
Abstract
DNA methylation is an important part of epigenetics, which can regulate gene expression, cell differentiation, growth and development in plants. In this study, the salt-sensitive variety Chuanqiao No.2 of Tartary buckwheat was used as experimental material. Different concentrations of 5-azaC were used to infect Tartary buckwheat leaves and combined with gene expression analysis, it is determined that the appropriate concentration of 5-azaC treatment is 100 µM. DNA methylation often occurs in the gene promoter region, causing gene transcription changes, and thus regulating the response to salt stress. By CpG island prediction and promoter region methylation site analysis of FtNHX1 gene, it was found that FtNHX1 gene expression was significantly regulated by DNA methylation. Moreover, after salt stress for 6 h, DNA methylation rate of FtNHX1 gene was increased. This increase of DNA methylation rate in a short time may inhibit over-expression of FtNHX1 gene.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Buckwheat is a dicotyledonous plant of Fagopyrum Mill., belonging to Polygonaceae. The main cultivated species are common buckwheat (F. esculentum Moench) and Tartary buckwheat (F. tataricum [L.] Gaertn.). Buckwheat is drought resistant and leanness tolerant, with adaptability and short growth cycle (80–90 days). Buckwheat is a crop species, but it is different from rice and wheat; not belonging to Poaceae family. Rather, it is a crop with edible and medicinal value. Buckwheat is rich in protein, dietary fiber, trace mineral elements and vitamins, as well as flavonoids and rutin. The latter two components are not found in many gramineous crops. They function in human health in lowering blood cholesterol and preventing cardiovascular sclerosis, that can also help treat hypertension and diabetes (Bonafaccia et al. 2003; Fabjan et al. 2003; Kim and Kim 2006).
Soil salinization is a worldwide problem. At present, the area of saline alkali land in the world has reached 1 × 109 hm2, the total area of soil salinization in China has reached 0.99 × 108 hm2, which is one of the largest saline alkali land countries in the world (Wang et al. 2020). Studies have shown that the damage of NaCl stress on cotton germination and seedling growth mainly includes three aspects: First, osmotic stress, in which the salt concentration in the soil is too high, and the osmotic potential will be reduced. So, the water potential of plant cells will be greater than that of soil solution, which will affect the water content of seeds, and the plants begin to dehydration or even die. The second is the ionic toxicity. Under salt stress, plant cells absorb a large amount of Na+, and the concentration of other nutrient ions (such as Mg2+ and Ca2+) required for plant growth decreases, causing damage to plants. This is the main damage caused by salt to plants. Third, salt inhibits the activity of oxidase and breaks the balance between the production and elimination of reactive oxygen species in plants. It can accumulate more reactive oxygen species in plants and cause irreversible damage to plant cell membrane (McCord 2000; Sun et al. 2000; Yang et al. 2006; Su 2012).
DNA methylation is a common epigenetic phenomenon and a widely studied epigenetic regulation mechanism. DNA methylation generally refers to the methylation process on the fifth carbon atom of CpG dinucleotide. According to the different bases before and after the carbon atom, it can be divided into CG, CHG and CHH. It is a product formed by transferring methyl to DNA cytosine through DNA methyltransferase, which is called 5-methylcytosine (Kinoshita et al. 2004; Zhang and Jacobsen 2006; Cokus et al. 2008). DNA methylation is conserved in mammals and plants. This precise conserved pattern plays an important role in the development of genomic DNA methylation (Zhang et al. 2018). Therefore, DNA methylation is an important step in many biological processes. It is found that the destruction of DNA methylation can lead to abnormal growth and development of animals and plants, such as immature tomato fruit and DNA death of mouse embryo (Robertson 2005; Cortellino et al. 2011; Lang et al. 2017).
Taking the salt-sensitive variety Chuanqiao No.2 of Tartary buckwheat as the material, the relationship between DNA methylation level and the expression of salt tolerance gene was analyzed from the perspective of DNA methylation, the effects of different DNA methylation levels on salt tolerance of Tartary buckwheat were understood, and the role of DNA methylation in salt stress adaptation of Tartary buckwheat was determined, so as to provide a certain basis for the study of the molecular regulation mechanism of plant salt tolerance.
