an elegant layer above transcriptional control for both fine-tuning and dramatically altering activity and output of cell signaling. In addition, miRNAs may serve as points of crosstalk between signaling pathways by integrating transcriptional inputs or by their functional regulatory output on different gene networks. The dysregulation of many short interfering RNAs (siRNAs) and miRNAs has been linked to the development and progression of disease and consequently, these non-coding RNAs have gained considerable attention as therapeutic targets. The aforementioned results highlight the use of anti-miR-33 therapies in the treatment of a number of metabolic disorders. Inhibition of miRNA expression can be achieved using anti-sense oligonucleotides, termed ‘antagomirs,’ or their chemically modified versions, 2′-O-methyl (2′-OMe)-oligonucleotides and locked nucleic acids (LNA), termed ‘anti-miRs.’ Additionally, the production of the mature forms can also be disrupted at the processing level [24, 25, 157, 179]. Using anti-miR-33 treatment to elevate ABCA1 levels and increase HDL levels would hold tremendous potential for the treatment and/or prevention of coronary artery disease (CAD), in which an underlying risk factor is low levels of HDL. Moreover, inhibition of miR-33 would result in increased fatty acid oxidation and reduced accumulation of fat stores in the liver, suggesting that antagonism of endogenous miR-33 may also be used to treat metabolic syndrome and non-alcoholic fatty liver disease (NAFLD). While there are still unanswered questions surrounding miRNA therapeutics, the promise demonstrated by the use of anti-miRs in preclinical studies raise the possibility that miR-33 may become a viable therapeutic target in the future.
References
Vance DE, Van den Bosch H (2000) Cholesterol in the year. Biochim Biophys Acta 1529(1–3):1–8
Fernandez C, Lobo Md Mdel V, Gomez-Coronado D, Lasuncion MA (2004) Cholesterol is essential for mitosis progression and its deficiency induces polyploid cell formation. Exp Cell Res 300(1):109–120
Fernandez C, Martin M, Gomez-Coronado D, Lasuncion MA (2005) Effects of distal cholesterol biosynthesis inhibitors on cell proliferation and cell cycle progression. J Lipid Res 46(5):920–929
Ikonen E (2006) Mechanisms for cellular cholesterol transport: defects and human disease. Physiol Rev 86(4):1237–1261
Maxfield FR, Tabas I (2005) Role of cholesterol and lipid organization in disease. Nature 438(7068):612–621
Bloch K (1992) Sterol molecule: structure, biosynthesis, and function. Steroids 57(8):378–383
Ponticorvo L, Rittenberg D, Bloch K (1949) The utilization of acetate for the synthesis of fatty acids, cholesterol, and protoporphyrin. J Biol Chem 179(2):839–842
Brown MS, Goldstein JL (1976) Receptor-mediated control of cholesterol metabolism. Science 191(4223):150–154
Brown MS, Goldstein JL (1986) A receptor-mediated pathway for cholesterol homeostasis. Science 232(4746):34–47
Brown MS, Goldstein JL (1997) The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 89(3):331–340
Goldstein JL, Brown MS (1990) Regulation of the mevalonate pathway. Nature 343(6257):425–430
Sakakura Y, Shimano H, Sone H, Takahashi A, Inoue N, Toyoshima H, Suzuki S, Yamada N (2001) Sterol regulatory element-binding proteins induce an entire pathway of cholesterol synthesis. Biochem Biophys Res Commun 286(1):176–183
Beaven SW, Tontonoz P (2006) Nuclear receptors in lipid metabolism: targeting the heart of dyslipidemia. Annu Rev Med 57:313–329
Peet DJ, Janowski BA, Mangelsdorf DJ (1998) The LXRs: a new class of oxysterol receptors. Curr Opin Genet Dev 8(5):571–575
Tontonoz P, Mangelsdorf DJ (2003) Liver X receptor signaling pathways in cardiovascular disease. Mol Endocrinol 17(6):985–993
Gerin I, Clerbaux LA, Haumont O, Lanthier N, Das AK, Burant CF, Leclercq IA, Macdougald OA, Bommer GT (2010) Expression of miR-33 from an SREBP2 intron inhibits cholesterol export and fatty acid oxidation. J Biol Chem 285(44):33652–33661
Horie T, Ono K, Horiguchi M, Nishi H, Nakamura T, Nagao K, Kinoshita M, Kuwabara Y, Marusawa H, Iwanaga Y, Hasegawa K, Yokode M, Kimura T, Kita T (2010) MicroRNA-33 encoded by an intron of sterol regulatory element-binding protein 2 (Srebp2) regulates HDL in vivo. Proc Natl Acad Sci USA 107(40):17321–17326
Marquart TJ, Allen RM, Ory DS, Baldan A (2010) miR-33 links SREBP-2 induction to repression of sterol transporters. Proc Natl Acad Sci USA 107(27):12228–12232
Najafi-Shoushtari SH, Kristo F, Li Y, Shioda T, Cohen DE, Gerszten RE, Naar AM (2010) MicroRNA-33 and the SREBP host genes cooperate to control cholesterol homeostasis. Science 328(5985):1566–1569
