
Abstract
A rapid method for quantitation of lytic polysaccharide monooxygenase (LPMO) in multienzyme cocktails produced by mutant strains of filamentous fungi is developed. In this method, 2,6-dimethoxyphenol and hydrogen peroxide are used as cosubstrates in a nonspecific LPMO-catalyzed reaction leading to the formation of a colored product. With this method, we determine the LPMO content in the multienzyme cocktails produced by Penicillium verruculosum recombinant strains that homologously express LPMO. The results agree closely with the findings obtained using a method based on the fine chromatographic fractionation of the cocktails followed by the qualitative and quantitative evaluation of their content by electrophoresis and mass-spectrometry and protein quantitation assays in fractions collected from chromatographic separation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Cellulose is the most widespread polymer of plant origin in nature. The yearly increase in plant biomass due to biosynthesis is estimated to be around 2 × 1011 t, with the cellulose fraction constituting around a third of this amount [1]. Cellulose chains assemble into a crystalline supramolecular structure: a few dozen cellulose molecules are bonded by hydrogen and van der Waals forces into a microfibril. In microfibrils, homogeneous, highly ordered regions alternate with inhomogeneous amorphous zones [2]. Amorphous cellulose has a loose structure and is therefore more accessible to enzymes. In contrast, crystalline, highly ordered cellulose is much more resistant to the destructive action of enzymes [3].
Filamentous fungi from the genera Trichoderma and Penicillium are producers of the most efficient multicomponent enzymatic cocktails (ECs) for the destruction of cellulose-containing raw materials [4–6]. The composition of their cellulolytic complexes includes the classical set of three types of hydrolases: endoglucanase, exo-cellobiohydrolase (CBH), and β-glucosidase. The effectiveness of cellulose enzymatic hydrolysis and the relative content of the resulting decomposition products depend on the composition balance of the cellulase complex and the activity of its individual components.
Lytic polysaccharide monooxygenases (LPMOs), enzymes that perform oxidative depolymerization of cellulose and other polysaccharides, have recently been discovered [7–10]. These are metal-dependent (specifically, Cu-dependent) enzymes of the oxidoreductase class (enzyme code 1.14.99.53-56). Typically, LPMOs that break down cellulose are not capable of the advanced destruction of the polymer; however, they can substantially enhance the efficiency of classical (hydrolytic) cellulases. With an LPMO attacking cellulose, the polymer chains at the crystal surface are oxidized, and new open ends of polymer molecules and oligosaccharides are formed, which serve as a substrate for CBH action. In addition, charged groups resulting from cellulose oxidation facilitate amorphization of cellulose and increase the availability of the substrate to cellulases, especially to endoglucanases [9, 10]. The functioning of an LPMO requires the presence of an oxygen molecule and an electron donor, and the latter can be represented by lignin biodegradation products, ascorbic and gallic acids, or other reductants including cellobiose dehydrogenase, a component enzyme of fungal enzymatic complexes [7–10].
Presently, there are very few methods for evaluating LPMO activity by measuring the initial rate of the enzymatic reaction because LPMOs per se exhibit quite modest catalytic rate constants. The LPMO activity is evaluated through the high-performance liquid chromatography (HPLC) identification of oxidative degradation products or by mass spectrometry [11–13]. However, generation of oxidized oligosaccharides in the quantities necessary for a chromatographic analysis requires many hours of enzymatic reaction, and mass spectrometry, while being a qualitative analytical technique, cannot provide the quantitative evaluation of the enzyme activity.
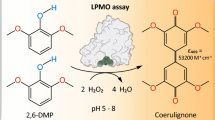
Around 2 years ago, it was found that hydrogen peroxide can also function as a cosubstrate in LPMO-catalyzed reactions, together with dioxygen [14]. The method for determining LPMO catalytic activity proposed in [15] is based on the peroxidase activity of the enzyme, with cellulose being replaced with a nonspecific model substrate, i.e., 2,6-dimethoxyphenol (2,6-DMP). In the presence of H2O2, two 2,6-DMP radicals dimerize to yield a colored product (Fig. 1).

LPMO-catalyzed oxidation of 2,6-DMP by hydrogen peroxide [15].