2 Material and methods
2.1 Plant material and treatment
Salt-sensitive variety Chuanqiao No.2 of Tartary buckwheat was used as the material. Full seeds were selected, shaken and disinfected with 1% sodium hypochlorite for 5 min, and washed with sterile water for 3 times, each time for no less than 2 min. Five seeds were evenly sown in each small plastic basin. After emergence, the seedlings were watered with 1/2 Hoagland nutrient solution, once every 3 days, with routine management, natural light, relative humidity of about 60%, and day and night temperature of 26 °C/18 °C. When the seedlings grew to the three leaf stage, the following five treatments were started: (1) 5-Azacytidine (5-azaC, a DNA methylation inhibitor) with different concentrations (10, 50, 100, 200, 500 µM) was used as the infection solution for 3 days; (2) 100 mM NaCl treatment for 2 days; (3) 100 mM NaCl treatment for 2 days and then 100 µM 5-azaC infection for 3 days; (4) 100 µM 5-azaC infection for 3 days; (5) 100 µM 5-azaC infection for 3 days and then 100 mM NaCl treatment for 2 days. Hoagland nutrient solution was used as control (CK). Each treatment was repeated 3 times.
2.2 Screening of DNA methylation related genes
Based on the transcriptome sequencing results of Tartary buckwheat, the functional annotation information of the four major databases was used to screen the genes that may be related to DNA methylation, and the promoter sequences of these genes were extracted from the transcriptome information. The gene promoter sequence was analyzed by Methyl Primer Express V1.0 software. Based on containing the CpG island (CpG island is a special fragment rich in CG dinuclear glycine contained in the biological genome. Generally, if the GC concentration is higher than 40%, it is defaulted as CpG island. The probability of CpG island in the gene promoter region or exon region is very high) combined with the changes of gene expression after demethylation, the genes studied in this experiment were screened.
2.3 Infection method
Prepare 5-azaC of corresponding concentration as infection solution (avoid light at 4 °C). Avoid light during infection. Before infection, 5 small holes were evenly punched on the leaves of Tartary buckwheat with a needle, and the whole leaf was immersed in the infection solution for 10–15 s. After infection, it was treated in dark for 8 h every day. After repeating this process for 3 days, the follow-up experiments can be carried out.
2.4 Total RNA extraction from Tartary buckwheat
RNA was extracted according to the method of Takara Plant Total RNA Extraction Kit. The quality was detected by electrophoresis and the RNA concentration was determined by spectrophotometer.
2.5 Reverse transcription method
gDNA was obtained (for subsequent fluorescence quantitative template): prepare several RNase-free centrifuge tubes, prepare the reagent according to Step 1 in Table S1, and finally add RNA samples. The PCR program was set to 42 °C for 3 min, and the reaction was terminated at 4 °C to remove genomic DNA. The reagent was then added according to the Step 2, and the reaction was briefly centrifuged and mixed. The PCR procedure was set at 42 °C for 15 min and 85 °C for 1 min, and the reaction was terminated at 4 °C. gDNA are obtained (all reagents and mixtures were prepared in ice bath). After the reaction, the whole system was taken out and diluted with 30 μL RNase-free dH2O for real-time quantitative PCR.
cDNA was obtained (for subsequent gene sequence amplification to detect mutation): prepare several RNase-free centrifuge tubes and prepare the reaction solution system of Step 1 according to Table S2. The reaction solution system of Step 1 was briefly centrifuged and mixed. The PCR program was set to 65 °C for 5 min. Cooling after ice bath can denature RNA and improve reverse transcription efficiency. The reaction solution system of Step 2 was then added to each test tube and briefly centrifuged and mixed. The PCR procedure was set to 42 °C for 60 min and 70 °C for 15 min. The reaction was terminated at 4 °C to obtaine cDNA (all reagents and the mixture were prepared in ice bath).