Rayner KJ, Suarez Y, Davalos A, Parathath S, Fitzgerald ML, Tamehiro N, Fisher EA, Moore KJ, Fernandez-Hernando C (2010) MiR-33 contributes to the regulation of cholesterol homeostasis. Science 328(5985):1570–1573
Davalos A, Goedeke L, Smibert P, Ramirez CM, Warrier NP, Andreo U, Cirera-Salinas D, Rayner K, Suresh U, Pastor-Pareja JC, Esplugues E, Fisher EA, Penalva LO, Moore KJ, Suarez Y, Lai EC, Fernandez-Hernando C (2011) miR-33a/b contribute to the regulation of fatty acid metabolism and insulin signaling. Proc Natl Acad Sci USA 108 (22):9232–9237
Gerin I, Clerbaux LA, Haumont O, Lanthier N, Das AK, Burant CF, Leclercq IA, MacDougald OA, Bommer GT (2010) Expression of miR-33 from an SREBP2 intron inhibits cholesterol export and fatty acid oxidation. J Biol Chem 285(44):33652–33661
Chen T, Huang Z, Wang L, Wang Y, Wu F, Meng S, Wang C (2009) MicroRNA-125a–5p partly regulates the inflammatory response, lipid uptake, and ORP9 expression in oxLDL-stimulated monocyte/macrophages. Cardiovasc Res 83(1):131–139
Elmen J, Lindow M, Schutz S, Lawrence M, Petri A, Obad S, Lindholm M, Hedtjarn M, Hansen HF, Berger U, Gullans S, Kearney P, Sarnow P, Straarup EM, Kauppinen S (2008) LNA-mediated microRNA silencing in non-human primates. Nature 452(7189):896–899
Esau C, Davis S, Murray SF, Yu XX, Pandey SK, Pear M, Watts L, Booten SL, Graham M, McKay R, Subramaniam A, Propp S, Lollo BA, Freier S, Bennett CF, Bhanot S, Monia BP (2006) miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab 3(2):87–98
Gerin I, Bommer GT, McCoin CS, Sousa KM, Krishnan V, MacDougald OA (2010) Roles for miRNA-378/378* in adipocyte gene expression and lipogenesis. Am J Physiol Endocrinol Metab 299 (2):E198–E206
Iliopoulos D, Drosatos K, Hiyama Y, Goldberg IJ, Zannis VI (2010) MicroRNA-370 controls the expression of microRNA-122 and Cpt1alpha and affects lipid metabolism. J Lipid Res 51 (6):1513–1523
Lin Q, Gao Z, Alarcon RM, Ye J, Yun Z (2009) A role of miR-27 in the regulation of adipogenesis. FEBS J 276(8):2348–2358
Nakanishi N, Nakagawa Y, Tokushige N, Aoki N, Matsuzaka T, Ishii K, Yahagi N, Kobayashi K, Yatoh S, Takahashi A, Suzuki H, Urayama O, Yamada N, Shimano H (2009) The up-regulation of microRNA-335 is associated with lipid metabolism in liver and white adipose tissue of genetically obese mice. Biochem Biophys Res Commun 385(4):492–496
Dietschy JM, Turley SD, Spady DK (1993) Role of liver in the maintenance of cholesterol and low-density lipoprotein homeostasis in different animal species, including humans. J Lipid Res 34(10):1637–1659
Bloch K (1987) Summing up. Annu Rev Biochem 56:1–19
Jasinska M, Owczarek J, Orszulak-Michalak D (2007) Statins: a new insight into their mechanisms of action and consequent pleiotropic effects. Pharmacol Rep 59(5):483–499
Kandutsch AA, Russell AE (1960) Preputial gland tumor sterols. 3. A metabolic pathway from lanosterol to cholesterol. J Biol Chem 235:2256–2261
Baumann NA, Sullivan DP, Ohvo-Rekila H, Simonot C, Pottekat A, Klaassen Z, Beh CT, Menon AK (2005) Transport of newly synthesized sterol to the sterol-enriched plasma membrane occurs via nonvesicular equilibration. Biochemistry 44(15):5816–5826
Chang TY, Chang CC, Cheng D (1997) Acyl-coenzyme A: cholesterol acyltransferase. Annu Rev Biochem 66:613–638
Brown MS, Goldstein JL (1980) Multivalent feedback regulation of HMG CoA reductase, a control mechanism coordinating isoprenoid synthesis and cell growth. J Lipid Res 21(5):505–517
Hooper NM (1999) Detergent-insoluble glycosphingolipid/cholesterol-rich membrane domains, lipid rafts and caveolae (review). Mol Membr Biol 16(2):145–156
Nwokoro NA, Wassif CA, Porter FD (2001) Genetic disorders of cholesterol biosynthesis in mice and humans. Mol Genet Metab 74(1–2):105–119
Smith DW, Lemli L, Opitz JM (1964) A newly recognized syndrome of multiple congenital anomalies. J Pediatr 64:210–217
Brunetti-Pierri N, Corso G, Rossi M, Ferrari P, Balli F, Rivasi F, Annunziata I, Ballabio A, Russo AD, Andria G, Parenti G (2002) Lathosterolosis, a novel multiple-malformation/mental retardation syndrome due to deficiency of 3beta-hydroxysteroid-delta5-desaturase. Am J Hum Genet 71(4):952–958
Dietschy JM, Turley SD (2001) Cholesterol metabolism in the brain. Curr Opin Lipidol 12(2):105–112
Jurevics H, Morell P (1995) Cholesterol for synthesis of myelin is made locally, not imported into brain. J Neurochem 64(2):895–901
Snipes GJ, Suter U (1997) Cholesterol and myelin. Subcell Biochem 28:173–204