The mutant strains of the Penicillium verruculosum fungus are highly active cellulase producers [5, 6]; however, the ECs derived from them do not usually contain LPMOs in significant amounts. In our previous work [16], in this fungus, we revealed and sequenced the silent gene lpmo1, which encodes an LPMO of the AA9 family of auxiliary activities for glucoside hydrolases (http://www.cazy.org/AA9.html). This gene was cloned in the P. verruculosum B1-537 auxotrophic strain under the control of a strong promoter of cbh1 gene. As a result, we obtained new recombinant strains of this producer that are able to secrete a complex of cellulolytic enzymes containing up to 57% LPMO of the total protein content in a culture liquid [16]. However, to evaluate the LPMO content in the EC, we had to resort to quite a laborious procedure based on fine chromatographic fractionation of the EC and involving the use of mass spectrometry and electrophoresis.
The aim of this study is to develop a rapid method for quantification of LPMO in cellulase multienzyme preparations using H2O2 and 2,6-DMP [15] as the substrates.
METHODS AND MATERIALS
Strains and ECs
The ECs used in this work were prepared by lyophilization of culture fluids based on P. verruculosum recombinant strains with a homologously expressed LPMO under the control of the cbh1 gene promoter in combination with either the CBH I signal sequence (sample series: sCBH1-8, sCBH1-14, and sCBH1-17) or the native signal sequence (sample series: sLPMO1-1, sLPMO1-4, sLPMO1-8, sLPMO1-9) [16].
Reagents
Buffer solutions were prepared using reagents supplied by Bio-Rad Laboratories (United States), Panreac (Germany), Helicon (Russia), and Reakhim (Russia). 2,6-DMP (Thermo Fisher Scientific, United States) and 3% hydrogen peroxide (OOO Tula Pharmaceutical Factory, Russia) were used as substrates.
LPMO Isolation and Purification
Chromatographic fractionation of the sLPMO1-9 EC was carried out using a NGC liquid chromatography system (Bio-Rad Laboratories, United States) equipped with a spectrophotometric detector. At the first stage, the EC (protein content, 10 mg) over-precipitated with ammonium sulfate was desalinated using a column filled with Biogel P4 (Bio-Rad Laboratories, United States) and then applied to a Source 15Q anion-exchange column (volume, 1 mL; Pharmacia, Sweden) pre-equilibrated with a 0.02 Bis-Tris-HCl buffer (pH 6.8). The proteins retained on the sorbent were gradient-eluted with NaCl from 0 to 0.4 M at a flow rate of 1 mL/min. While stirring, dry crystalline (NH4)2SO4 was added to the LPMO-containing fraction to a concentration of 1.7 M, and the protein was then purified using hydrophobic interaction chromatography on a Source 15 Isopropyl column (volume, 1 mL; Pharmacia, Sweden) pre-equilibrated with a 0.02 M sodium acetate buffer containing 1.7 M (NH4)2SO4. Elution was carried out at a rate of 0.5 mL/min using a decreasing gradient: the (NH4)2SO4 content decreased from 1.7 to 0 M.
The protein concentration in the initial EC and fractions collected from chromatographic separation were determined by a modified Lowry method [17]. The purity of isolated enzyme was evaluated by electrophoresis in 12% polyacrylamide gel (PAGE) containing sodium dodecyl sulfate (SDS) The electrophoretic separation was carried out on a Mini Protean II apparatus (Bio-Rad Laboratories, United States) according to the procedure provided by the manufacturer.
LPMO Activity Assays and Enzyme Concentration Measurements
The LPMO activity was assayed using 2,6-DMP as a model substrate according to the procedure described in [15]. Specifically, 860 μL of a 0.1 M Tris-HCl buffer (pH 7.5), 100 μL of a stock solution of 2,6‑DMP (10 mM) in the same buffer, and 20 μL of a 5 mM H2O2 solution were combined in a 2 mL test tube and preliminarily incubated at 30°C for 10 min. Subsequently, 20 μL of enzyme solution diluted beforehand to a specific concentration was added to the test tube, which marked the start of the enzymatic reaction. After 5 min, the optical absorption of the solution was measured at λ = 469 nm versus a blank solution containing the same components except for the enzyme (20 μL of the buffer was added in place of the enzyme). In calculating the concentration of the colored product, the extinction coefficient was taken to be 53 200 M–1 cm–1 [15].