2.6 Total DNA extraction from Tartary buckwheat
The total DNA of Tartary buckwheat leaves was extracted by improved CTAB method.
2.7 DNA bisulfite transformation of Tartary buckwheat
It was added 1 mL ddH2O, 200 μL CT conversion Diluent and 100 μL CT conversion Buffer into a tube of CT conversion Powder, mixing and rotating for at least 1 min until dissolved, and prepared solution CT Conversion Mix (one tube can be used for 10 reactions, preferably available at any time).
The reaction solution (Table S3) was added to 200 μL sterilized PCR tube, mixed and briefly centrifuged to collect the reaction solution. After the reaction, the product was purified for subsequent experiments.
2.8 BPS technology primer design principles
In view of the particularity and difficulty of methylation amplification in promoter region, the fragment amplified each time should not exceed 600 bp. If the gene sequence is long, it is suggested to design primers in sections for amplification.
The segmentation of gene sequence can be analyzed by software to ensure that the segmented sequence contains at least 3 CG loci.
The designed primer sequence should not cross the CG site as far as possible, and the length should not be short.
The GC concentration of segmented detection sequence should be guaranteed, generally not less than 35%, otherwise it will affect the amplification efficiency.
2.9 Real-time fluorescence quantitative PCR technology (RT-qPCR)
RT-qPCR was used to detect the changes of gene expression. The research gene sequence was extracted from the transcriptome data of Tartary buckwheat, and the quantitative primer design was completed on the NCBI website. The following design principles were followed: blasting the sequence to determine the location of the conserved region. Generally, the product length is 200–300 bp. The primer length is generally 18–25 bp. The GC concentration is usually between 45 and 55%. Prepare the reaction solution system according to the RT-qPCR kit provided by Novizan (Table S4), and the primers for RT-qPCR are shown in Table S5. The prepared reaction solution system was dispersed and mixed, centrifuged briefly, and then put into RT-qPCR instrument for detection (the whole process was carried out in ice bath and avoided light). The relative expression of genes was calculated by 2−∆∆CT method (Livak and Schmittgen 2001). The instrument procedure was set as follows: 95 °C for 10 s, 40 cycles, including 95 °C for 5 s, 55 °C for 30 s and 72 °C for 20 s. The melting process includes 95 °C for 1 min, 55 °C for 30 s and 95 °C for 30 s, and then 25 °C for 1 min. After cooling, it was stored.
2.10 Data analysis
Different concentrations of 5-azaC treatment and different NaCl and 5-azaC treatment time were subjected to a TWO-WAY analysis of variance (TWO-WAY ANOVA) and mean values compared by Tukey test (Wang et al. 2010). All statistical analyses were performed using SPSS 17.0 software.
3 Results
3.1 Determination of 5-azaC infection method
5-azaC, full known as 5-azacytosine nucleoside, is a cytosine analog. It can directly act on 5-mC in DNA or RNA and take away methyl to achieve demethylation, to reduce the methylation level of genomic DNA, which is called methylation inhibitor. In this study, seedlings were treated with 5-azaC smear, injection and infection, and the gene expression level was measured. The gene expression levels of Tartary buckwheat seedlings treated by smear method did not change compared with the control, indicating that the smear method can not make 5-azaC enter the plant to play the role of demethylation. Compared with the control group, the gene expression of seedlings treated with injection was changed. However, due to the small and thin leaves of seedlings, it was difficult to inject, and seedlings were easy to be damaged. When the seedlings were treated with infection method, only a small needle was needed to prick a few holes in the leaves, which could not only reduce the damage to the seedlings, but also change compared with the control group from the result of gene expression. Therefore, the seedlings were finally treated by infection.