Fernandez-Hernando C, Suarez Y, Lasuncion MA (2005) Lovastatin-induced PC-12 cell differentiation is associated with RhoA/RhoA kinase pathway inactivation. Mol Cell Neurosci 29(4):591–602
Hayashi H, Campenot RB, Vance DE, Vance JE (2004) Glial lipoproteins stimulate axon growth of central nervous system neurons in compartmented cultures. J Biol Chem 279(14):14009–14015
Holtzman DM, Pitas RE, Kilbridge J, Nathan B, Mahley RW, Bu G, Schwartz AL (1995) Low-density lipoprotein receptor-related protein mediates apolipoprotein E-dependent neurite outgrowth in a central nervous system-derived neuronal cell line. Proc Natl Acad Sci USA 92(21):9480–9484
Block RC, Dorsey ER, Beck CA, Brenna JT, Shoulson I Altered cholesterol and fatty acid metabolism in Huntington disease. J Clin Lipidol 4 (1):17-23
Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA (1993) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261(5123):921–923
Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, Roses AD (1993) Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci USA 90(5):1977–1981
Goldstein JL, Brown MS (1982) The LDL receptor defect in familial hypercholesterolemia. Implications for pathogenesis and therapy. Med Clin North Am 66 (2):335-362
Grundy SM (1983) Absorption and metabolism of dietary cholesterol. Annu Rev Nutr 3:71–96
Linder MD, Uronen RL, Holtta-Vuori M, van der Sluijs P, Peranen J, Ikonen E (2007) Rab8-dependent recycling promotes endosomal cholesterol removal in normal and sphingolipidosis cells. Mol Biol Cell 18(1):47–56
Holtta-Vuori M, Tanhuanpaa K, Mobius W, Somerharju P, Ikonen E (2002) Modulation of cellular cholesterol transport and homeostasis by Rab11. Mol Biol Cell 13(9):3107–3122
Ory DS (2004) The Niemann–Pick disease genes; regulators of cellular cholesterol homeostasis. Trends Cardiovasc Med 14(2):66–72
Wang ML, Motamed M, Infante RE, Abi-Mosleh L, Kwon HJ, Brown MS, Goldstein JL (2010) Identification of surface residues on Niemann–Pick C2 essential for hydrophobic handoff of cholesterol to NPC1 in lysosomes. Cell Metab 12(2):166–173
Lebrand C, Corti M, Goodson H, Cosson P, Cavalli V, Mayran N, Faure J, Gruenberg J (2002) Late endosome motility depends on lipids via the small GTPase Rab7. EMBO J 21(6):1289–1300
Ganley IG, Pfeffer SR (2006) Cholesterol accumulation sequesters Rab9 and disrupts late endosome function in NPC1-deficient cells. J Biol Chem 281(26):17890–17899
Maxfield FR, Wustner D (2002) Intracellular cholesterol transport. J Clin Invest 110(7):891–898
Maxfield FR, Mondal M (2006) Sterol and lipid trafficking in mammalian cells. Biochem Soc Trans 34(Pt 3):335–339
Soccio RE, Breslow JL (2004) Intracellular cholesterol transport. Arterioscler Thromb Vasc Biol 24(7):1150–1160
Soccio RE, Breslow JL (2003) StAR-related lipid transfer (START) proteins: mediators of intracellular lipid metabolism. J Biol Chem 278(25):22183–22186
Seedorf U, Ellinghaus P, Roch Nofer J (2000) Sterol carrier protein-2. Biochim Biophys Acta 1486(1):45–54
Gallegos AM, Atshaves BP, Storey SM, Starodub O, Petrescu AD, Huang H, McIntosh AL, Martin GG, Chao H, Kier AB, Schroeder F (2001) Gene structure, intracellular localization, and functional roles of sterol carrier protein-2. Prog Lipid Res 40(6):498–563
Prinz WA (2007) Non-vesicular sterol transport in cells. Prog Lipid Res 46(6):297–314
Martin S, Parton RG (2005) Caveolin, cholesterol, and lipid bodies. Semin Cell Dev Biol 16(2):163–174
Parton RG, Simons K (2007) The multiple faces of caveolae. Nat Rev Mol Cell Biol 8(3):185–194
Uittenbogaard A, Smart EJ (2000) Palmitoylation of caveolin-1 is required for cholesterol binding, chaperone complex formation, and rapid transport of cholesterol to caveolae. J Biol Chem 275(33):25595–25599
Uittenbogaard A, Ying Y, Smart EJ (1998) Characterization of a cytosolic heat-shock protein-caveolin chaperone complex. Involvement in cholesterol trafficking. J Biol Chem 273(11):6525–6532
Heino S, Lusa S, Somerharju P, Ehnholm C, Olkkonen VM, Ikonen E (2000) Dissecting the role of the Golgi complex and lipid rafts in biosynthetic transport of cholesterol to the cell surface. Proc Natl Acad Sci U S A 97(15):8375–8380
Kandutsch AA, Shown EP (1981) Assay of oxysterol-binding protein in a mouse fibroblast, cell-free system. Dissociation constant and other properties of the system. J Biol Chem 256(24):13068–13073
Wang C, JeBailey L, Ridgway ND (2002) Oxysterol-binding-protein (OSBP)-related protein 4 binds 25-hydroxycholesterol and interacts with vimentin intermediate filaments. Biochem J 361(Pt 3):461–472
Ngo M, Ridgway ND (2009) Oxysterol binding protein-related Protein 9 (ORP9) is a cholesterol transfer protein that regulates Golgi structure and function. Mol Biol Cell 20(5):1388–1399