RESULTS AND DISCUSSION
LPMOs that have been discovered relatively recently [7–10] as part of multienzyme systems secreted by filamentous fungi and bacteria exhibit a marked synergy when combined with hydrolytic-type cellulases, which made them an important component of commercial ECs produced for use in the bioconversion of cellulose-containing raw materials [10, 18]. In our laboratory, research into cellulolytic enzymes produced by the fungus P. verruculosum has been carried out for nearly 30 years [6, 19]. They, however, did not typically display a notable LPMO-like activity (this pertains to B1-537 as well, one key cellulolytic strain of P. verruculosum), despite the fact that ECs based on modern mutant strains of this producer are efficient in hydrolyzing cellulose-containing substrates [19]. Previously, using B1-537 as a host for cloning and enhanced homologous expression of its native silent lpmo1 gene [16], we obtained new recombinant P. verruculosum strains capable of secreting a complex of cellulolytic enzymes with the LPMO content at 9 to 57% of the total protein content (see Table 1). However, as mentioned above, the available method for assaying the LPMO content in the resulting ECs is quite laborious and includes fine chromatographic fractionation of the EC. Although the LPMO content can be evaluated qualitatively from the electrophoresis data by the intensity of the protein band associated with the considered enzyme (Fig. 2), in our case, the assay was complicated by the fact that some quantity of endoglucanase II of P. verruculosum, which has the molecular weight similar to that of LPMO, was also present in the same band.

SDS-PAGE electrophoresis of the prepared ECs: (1) sLPMO1-1, (2) sLPMO1-4, (3) sLPMO1-8, and (4) sLPMO1-9. M: molecular markers (molecular weight of reference proteins (kDa)). The protein band corresponding to the LPMO is indicated with an arrow (contains some quantity of endoglucanase II, as an impurity).
Recently, a rapid and sensitive spectrophotometric method for the evaluation of the nonspecific peroxidase activity of LPMO, in which 2,6-DMP is used as the substrate, has been proposed [15]. Here, we used this method to evaluate the LPMO content of P. verruculosum multienzyme cocktails.
The reference preparation (i.e., a cocktail) based on the B1-537 strain did not exhibit notable activity with respect to 2,6-DMP in the presence of hydrogen peroxide, whereas in the case of EC containing homologous LPMO, the activity varied within the range of 6 to 64 U/g protein (Table 1). To use these data in order to estimate the percentage of LPMO in the EC, we must know the specific activity of the pure enzyme, i.e., free of any impurity proteins. For this, we isolated the LPMO from sLPMO1-9 EC and purified it until it became homogeneous. The protein’s elution profile registered during anion-exchange chromatography on a Source 15Q column is shown in Fig. 3, and the results of the PAGE electrophoresis of the purified enzyme are presented in Fig. 4. The activity of homogeneous LPMO with respect to 2,6-DMP was found to be 103 U/g. The LPMO content in the ECs under study was calculated as the ratio of the EC activity to the activity of homogeneous LPMO (Table 1). The findings obtained with the new method agree closely with the results obtained using the complicated procedure for assaying the LPMO content in the same ECs, which is based on fine chromatographic fractionation followed by qualitative and quantitative evaluation of the cocktail content by electrophoresis and mass spectrometry and protein assays in the fractions collected from chromatographic separation. The correlation coefficient for the data obtained by the two methods is 0.9832 (R2 = 0.9667). Considering that the two methods produced similar results, the more facile rapid method, which is based on measuring the peroxidase-like activity of the LPMO in reaction with 2,6-DMP (i.e., the substrate), can be used for the quantitative assaying of this enzyme in multicomponent ECs based on the P. verruculosum recombinant strains and probably other microbial producers of glycoside hydrolases.

Fractionation of sLPMO1-9 EC on a Source 15Q anion-exchange column: (1) protein elution profile and (2) NaCl gradient. The LPMO peak is indicated with an arrow.

SDS-PAGE electrophoresis of purified LPMO. M: molecular markers (molecular weight of reference proteins (kDa)).
REFERENCES
Tiunova, N.A. and Kretovich, V.L., in Tsellyulazy mikroorganizmov (Cellulases of Microorganisms), Moscow: Nauka, 1981, p. 40.
Kirk-Othmer Encyclopedia of Chemical Technology, New York: Wiley, 2001, vol. 13, p. 866.