3.2 Determination of 5-azaC concentration and effect of DNA demethylation on FtNHX1 gene expression
Transcriptome results showed that FtNHX1 gene had high homology with many plant NHX amino acid sequences. It was a Na+/H+ antiporter, and its expression was significantly induced by salt stress. After the leaves were infected with DNA methyl inhibitor, the expression of FtNHX1 gene was analyzed. Figure 1 shows the changes of FtNHX1 gene expression after five concentrations of 5-azaC infected Chuanqiao No.2 seedlings. The expression level of FtNHX1 gene increased after 10, 50, 100, 200 and 500 μM 5-azaC treatment. Compared with the control, the expression level of 5-azaC treatment was increased by 340.75%, 532.87%, 399.44%, 714.52% and 38.62%, respectively. When the concentration of 5-azaC reached to 500 µM, the expression level of FtNHX1 gene began to decrease rapidly.

Changes of FtNHX1 gene expression of Tartary buckwheat after demethylation treatment. Data are the means ± standard deviation (SD) (n = 3). * indicates significant difference (P < 0.05) and ** indicates especially significant difference (P < 0.01)
5-azaC is an inhibitor of DNA methylation. In recent years, many researchers have applied 5-azaC to plant demethylation in order to study the role of 5-azac in plant growth and development. Studies showed that using 100 μM (or above) 5-azaC can affect the growth and development of wheat, such as increasing grain mass and tiller number (Chen and Wang 2011). Wu (2017) sprayed Rhododendron hybridum with different concentrations of 5-azaC for a week, and found that the flower buds broke the dormant period, differentiated and blossomed earlier, and some physiological indexes changed significantly.
As shown in Fig. 1, after 200 μM 5-azaC treatment, the expression of FtNHX1 gene was increased the most. However, combined with the growth status of Tartary buckwheat seedlings treated with different concentrations of 5-azaC (Fig. 2), when the concentration of 5-azaC exceeded 100 μM, most seedlings withered or even died. Therefore, 100 μM was selected and used as the treatment concentration of 5-azaC for subsequent experiments.

Effect of 5-azaC with different concentrations on seedling growth of Tartary buckwheat
3.3 Cloning of promoter region
This paper mainly analyzed whether DNA methylation in gene promoter region could regulate gene expression and affect the response of Tartary buckwheat to salt stress. The reasons for studying DNA methylation in gene promoter region are as follows: (1) Promoter sequence is not a DNA sequence that can be accurately located.
At present, it is generally believed that the promoter sequence is about 2000 bp upstream of the gene transcription initiation site. (2) There are conserved transcriptional sequences in the promoter sequence. The promoter acts as a switch to control and affect some transcription factors and regulate the transcriptional activity of genes. (3) The gene sequence of any species will contain multiple discontinuous CpG islands. CpG islands are usually hypomethylated without external influence. If the methylation of CpG islands changes, gene expression will change. Studies found that CpG islands exist near the transcription initiation sites of many genes (Zhang et al. 2019a; Hughes et al. 2020), so researchers will focus on the promoter region of gene transcription initiation site.
This study showed that the salt tolerant gene FtNHX1 underwent DNA demethylation after being interfered by exogenous inhibitors. Next, it is necessary to study whether DNA methylation in the promoter region of the gene regulates gene expression through transcription initiation sites, so as to make Tartary buckwheat respond to salt stress quickly. Combined with the sequencing results of Tartary buckwheat transcriptome and the genomic information of Zhang et al. (2017), the 2000 bp sequence upstream of the transcription start site of Tagopyrum tartaris was extracted as the promoter sequence, and the promoter sequence was piecemeal amplified by PCR (PCR primers are shown in Table S6) to ensure the sequence accuracy of the sequence and check whether the sequence base was mutated for subsequent research.
Figures 3 and 4 show the sequencing results of PCR amplification containing only CpG island sequence in the gene promoter region (due to the results of many segmented amplification, only the sequencing results containing CpG island sequence are shown here, and the other two sequences have no base mutation). Figure 3 shows the electrophoresis results, and Fig. 4 shows the sequencing results of the 1270–1833 bp sequence of FtNHX1 gene, with its CpG island located at 1306–1790 bp. After PCR amplification, agarose gel electrophoresis showed that the bands were bright and no heterozygosity. Sequencing showed that there was no base mutation in the whole sequence. These results indicated that the extracted promoter sequence can be used as a control sequence to compare the changes of DNA methylation sites with the sequencing results under salt stress.