Johansson M, Bocher V, Lehto M, Chinetti G, Kuismanen E, Ehnholm C, Staels B, Olkkonen VM (2003) The two variants of oxysterol binding protein-related protein-1 display different tissue expression patterns, have different intracellular localization, and are functionally distinct. Mol Biol Cell 14(3):903–915
Du X, Kumar J, Ferguson C, Schulz TA, Ong YS, Hong W, Prinz WA, Parton RG, Brown AJ, Yang H (2011) A role for oxysterol-binding protein-related protein 5 in endosomal cholesterol trafficking. J Cell Biol 192(1):121–135
Ponting CP, Aravind L (1999) START: a lipid-binding domain in StAR, HD-ZIP and signalling proteins. Trends Biochem Sci 24(4):130–132
Stocco DM (2001) StAR protein and the regulation of steroid hormone biosynthesis. Annu Rev Physiol 63:193–213
Hanada K, Kumagai K, Yasuda S, Miura Y, Kawano M, Fukasawa M, Nishijima M (2003) Molecular machinery for non-vesicular trafficking of ceramide. Nature 426 (6968):803–809
Rigotti A, Cohen DE, Zanlungo S (2010) STARTing to understand MLN64 function in cholesterol transport. J Lipid Res 51(8):2015–2017
Charman M, Kennedy BE, Osborne N, Karten B (2010) MLN64 mediates egress of cholesterol from endosomes to mitochondria in the absence of functional Niemann–Pick Type C1 protein. J Lipid Res 51(5):1023–1034
Glomset JA, Norum KR (1973) The metabolic role of lecithin: cholesterol acyltransferase: perspectives form pathology. Adv Lipid Res 11:1–65
Cuchel M, Rader DJ (2006) Macrophage reverse cholesterol transport: key to the regression of atherosclerosis? Circulation 113(21):2548–2555
Oram JF, Yokoyama S (1996) Apolipoprotein-mediated removal of cellular cholesterol and phospholipids. J Lipid Res 37(12):2473–2491
Yokoyama S (1998) Apolipoprotein-mediated cellular cholesterol efflux. Biochim Biophys Acta 1392(1):1–15
Yokoyama S (2000) Release of cellular cholesterol: molecular mechanism for cholesterol homeostasis in cells and in the body. Biochim Biophys Acta 1529(1–3):231–244
Jonas A (2000) Lecithin cholesterol acyltransferase. Biochim Biophys Acta 1529(1–3):245–256
Ji Y, Jian B, Wang N, Sun Y, Moya ML, Phillips MC, Rothblat GH, Swaney JB, Tall AR (1997) Scavenger receptor BI promotes high-density lipoprotein-mediated cellular cholesterol efflux. J Biol Chem 272(34):20982–20985
Tanigawa H, Billheimer JT, Tohyama J, Fuki IV, Ng DS, Rothblat GH, Rader DJ (2009) Lecithin: cholesterol acyltransferase expression has minimal effects on macrophage reverse cholesterol transport in vivo. Circulation 120(2):160–169
Calabresi L, Favari E, Moleri E, Adorni MP, Pedrelli M, Costa S, Jessup W, Gelissen IC, Kovanen PT, Bernini F, Franceschini G (2009) Functional LCAT is not required for macrophage cholesterol efflux to human serum. Atherosclerosis 204(1):141–146
Linsel-Nitschke P, Tall AR (2005) HDL as a target in the treatment of atherosclerotic cardiovascular disease. Nat Rev Drug Discov 4(3):193–205
Berge KE, Tian H, Graf GA, Yu L, Grishin NV, Schultz J, Kwiterovich P, Shan B, Barnes R, Hobbs HH (2000) Accumulation of dietary cholesterol in sitosterolemia caused by mutations in adjacent ABC transporters. Science 290(5497):1771–1775
Bhattacharyya AK, Connor WE (1974) Beta-sitosterolemia and xanthomatosis. A newly described lipid storage disease in two sisters. J Clin Invest 53(4):1033–1043
Miettinen TA (1980) Phytosterolaemia, xanthomatosis and premature atherosclerotic arterial disease: a case with high plant sterol absorption, impaired sterol elimination and low cholesterol synthesis. Eur J Clin Invest 10(1):27–35
Yokoyama S (2006) ABCA1 and biogenesis of HDL. J Atheroscler Thromb 13(1):1–15
Tall AR, Yvan-Charvet L, Terasaka N, Pagler T, Wang N (2008) HDL, ABC transporters, and cholesterol efflux: implications for the treatment of atherosclerosis. Cell Metab 7(5):365–375
Brooks-Wilson A, Marcil M, Clee SM, Zhang LH, Roomp K, van Dam M, Yu L, Brewer C, Collins JA, Molhuizen HO, Loubser O, Ouelette BF, Fichter K, Ashbourne-Excoffon KJ, Sensen CW, Scherer S, Mott S, Denis M, Martindale D, Frohlich J, Morgan K, Koop B, Pimstone S, Kastelein JJ, Genest J Jr, Hayden MR (1999) Mutations in ABC1 in Tangier disease and familial high-density lipoprotein deficiency. Nat Genet 22(4):336–345
Neufeld EB, Remaley AT, Demosky SJ, Stonik JA, Cooney AM, Comly M, Dwyer NK, Zhang M, Blanchette-Mackie J, Santamarina-Fojo S, Brewer HB Jr (2001) Cellular localization and trafficking of the human ABCA1 transporter. J Biol Chem 276(29):27584–27590