Sinitsyn, A.P., Gusakov, A.V., and Chernoglazov, V.M., in Biokonversiya lignotsellyuloznykh materialov (Bioconversion of Lignocellulosic Materials), Moscow: Mosk. Gos. Univ., 1995.
Cherry, J.R. and Fidantsef, A.L., Curr. Opin. Biotechnol., 2003, vol. 14, p. 438.
Gusakov, A.V., Trends Biotechnol., 2011, vol. 29, p. 419.
Gusakov, A.V. and Sinitsyn, A.P., Biofuels, 2012, vol. 3, no. 4, p. 463.
Vaaje-Kolstad, G., Westereng, B., Horn, S.J., Liu, Z., Zhai, H., Sørlie, M., and Eijsink, V.G.H., Science, 2010, vol. 330, p. 219.
Zifcalkova, L. and Baldrian, P., Fungal Ecol., 2012, no. 5, p. 481.
Kumar, G.S. and Murthy, D., Biotechnol. Biofuels, 2013, vol. 6, p. 63.
Horn, S.J., Vaaje-Kolstad, G., Westereng, B., and Eijsink, V.G.H., Biotechnol. Biofuels, 2012, vol. 5, p. 45.
Westereng, B., Ishida, T., Vaaje-Kolstad, G., Wu, M., Eijsink, V.G.H., Igarashi, K., Samejima, M., Stahlberg, J., Horn, S.J., and Sandgren, M., PLoS One, 2011, no. 6, E27807.
Westereng, B., Agger, J.W., Horn, S.J., Vaaje-Kolstad, G., Aachmann, F.L., Stenstrøm, Y.H., and Eijsink, V.G.H., J. Chromatogr. A, 2013, no. 1271, p. 144.
Bey, M., Zhou, S., Poidevin, L., Henrissat, B., Coutinho, P.M., Berrin, J.-G., and Sigoillota, J.-C., Appl. Environ. Microbiol., 2013, vol. 79, p. 488.
Bissaro, B., Rohr, A.K., Muller, G., Chylenski, P., Skaugen, M., Forsberg, Z., Horn, S.J., Vaaje-Kolstad, G., and Eijsink, V.G.H., Nat. Chem. Biol., 2017, vol. 13, p. 1123.
Breslmayr, E., Hanzek, M., Hanrahan, A., Leitner, C., Kittl, R., Santek, B., Oostenbrink, C., and Ludwig, R., Biotechnol. Biofuels, 2018, vol. 11, p. 79.
Semenova, M.V., Gusakov, A.V., Volkov, P.V., Matys, V.Y., Nemashkalov, V.A., Telitsin, V.D., Rozhkova, A.M., and Sinitsyn, A.P., Mol. Biol. Rep., 2019, vol. 46, no. 2, p. 2363.
Peterson, G.L., Anal. Biochem., 1979, vol. 100, p. 201.
Cannella, D. and Jørgensen, H., Biotechnol. Bioeng., 2014, vol. 111, p. 59.
Sinitsyn, A.P., Osipov, D.O., Rozhkova, A.M., Bushina, E.V., Dotsenko, G.S., Sinitsyna, O.A., Kondrat’eva, E.G., Zorov, I.N., Okunev, O.N., Nemashkalov, V.A., Matys, V.Y., and Koshelev, A.V., Appl. Biochem. Microbiol., 2014, vol. 50, no. 8, p. 761.
Funding
The study was supported within the research program “Molecular Design, Structure–Function Analysis and Regulation of Enzyme Systems, Cellular Constructs, and Bionanomaterials: Fundamentals and Application in Technology, Medicine, and Environmental Protection” (state registration no. АААА-А16-116052010081-5).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
SUPPLEMENTARY INFORMATION
No supplementary information is provided.
Additional information
Translated by A. Kukharuk
Abbreviations: LPMO, lytic polysaccharide monooxygenase; EC, enzyme cocktail; 2,6-DMP, 2,6-dimethoxyphenol; CBH, cellobiohydrolase.
About this article

Cite this article
Telitsin, V.D., Semenova, M.V., Osipov, D.O. et al. 2,6-Dimethoxyphenol-Based Assay for Quantitation of Polysaccharide Monooxygenase in Multienzyme Cocktails. Moscow Univ. Chem. Bull. 75, 96–100 (2020). https://doi.org/10.3103/S0027131420020157
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.3103/S0027131420020157