FtNHX1 gene sequencing result of CpG island sequences of Tartary buckwheat

FtNHX1 gene electrophoresis result of CpG island sequences
3.4 Expression analysis of FtNHX1 gene
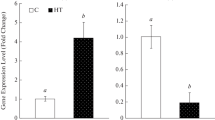
NHX1 was first identified as a Na+/H+ antiporter gene in yeast and has been proved to be directly related to plant salt tolerance (Yang et al. 2005). At present, the gene has been cloned in many plants, such as Arabidopsis AtNHX1, wheat TaNHX1, rice OsNHX1 and alfalfa MsNHX1 (Nass et al. 1997). Liu et al. (2017) cloned the FtNHX1 gene of Tartary buckwheat and analyzed it bioinformatics. Figure 5 showed that the expression of FtNHX1 gene in Chuanqiao No. 2 reached the maximum at 12 h under NaCl stress, which was increased by 4127.26% compared with the control. After NaCl + 5-azaC treatment, the expression of FtNHX1 gene decreased first and then increased after 12 h. After 5-azaC treatment, the expression of FtNHX1 gene in Chuanqiao No.2 increased significantly at 6 h and reached the highest expression at 48 h, which was increased by 96.82% compared with the control. After 5-azaC + NaCl treatment, compared with 5-azaC treatment, the expression of FtNHX1 gene in Chuanqiao No.2 was increased by 1359.10% at 12 h.

Relative expression of FtNHX1 gene of Tartary buckwheat after demethylation treatment. Data are the means ± standard deviation (SD) (n = 3). Values followed by different letters indicate significant difference (P < 0.05)
3.5 DNA methylation analysis of FtNHX1 gene under salt stress
Using MethPrimer software (Li 2007) (http://www.urogene.org/methprimer/), the distribution of CG sites in the promoter region of FtNHX1 gene and the regional distribution of CpG island (parameter setting CG concentration≧50%) were analyzed. As shown in Fig. 6, the CG high enrichment region of the gene is 1450–1600 bp. Further analysis showed that the CpG island was close to one end of the gene ATG (translation start site), and is distributed at 1306–1790 bp in the promoter region. We found a high enrichment region of CG, at about 500 to 1000 bp. BSP sequencing analysis was performed according to the location of CpG island. Table 1 shows the BSP sequencing primers.

CG enrichment and BSP detection in FtNHX1 promoter region
According to the enrichment degree of CG sites, the whole promoter sequence was divided into three segments (detection regions 1, 2 and 3). Each segment contained a CG enrichment region of no more than 600 bp, which was amplified by PCR after bisulfite transformation. Table 2 shows the DNA methylation in the promoter region of FtNHX1 gene. After 6 h of salt stress, the methylation rate of Chuanqiao No.2 in detection region 1 did not change. The DNA methylation rate of Chuanqiao No.2 in detection region 2 increased significantly, which was 44.76% higher than that of the control, indicating that it has obvious hypermethylation. The fragment of detection region 3 was close to the gene translation initiation site, and the methylation ratio of Chuanqiao No.2 did not change after salt treatment. Through the comparison and analysis of sequencing results, the increase of methylation rate under salt stress was due to the hypermethylation of many cytosine (C) sites in the gene sequence. The DNA methylation pattern of Chuanqiao No.2 changed in the detection regions 1 and 3, but did not affect the total DNA methylation rate.
4 Discussion
Since DNA methylation mainly occurs in cytosine (C), CG in CpG islands is very high, and 70– 80% of CpG islands in the genome are methylated, affecting various physiological processes of plants (Chen 2017; Choi 2020; Galbally et al. 2020). According to the annotations of four databases, the genes related to DNA methylation were preliminarily screened. The CpG island in the gene promoter region was analyzed by Methy Primer software, and FtNHX1 was determined as the gene studied in this paper.