Neufeld EB, Stonik JA, Demosky SJ Jr, Knapper CL, Combs CA, Cooney A, Comly M, Dwyer N, Blanchette-Mackie J, Remaley AT, Santamarina-Fojo S, Brewer HB Jr (2004) The ABCA1 transporter modulates late endocytic trafficking: insights from the correction of the genetic defect in Tangier disease. J Biol Chem 279(15):15571–15578
Takahashi Y, Smith JD (1999) Cholesterol efflux to apolipoprotein AI involves endocytosis and resecretion in a calcium-dependent pathway. Proc Natl Acad Sci USA 96(20):11358–11363
Choi HY, Karten B, Chan T, Vance JE, Greer WL, Heidenreich RA, Garver WS, Francis GA (2003) Impaired ABCA1-dependent lipid efflux and hypoalphalipoproteinemia in human Niemann–Pick type C disease. J Biol Chem 278(35):32569–32577
Haidar B, Kiss RS, Sarov-Blat L, Brunet R, Harder C, McPherson R, Marcel YL (2006) Cathepsin D, a lysosomal protease, regulates ABCA1-mediated lipid efflux. J Biol Chem 281(52):39971–39981
Nandi S, Ma L, Denis M, Karwatsky J, Li Z, Jiang XC, Zha X (2009) ABCA1-mediated cholesterol efflux generates microparticles in addition to HDL through processes governed by membrane rigidity. J Lipid Res 50(3):456–466
Vaughan AM, Oram JF (2003) ABCA1 redistributes membrane cholesterol independent of apolipoprotein interactions. J Lipid Res 44(7):1373–1380
Vedhachalam C, Duong PT, Nickel M, Nguyen D, Dhanasekaran P, Saito H, Rothblat GH, Lund-Katz S, Phillips MC (2007) Mechanism of ATP-binding cassette transporter A1-mediated cellular lipid efflux to apolipoprotein A-I and formation of high-density lipoprotein particles. J Biol Chem 282(34):25123–25130
Remaley AT, Stonik JA, Demosky SJ, Neufeld EB, Bocharov AV, Vishnyakova TG, Eggerman TL, Patterson AP, Duverger NJ, Santamarina-Fojo S, Brewer HB Jr (2001) Apolipoprotein specificity for lipid efflux by the human ABCAI transporter. Biochem Biophys Res Commun 280(3):818–823
van der Velde AE (2010) Reverse cholesterol transport: from classical view to new insights. World J Gastroenterol 16(47):5908–5915
Gelissen IC, Harris M, Rye KA, Quinn C, Brown AJ, Kockx M, Cartland S, Packianathan M, Kritharides L, Jessup W (2006) ABCA1 and ABCG1 synergize to mediate cholesterol export to apoA-I. Arterioscler Thromb Vasc Biol 26(3):534–540
Vaughan AM, Oram JF (2006) ABCA1 and ABCG1 or ABCG4 act sequentially to remove cellular cholesterol and generate cholesterol-rich HDL. J Lipid Res 47(11):2433–2443
Hirsch-Reinshagen V, Zhou S, Burgess BL, Bernier L, McIsaac SA, Chan JY, Tansley GH, Cohn JS, Hayden MR, Wellington CL (2004) Deficiency of ABCA1 impairs apolipoprotein E metabolism in brain. J Biol Chem 279(39):41197–41207
Wahrle SE, Jiang H, Parsadanian M, Hartman RE, Bales KR, Paul SM, Holtzman DM (2005) Deletion of Abca1 increases Abeta deposition in the PDAPP transgenic mouse model of Alzheimer disease. J Biol Chem 280(52):43236–43242
Wahrle SE, Jiang H, Parsadanian M, Kim J, Li A, Knoten A, Jain S, Hirsch-Reinshagen V, Wellington CL, Bales KR, Paul SM, Holtzman DM (2008) Overexpression of ABCA1 reduces amyloid deposition in the PDAPP mouse model of Alzheimer disease. J Clin Invest 118(2):671–682
Horton JD, Goldstein JL, Brown MS (2002) SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest 109(9):1125–1131
Horton JD, Shimomura I (1999) Sterol regulatory element-binding proteins: activators of cholesterol and fatty acid biosynthesis. Curr Opin Lipidol 10(2):143–150
Tontonoz P, Kim JB, Graves RA, Spiegelman BM (1993) ADD1: a novel helix-loop-helix transcription factor associated with adipocyte determination and differentiation. Mol Cell Biol 13(8):4753–4759
Duncan EA, Brown MS, Goldstein JL, Sakai J (1997) Cleavage site for sterol-regulated protease localized to a leu-Ser bond in the lumenal loop of sterol regulatory element-binding protein-2. J Biol Chem 272(19):12778–12785
Hua X, Yokoyama C, Wu J, Briggs MR, Brown MS, Goldstein JL, Wang X (1993) SREBP-2, a second basic-helix-loop-helix-leucine zipper protein that stimulates transcription by binding to a sterol regulatory element. Proc Natl Acad Sci USA 90(24):11603–11607
Yang T, Espenshade PJ, Wright ME, Yabe D, Gong Y, Aebersold R, Goldstein JL, Brown MS (2002) Crucial step in cholesterol homeostasis: sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell 110(4):489–500
Radhakrishnan A, Ikeda Y, Kwon HJ, Brown MS, Goldstein JL (2007) Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: oxysterols block transport by binding to Insig. Proc Natl Acad Sci USA 104(16):6511–6518
Sun LP, Seemann J, Goldstein JL, Brown MS (2007) Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: Insig renders sorting signal in Scap inaccessible to COPII proteins. Proc Natl Acad Sci USA 104(16):6519–6526