DNA methylation and demethylation play an important role in plant growth, development and resistance to various stresses. Studies have shown that Arabidopsis DNA demethylation could activate disease resistance genes and improve disease resistance in fungal infected plants (Gehring et al. 2006; Penterman et al. 2007). Demethylation can be divided into active demethylation and passive demethylation. Active demethylation is an active behavior in plants, which can be demethylated by 5-azaC treatment (Choi et al. 2002; Zhang et al. 2019b). In these studies, it is not enough to only rely on the change of gene expression as research evidence, but also need the change of gene sequence to improve the accuracy. The techniques used in this part include MSAP, MSP and BSP. MSAP technology can analyze the pattern of DNA methylation through the obtained bands, but it cannot analyze the cytosine changes at CG sites of specific genes (Peng et al. 2018). MSP and BSP were transformed into DNA by bisulfite and then amplified by PCR. The main differences between them are as follows: First, the design principles of primers are different. Secondly, MSP technology can only judge whether there is DNA methylation in the fragment by amplifying strip, while BSP technology can analyze the changes of CG sites by sequencing (Fu et al. 2012; Stenz et al. 2018; Zeng et al. 2018). We found that the expression of FtNHX1 gene in Chuanqiao No.2 was strongly induced by 5-azaC, indicating that the expression of FtNHX1 gene in Tartary buckwheat was obviously regulated by the level of DNA methylation.
In this paper, the changes of cytosine (C) in DNA sequence after salt stress for 6 h were detected by BSP technology as further evidence. Under salt stress, the promoter detection region 2 of FtNHX1 gene in Chuanqiao No.2 showed hypermethylation, which may inhibit the high expression of salt stress related genes, so it is more sensitive to salt stress. However, whether this DNA hypermethylation phenomenon will affect the expression of FtNHX1 gene needs further experiments.
References
Bonafaccia G, Marocchini M, Kreft I (2003) Composition and technological properties of the flour and bran from common and tartary buckwheat. Food Chem 80(1):9–15. https://doi.org/10.1016/S0308-8146(02)00228-5
Chen F, Wang ZC (2011) Effects of 5-azaC on development and DNA methylation in wheat. J Henan Univ 41(1):61–66. https://doi.org/10.3969/j.issn.1003-4978.2011.01.013
Chen FS (2017) Investigating the correlation of CpG density and methylation. Dalian Med University
Choi Y, Gehring M, Johnson L, Hannon M, Harada JJ, Goldberg RB, Jacobsen SE, Fischer RL (2002) Demeter, a DNA glycosylase domain protein, is required for endosperm gene imprinting and seed viability in Arabidopsis. Cell 110(1):33–42. https://doi.org/10.1016/S0092-8674(02)00807-3
Choi J, Lyons DB, Kim MY, Moore JD, Zilberman D (2020) DNA methylation and histone H1 jointly repress transposable elements and aberrant intragenic transcripts. Mol Cell 77(2):310–323. https://doi.org/10.1016/j.molcel.2019.10.011
Cokus SJ, Feng S, Zhang X, Chen Z, Merriman B, Haudenschild CD, Pradhan S, Nelson SF, Pellegrini M, Jacobsen SE (2008) Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 452(7184):215–219. https://doi.org/10.1038/nature06745
Cortellino S, Xu J, Sannai M, Moore R, Caretti E, Cigliano A, Coz ML, Devarajan K, Wessels A, Soprano D, Abramowitz LK, Bartolomei MS, Rambow F, Bassi MR, Bruno T, Fanciulli M, Renner C, Klein-Szanto AJ, Matsumoto Y, Kobi D, Davidson I, Alberti C, Larue L, Bellacosa A (2011) Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell 146(1):67–79. https://doi.org/10.1016/j.cell.2011.06.020