Osborne TF (2000) Sterol regulatory element-binding proteins (SREBPs): key regulators of nutritional homeostasis and insulin action. J Biol Chem 275(42):32379–32382
Goldstein JL, DeBose-Boyd RA, Brown MS (2006) Protein sensors for membrane sterols. Cell 124(1):35–46
Espenshade PJ (2006) SREBPs: sterol-regulated transcription factors. J Cell Sci 119(Pt 6):973–976
Brown MS, Goldstein JL (1999) A proteolytic pathway that controls the cholesterol content of membranes, cells, and blood. Proc Natl Acad Sci USA 96(20):11041–11048
Goldstein JL, Rawson RB, Brown MS (2002) Mutant mammalian cells as tools to delineate the sterol regulatory element-binding protein pathway for feedback regulation of lipid synthesis. Arch Biochem Biophys 397(2):139–148
Eberle D, Hegarty B, Bossard P, Ferre P, Foufelle F (2004) SREBP transcription factors: master regulators of lipid homeostasis. Biochimie 86(11):839–848
Hegarty BD, Bobard A, Hainault I, Ferre P, Bossard P, Foufelle F (2005) Distinct roles of insulin and liver X receptor in the induction and cleavage of sterol regulatory element-binding protein-1c. Proc Natl Acad Sci USA 102(3):791–796
Repa JJ, Liang G, Ou J, Bashmakov Y, Lobaccaro JM, Shimomura I, Shan B, Brown MS, Goldstein JL, Mangelsdorf DJ (2000) Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRalpha and LXRbeta. Genes Dev 14(22):2819–2830
Hirano Y, Yoshida M, Shimizu M, Sato R (2001) Direct demonstration of rapid degradation of nuclear sterol regulatory element-binding proteins by the ubiquitin-proteasome pathway. J Biol Chem 276(39):36431–36437
Hirano Y, Murata S, Tanaka K, Shimizu M, Sato R (2003) Sterol regulatory element-binding proteins are negatively regulated through SUMO-1 modification independent of the ubiquitin/26 S proteasome pathway. J Biol Chem 278(19):16809–16819
Walker AK, Yang F, Jiang K, Ji JY, Watts JL, Purushotham A, Boss O, Hirsch ML, Ribich S, Smith JJ, Israelian K, Westphal CH, Rodgers JT, Shioda T, Elson SL, Mulligan P, Najafi-Shoushtari H, Black JC, Thakur JK, Kadyk LC, Whetstine JR, Mostoslavsky R, Puigserver P, Li X, Dyson NJ, Hart AC, Naar AM (2010) Conserved role of SIRT1 orthologs in fasting-dependent inhibition of the lipid/cholesterol regulator SREBP. Genes Dev 24(13):1403–1417
Willy PJ, Umesono K, Ong ES, Evans RM, Heyman RA, Mangelsdorf DJ (1995) LXR, a nuclear receptor that defines a distinct retinoid response pathway. Genes Dev 9(9):1033–1045
Repa JJ, Mangelsdorf DJ (2000) The role of orphan nuclear receptors in the regulation of cholesterol homeostasis. Annu Rev Cell Dev Biol 16:459–481
Attie AD, Kastelein JP, Hayden MR (2001) Pivotal role of ABCA1 in reverse cholesterol transport influencing HDL levels and susceptibility to atherosclerosis. J Lipid Res 42(11):1717–1726
Yu L, Hammer RE, Li-Hawkins J, Von Bergmann K, Lutjohann D, Cohen JC, Hobbs HH (2002) Disruption of Abcg5 and Abcg8 in mice reveals their crucial role in biliary cholesterol secretion. Proc Natl Acad Sci USA 99(25):16237–16242
Bradley MN, Hong C, Chen M, Joseph SB, Wilpitz DC, Wang X, Lusis AJ, Collins A, Hseuh WA, Collins JL, Tangirala RK, Tontonoz P (2007) Ligand activation of LXR beta reverses atherosclerosis and cellular cholesterol overload in mice lacking LXR alpha and apoE. J Clin Invest 117(8):2337–2346
Repa JJ, Turley SD, Lobaccaro JA, Medina J, Li L, Lustig K, Shan B, Heyman RA, Dietschy JM, Mangelsdorf DJ (2000) Regulation of absorption and ABC1-mediated efflux of cholesterol by RXR heterodimers. Science 289(5484):1524–1529
Schultz JR, Tu H, Luk A, Repa JJ, Medina JC, Li L, Schwendner S, Wang S, Thoolen M, Mangelsdorf DJ, Lustig KD, Shan B (2000) Role of LXRs in control of lipogenesis. Genes Dev 14(22):2831–2838
Song BL, Javitt NB, DeBose-Boyd RA (2005) Insig-mediated degradation of HMG CoA reductase stimulated by lanosterol, an intermediate in the synthesis of cholesterol. Cell Metab 1(3):179–189
Song BL, DeBose-Boyd RA (2004) Ubiquitination of 3-hydroxy-3-methylglutaryl-CoA reductase in permeabilized cells mediated by cytosolic E1 and a putative membrane-bound ubiquitin ligase. J Biol Chem 279(27):28798–28806
Song BL, Sever N, DeBose-Boyd RA (2005) Gp78, a membrane-anchored ubiquitin ligase, associates with Insig-1 and couples sterol-regulated ubiquitination to degradation of HMG CoA reductase. Mol Cell 19(6):829–840
Sever N, Song BL, Yabe D, Goldstein JL, Brown MS, DeBose-Boyd RA (2003) Insig-dependent ubiquitination and degradation of mammalian 3-hydroxy-3-methylglutaryl-CoA reductase stimulated by sterols and geranylgeraniol. J Biol Chem 278(52):52479–52490
Gill S, Stevenson J, Kristiana I, Brown AJ (2011) Cholesterol-dependent degradation of squalene monooxygenase, a control point in cholesterol synthesis beyond HMG-CoA reductase. Cell Metab 13 (3):260–273
Beg ZH, Allmann DW, Gibson DM (1973) Modulation of 3-hydroxy-3-methylglutaryl coenzyme A reductase activity with cAMP and wth protein fractions of rat liver cytosol. Biochem Biophys Res Commun 54(4):1362–1369
Clarke PR, Hardie DG (1990) Regulation of HMG-CoA reductase: identification of the site phosphorylated by the AMP-activated protein kinase in vitro and in intact rat liver. EMBO J 9(8):2439–2446
Hardie DG, Pan DA (2002) Regulation of fatty acid synthesis and oxidation by the AMP-activated protein kinase. Biochem Soc Trans 30(Pt 6):1064–1070
Sato R, Goldstein JL, Brown MS (1993) Replacement of serine-871 of hamster 3-hydroxy-3-methylglutaryl-CoA reductase prevents phosphorylation by AMP-activated kinase and blocks inhibition of sterol synthesis induced by ATP depletion. Proc Natl Acad Sci USA 90(20):9261–9265
Omkumar RV, Darnay BG, Rodwell VW (1994) Modulation of Syrian hamster 3-hydroxy-3-methylglutaryl-CoA reductase activity by phosphorylation. Role of serine 871. J Biol Chem 269(9):6810–6814
Benjannet S, Rhainds D, Essalmani R, Mayne J, Wickham L, Jin W, Asselin MC, Hamelin J, Varret M, Allard D, Trillard M, Abifadel M, Tebon A, Attie AD, Rader DJ, Boileau C, Brissette L, Chretien M, Prat A, Seidah NG (2004) NARC-1/PCSK9 and its natural mutants: zymogen cleavage and effects on the low-density lipoprotein (LDL) receptor and LDL cholesterol. J Biol Chem 279(47):48865–48875
Maxwell KN, Breslow JL (2004) Adenoviral-mediated expression of Pcsk9 in mice results in a low-density lipoprotein receptor knockout phenotype. Proc Natl Acad Sci USA 101(18):7100–7105
Rashid S, Curtis DE, Garuti R, Anderson NN, Bashmakov Y, Ho YK, Hammer RE, Moon YA, Horton JD (2005) Decreased plasma cholesterol and hypersensitivity to statins in mice lacking Pcsk9. Proc Natl Acad Sci USA 102(15):5374–5379
Chan JC, Piper DE, Cao Q, Liu D, King C, Wang W, Tang J, Liu Q, Higbee J, Xia Z, Di Y, Shetterly S, Arimura Z, Salomonis H, Romanow WG, Thibault ST, Zhang R, Cao P, Yang XP, Yu T, Lu M, Retter MW, Kwon G, Henne K, Pan O, Tsai MM, Fuchslocher B, Yang E, Zhou L, Lee KJ, Daris M, Sheng J, Wang Y, Shen WD, Yeh WC, Emery M, Walker NP, Shan B, Schwarz M, Jackson SM (2009) A proprotein convertase subtilisin/kexin type 9 neutralizing antibody reduces serum cholesterol in mice and nonhuman primates. Proc Natl Acad Sci USA 106(24):9820–9825
Zelcer N, Hong C, Boyadjian R, Tontonoz P (2009) LXR regulates cholesterol uptake through Idol-dependent ubiquitination of the LDL receptor. Science 325(5936):100–104
Hong C, Duit S, Jalonen P, Out R, Scheer L, Sorrentino V, Boyadjian R, Rodenburg KW, Foley E, Korhonen L, Lindholm D, Nimpf J, van Berkel TJ, Tontonoz P, Zelcer N (2010) The E3 ubiquitin ligase IDOL induces the degradation of the low-density lipoprotein receptor family members VLDLR and ApoER2. J Biol Chem 285 (26):19720–19726
Ambros V (2003) MicroRNA pathways in flies and worms: growth, death, fat, stress, and timing. Cell 113(6):673–676
Ambros V (2004) The functions of animal microRNAs. Nature 431(7006):350–355
Bartel DP (2009) MicroRNAs: target recognition and regulatory functions. Cell 136(2):215–233
Filipowicz W, Bhattacharyya SN, Sonenberg N (2008) Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet 9(2):102–114
Elmen J, Lindow M, Silahtaroglu A, Bak M, Christensen M, Lind-Thomsen A, Hedtjarn M, Hansen JB, Hansen HF, Straarup EM, McCullagh K, Kearney P, Kauppinen S (2008) Antagonism of microRNA-122 in mice by systemically administered LNA-antimiR leads to up-regulation of a large set of predicted target mRNAs in the liver. Nucleic Acids Res 36(4):1153–1162
Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Lendeckel W, Tuschl T (2002) Identification of tissue-specific microRNAs from mouse. Curr Biol 12(9):735–739
Lanford RE, Hildebrandt-Eriksen ES, Petri A, Persson R, Lindow M, Munk ME, Kauppinen S, Orum H (2010) Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science 327(5962):198–201
Esau C, Kang X, Peralta E, Hanson E, Marcusson EG, Ravichandran LV, Sun Y, Koo S, Perera RJ, Jain R, Dean NM, Freier SM, Bennett CF, Lollo B, Griffey R (2004) MicroRNA-143 regulates adipocyte differentiation. J Biol Chem 279(50):52361–52365
Rayner KJ, Sheedy FJ, Esau CC, Hussain FN, Temel RE, Parathath S, van Gils JM, Rayner AJ, Chang AN, Suarez Y, Fernandez-Hernando C, Fisher EA, Moore KJ (2011) Antagonism of miR-33 in mice promotes reverse cholesterol transport and regression of atherosclerosis. J Clin Invest 121 (7):2921–2931