Fabjan N, Rode J, Kosir IJ, Wang Z, Kreft I (2003) Tartary buckwheat (Fagopyrum tataricum Gaertn.) as a source of dietary rutin and quercitin. J Agric Food Chem 51(22):6452–6455. https://doi.org/10.1021/jf034543e
Fu L, Xia Y, He J, Liu X, Chen X, Wang Y, Ding Y (2012) Data analysis and its analytical softs application on DNA methylation in tumor research. Chongqing Med. https://doi.org/10.3969/j.issn.1671-8348.2012.17.018
Galbally M, Watson SJ, van IJzendoorn M, Saffery R, Ryan J, de Kloet ER, Oberlander TF, Lappas M, Lewis AJ (2020) The role of glucocorticoid and mineralocorticoid receptor DNA methylation in antenatal depression and infant stress regulation. Psychoneuroendocrino 115:104611. https://doi.org/10.1016/j.psyneuen.2020.104611
Gehring M, Jin HH, Hsieh TF, Penterman J, Choi Y, Harada JJ, Goldberg RB, Fischer RL (2006) DEMETER DNA glycosylase establishes MEDEA polycomb gene self-imprinting by allele-specific demethylation. Cell 124:495–506. https://doi.org/10.1016/j.cell.2005.12.034
Hughes AL, Kelley JR, Klose RJ (2020) Understanding the interplay between CpG island-associated gene promoters and H3K4 methylation. BBA-Gene Regul Mech 1863(8):194567. https://doi.org/10.1016/j.bbagrm.2020.194567
Kim JK, Kim SK (2006) Physiochemical properties of buckwheat starches from different areas. Korean J Food Sci Technol 36(4):598–603. https://doi.org/10.1134/S0020168513080165
Kinoshita T, Miura A, Choi Y, Kinoshital Y, Cao X, Jacobsen SE, Fischer RL, Kakutani T (2004) One-way control of FWA imprintingin Arabidopsis endosperm by DNA methylation. Science 303(5657):521–523. https://doi.org/10.1126/science.1089835
Lang Z, Wang Y, Tang K, Tang D, Datsenka T, Cheng J, Zhang Y, Handa AK, Zhu JK (2017) Critical roles of DNA demethylation in the activation of ripening-induced genes and inhibition of ripening-repressed genes in tomato fruit. P Natl Acad Sci USA 114(22):E4511–E4519. https://doi.org/10.1073/pnas.1705233114
Li LC (2007) Designing PCR primer for DNA methylation mapping. Methods Mol Biol. https://doi.org/10.1007/978-1-59745-528-2_19
Liu X, Song J, Zhang Y, Hou L, Yu Y, Zhao F, Liu C, Dong C, Yang H (2017) Cloning and expression analysis of FtNHX1 in Tartary buckwheat. Acta Agric Boreali-Sin 32(4):49–54. https://doi.org/10.7668/hbnxb.2017.04.008
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2-△△CT method. Methods 25:402–408. https://doi.org/10.1006/meth.2001.1262
McCord JM (2000) The evolution of free radicals and oxidative stress. Am J Med 108(8):652–659. https://doi.org/10.1016/S0002-9343(00)00412-5
Nass R, Cunningham KW, Rao R (1997) Intracellular sequestration of sodium by a novel Na+/H+ exchanger in yeast is enhanced by mutations in the plasma membrane H+-ATPase. J Biol Chem 272(42):26145–26152. https://doi.org/10.1074/JBC.272.42.26145
Peng C, Sui Z, Zhou W, Hu Y, Mi P, Jiang M, Li X, Ruan X (2018) Analysis of DNA methylation of Gracilariopsis lemaneiformis under temperature stress using the methylation sensitive amplification polymorphism (MSAP) technique. J Ocean Univ China 17(3):623–631. https://doi.org/10.1007/s11802-018-3426-9
Penterman J, Zilberman D, Huh JH, Ballinger T, Henikoff S, Fischer RL (2007) DNA demethylation in the Arabidopsis genome. P Natl Acad Sci USA 104(16):6752–6757. https://doi.org/10.1073/pnas.0701861104
Robertson KD (2005) DNA methylation and human disease. Nat Rev Genet 6(8):597–610. https://doi.org/10.1038/nrg1655
Stenz L, Schechter DS, Serpa SR, Paoloni-Giacobino A (2018) Intergenerational transmission of DNA methylation signatures associated with early life stress. Curr Genomics 19(8):665–675. https://doi.org/10.2174/1389202919666171229145656
Su D (2012) Studies on physiological and biochemical responses of Halocnermum strobilaceum to salt stress and the cloning of its betaine aldehyde dehydrogenase gene. Gansu Agric Univ
Sun XF, Zheng QS, Liu YL (2000) Salinity injury to germination and growth of cotton (Gossypium hirsutum L.) at emergence and seedling stages. J Plant Resour Environ 9(3):22–25. https://doi.org/10.3969/j.issn.1674-7895.2000.03.007
Wang XA, Yang HB, Qiu NW (2010) Application of paired-sample design in plant physiology experiment. Plant Physiol Commun 46:161–164
Wang D, Zhao Y, Zhang F (2020) Screening and identification of salt-tolerant plant growth-promoting bacteria and its promotion effect on wheat seedling under salt stress. J Tritic Crops 40(1):110–117. https://doi.org/10.7606/j.issn.1009-1041.2020.01.13
Wu Y (2017) Effects of 5-azaC on flowering time and physiological indexes of Rhododendron occidentalis. Fujian Agric Forest Univ
Yang Q, Wu M, Wang P, Kang J, Zhou X (2005) Cloning and expression analysis of a vacuolar Na+/H+ antiporter gene from alfalfa. DNA Seq 16(5):352–357. https://doi.org/10.1080/10425170500272742
Yang S, Ji J, Wang G (2006) Effects of salt stress on plants and the mechanism of salt resistance in plants. World Sci Technol Res Dev 28(4):70–76
Zeng Y, Wu N, Wang NN (2018) Research progress on gene expression variation in rice under heavy metal stress. Mol Pant Beed 16(24):8179–8184
Zhang X, Jacobsen SE (2006) Genetic analyses of DNA methyltransferases in Arabidopsis thaliana. Cold Spr Harb Symp Quant Biol 71:439–447. https://doi.org/10.1101/sqb.2006.71.047
Zhang L, Li X, Ma B, Gao Q, Du H, Han Y, Li Y, Cao Y, Qi M, Zhu Y, Lu H, Ma M, Liu L, Zhou J, Nan C, Qin Y, Wang J, Cui L, Liu H, Liang C, Qiao Z (2017) The Tartary buckwheat genome provides insights into rutin biosynthesis and abiotic stress tolerance. Mol Plant 10(9):1224–1237. https://doi.org/10.1016/j.molp.2017.08.013
Zhang H, Lang Z, Zhu JK (2018) Dynamics and function of DNA methylation in plants. Nat Rev Mol Cell Biol 19(8):489–506. https://doi.org/10.1038/s41580-018-0016-z
Zhang F, Zhang Y, Yuan F, Liu Y, Chen Y, Yilidana S, Liu L, Li H (2019a) The clinical significance and methylation level of RASSF1A and survivin promoter CpG island in Kazakh esophageal squa-mous cell carcinoma. J Shanxi Med Univ 50(9):1210–1213
Zhang Y, Ruan Y, Zhao C, Xue M, Li B, Wang J, Liu Y, Wang K, Wang H (2019b) Analysis of genomic DNA methylation level in foxtail millet by Methylation Sensitive Amplified Polymorphism. J Biol Eng 35(2):263–269
Acknowledgements
We acknowledge the financial support from the National Natural Science Foundation of China (Grant No. 31371552). The research material Chuanqiao No.2 was furnished by the Research Station of Alpine Crop, Department of Agriculture and Science in Liangshan State, Sichuan Province, China.
Author information
Authors and Affiliations
Contributions
WYQ analyzed the DNA methylation and wrote the manuscript. SJN determined the relative expression of FtNHX1 gene. YHB designed the experiment and revised the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Wang, YQ., Song, JN. & Yang, HB. DNA methylation regulates the expression of salt tolerance gene FtNHX1 in Tartary buckwheat. Theor. Exp. Plant Physiol. 34, 185–195 (2022). https://doi.org/10.1007/s40626-022-00241-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40626-022-00241-5