Fish JE, Santoro MM, Morton SU, Yu S, Yeh RF, Wythe JD, Ivey KN, Bruneau BG, Stainier DY, Srivastava D (2008) miR-126 regulates angiogenic signaling and vascular integrity. Dev Cell 15(2):272–284
Wang S, Aurora AB, Johnson BA, Qi X, McAnally J, Hill JA, Richardson JA, Bassel-Duby R, Olson EN (2008) The endothelial-specific microRNA miR-126 governs vascular integrity and angiogenesis. Dev Cell 15(2):261–271
Harris TA, Yamakuchi M, Ferlito M, Mendell JT, Lowenstein CJ (2008) MicroRNA-126 regulates endothelial expression of vascular cell adhesion molecule 1. Proc Natl Acad Sci USA 105(5):1516–1521
Zhang Q, Kandic I, Kutryk MJ (2010) Dysregulation of angiogenesis-related microRNAs in endothelial progenitor cells from patients with coronary artery disease. Biochem Biophys Res Commun 405 (1):42–46
Boettger T, Beetz N, Kostin S, Schneider J, Kruger M, Hein L, Braun T (2009) Acquisition of the contractile phenotype by murine arterial smooth muscle cells depends on the Mir143/145 gene cluster. J Clin Invest 119(9):2634–2647
Cordes KR, Sheehy NT, White MP, Berry EC, Morton SU, Muth AN, Lee TH, Miano JM, Ivey KN, Srivastava D (2009) miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature 460(7256):705–710
Elia L, Quintavalle M, Zhang J, Contu R, Cossu L, Latronico MV, Peterson KL, Indolfi C, Catalucci D, Chen J, Courtneidge SA, Condorelli G (2009) The knockout of miR-143 and -145 alters smooth muscle cell maintenance and vascular homeostasis in mice: correlates with human disease. Cell Death Differ 16(12):1590–1598
Xin M, Small EM, Sutherland LB, Qi X, McAnally J, Plato CF, Richardson JA, Bassel-Duby R, Olson EN (2009) MicroRNAs miR-143 and miR-145 modulate cytoskeletal dynamics and responsiveness of smooth muscle cells to injury. Genes Dev 23(18):2166–2178
Raitoharju E, Lyytikainen LP, Levula M, Oksala N, Mennander A, Tarkka M, Klopp N, Illig T, Kahonen M, Karhunen PJ, Laaksonen R, Lehtimaki T (2011) miR-21, miR-210, miR-34a, and miR-146a/b are up-regulated in human atherosclerotic plaques in the Tampere Vascular Study. Atherosclerosis
Mitchell PS, Parkin RK, Kroh EM, Fritz BR, Wyman SK, Pogosova-Agadjanyan EL, Peterson A, Noteboom J, O’Briant KC, Allen A, Lin DW, Urban N, Drescher CW, Knudsen BS, Stirewalt DL, Gentleman R, Vessella RL, Nelson PS, Martin DB, Tewari M (2008) Circulating microRNAs as stable blood-based markers for cancer detection. Proc Natl Acad Sci USA 105(30):10513–10518
Zomer A, Vendrig T, Hopmans ES, van Eijndhoven M, Middeldorp JM, Pegtel DM (2010) Exosomes: Fit to deliver small RNA. Commun Integr Biol 3 (5):447–450
Fichtlscherer S, De Rosa S, Fox H, Schwietz T, Fischer A, Liebetrau C, Weber M, Hamm CW, Roxe T, Muller-Ardogan M, Bonauer A, Zeiher AM, Dimmeler S (2010) Circulating microRNAs in patients with coronary artery disease. Circ Res 107 (5):677–684
Zampetaki A, Kiechl S, Drozdov I, Willeit P, Mayr U, Prokopi M, Mayr A, Weger S, Oberhollenzer F, Bonora E, Shah A, Willeit J, Mayr M (2010) Plasma microRNA profiling reveals loss of endothelial miR-126 and other microRNAs in type 2 diabetes. Circ Res 107 (6):810–817
Vickers KC, Palmisano BT, Shoucri BM, Shamburek RD, Remaley AT (2011) MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat Cell Biol 13 (4):423–433
Fazi F, Rosa A, Fatica A, Gelmetti V, De Marchis ML, Nervi C, Bozzoni I (2005) A minicircuitry comprised of microRNA-223 and transcription factors NFI-A and C/EBPalpha regulates human granulopoiesis. Cell 123(5):819–831
Izumi B, Nakasa T, Tanaka N, Nakanishi K, Kamei N, Yamamoto R, Nakamae T, Ohta R, Fujioka Y, Yamasaki K, Ochi M (2011) MicroRNA-223 expression in neutrophils in the early phase of secondary damage after spinal cord injury. Neurosci Lett 492 (2):114–118
Sugatani T, Hruska KA (2007) MicroRNA-223 is a key factor in osteoclast differentiation. J Cell Biochem 101(4):996–999
Krutzfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, Stoffel M (2005) Silencing of microRNAs in vivo with ‘antagomirs’. Nature 438(7068):685–689
Acknowledgments
The authors would like to thank Dr. Yajaira Suárez for her helpful comments and stimulating discussions. C.F.-H. laboratory is supported by grants from the National Institute of Health (R01HL106063 and R01HL107953). We apologize to those whose work could not be cited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Goedeke, L., Fernández-Hernando, C. Regulation of cholesterol homeostasis. Cell. Mol. Life Sci. 69, 915–930 (2012). https://doi.org/10.1007/s00018-011-0857-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-011-0857-5