
Abstract
Peptidase clan is the largest group of proteases with common ancestry as identified by structural homology. A peptidase with unknown catalytic type is referred to as an unassigned peptidase clan, which can be classified into 8 peptidase families (U32, U40, U49, U56, U57, U62, U69, and U72). The members of this clan are widely dispersed in diverse microbial pathogens and their apparent involvement in the microbial virulence is not yet known for antibiotic drug discovery. In the present study, we have analyzed their common structural and functional characteristics using evolutionary genetic analysis. As shown by our analysis, the molecular and functional characteristics of this clan diverged across the microbial pathogens. It also indicates that the members of each family might have evolved independently and a peptidase core converged to interconnect the unassigned peptidase clan. Several evolutionary constraints have been identified as selective measures from this clan that inferred on their functional evolution and divergence. Genetic diversity analysis described that many members of this clan have evolved as new molecular functions across the microbial pathogens by imposing the Darwinian positive selection. Structural analysis of this study indicates that members of this clan have a conserved fold, convergence in functional parts, and divergence in spatial structural arrangements. As the results of our study, the neofunctionalization of several unassigned peptidases provides a full virulence for microbial pathogens occupying at the different pathophysiological niche.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Molecular virulence of some bacteria and parasites can survive and reproduce inside phagocytic cells. Cell surface proteins, toxic proteins, and hydrolytic enzymes are some of the common virulence factors produced by the pathogenic bacteria. Microbial infections depend on a combination of virulence factors, the immune status of the host, and the innate resistance of the host. Molecular virulence of any pathogenic bacteria is usually determined by the proteins in nature (Henderson and Nataro 2001). Proteinase is a hydrolytic enzyme that breaks down peptide bonds in proteins and peptides. It can be classified into 8 main clans based on the catalytic residues (Rawlings and Barrett 1993; Rawlings et al. 2004, 2006) and further grouped into 225 families with 71 peptide inhibitors (Rawlings et al. 2012).
A peptidase with the unknown active site and the catalytic site is grouped as an unassigned peptidase clan (Hicks et al. 2001). A clan is a group of families that are thought to share a common ancestor. According to the MEROPS 9.5, 8 peptidase families (U32, U40, U49, U56, U57, U62, U69, and U72) belong to unassigned peptidase clan. These family members are widely distributed in diverse microbial pathogens (Rawlings 2010, Rawlings et al. 2012) and play important roles in the microbe-microbe and microbe-host interactions. Some members of this clan allow a pathogen to evade the host’s immune response, and degrade some extracellular components in the host tissues (Popoff and Bouvet 2009). Consequently, a redox potential has been lowered in the infected tissues due to the buildup of metabolites from protein degradation (Mittal et al. 2014). It ensures a competitive advantage to the pathogens to possibly implicate for microbial infections and diseases. However, it is still debated to evaluate potential associations between unassigned peptidases and microbial virulence in contrast to the mammalian counterparts.
To date, crystallographic structures have been solved only for the Thermotoga maritima DNA gyrase, a member of the unassigned peptidase U62 family (Rife et al. 2005). The specificity, structures, and biochemical functions of microbial unassigned peptidases are not yet known in detail compared to those found in humans (Duarte et al. 2016). Most recently, apparent involvements of the members of this clan were extensively analyzed to understand the microbial pathogenesis and virulence mechanisms based on the evolutionary aspects of their structures and functions (Sharov 2014; Chellapandi and Prisilla 2017).
Several evolutionary constraints have been identified from the structures and functions of some virulence proteins or protein families (Chellapandi et al. 2013, 2018, 2019; Prathiviraj et al. 2016; Prisilla et al. 2016; 2017). A common functional core is conserved and substantially established across unassigned peptidases of microbial pathogens by evolutionary pressure (Barrett and Rawlings 2007). In this study, evolutionary genetic analysis was carried out to understand the molecular involvement of unassigned peptidases in the microbial virulence and pathogenesis. Current bioinformatics resources can ensure our knowledge of explicating the molecular hypothesis of their structures, functions, and evolution behind the development of human therapeutics.
Materials and methods
Dataset
The unassigned peptidase’s sequences were retrieved from the UniProt database (http://www.uniprot.org/). These sequences were used as templates for the identification of respective similarity hits by searching against microbial pathogens using the National Center for Biotechnology Information-Delta-Blast program (Boratyn et al. 2012). A topology of conserved domains in each target peptidase was identified with the Simple Modular Architecture Research Tool 8.0 server (Letunic and Bork 2018). The sequences with low sequence similarity, and partially conserved domain removed from the dataset (Table S1).
Evolutionary genetic analysis
Multiple sequence alignment was separately carried out for each family by ClustalX 2.0 software (Thompson et al. 1997). The aligned sequences were inspected manually and refined to obtain a significant alignment. Homogeneous patterns among all lineages were searched with a neighbor-joining algorithm using the Molecular Evolutionary Genetics Analysis (MEGA) X software (Kumar et al. 2018). The final phylogenetic tree was manually corrected and constructed with 1000 bootstrapping replicates. A supertree was constructed for the overall clan members with the COBALT server using a Fast Minimum Evolution algorithm (Papadopoulos and Agarwala 2007). Phylogenetic divergence across each family was calculated from corresponding trees by SplitsTree 4.0 software (Huson and Bryant 2006) using the BioNJ algorithm. Evolutionary genetic analyses were performed with MEGA software. Recombination frequency and population-scaled mutation rate were calculated by Recombination Detection Program 3.0 software using the Recomb2007 method (Martin et al. 2010). Coefficients for the type-I (θI) and type-II (θII) functional divergence were examined with DIVERGE 3.0 software (Gu et al. 2013) using the Kimura model with 100 bootstraps. Synonymous and non-synonymous substitution sites subject to the Darwinian selection were calculated by a selective strength (Ka/Ks ratio, ω) using the Ka/Ks calculator 2.0 (Wang et al. 2010) and HyPhy 2.2.1 program (Pond et al. 2005). The evolution rate of each family was calculated by Rate4Site 2.01 software (Mayrose et al. 2004). Evolutionary patterns were inferred between two similarity sequences of microbial pathogens by EMBOSS ALIGN (http://www.ebi.ac.uk/Tools/psa/emboss_needle/) using different amino acid substitution matrices.
Structural and functional analysis
The structural homologs were searched in the protein databank with the Position-Specific Iterated-Basic Local Alignment Search Tool (Altschul et al. 1997) and then used as structural templates for the prediction of 3D structures from the target sequences using the SWISS-MODEL (Biasini et al. 2014). The predicted structures were analyzed for their quality and accuracy by Structural Analysis and Verification Server (https://services.mbi.ucla.edu/SAVES/). The protein folding rates of all structural classes were predicted from the target sequences by the FoldRate server using a multiple regression program (Gromiha et al. 2006). The sequentially conserved motifs and regular expression patterns across unassigned peptidase families were discovered by MEME Suite 5.1.1 (Bailey et al. 2015). The PROSITE signature matches and ProRule-associated functional and structural residues were detected using the ScanProsite (de Castro et al. 2006).
Results
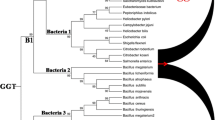
The members of this clan are found in diverse pathogenic bacteria, and many of them belong to the Enterobacteriaceae family (Table 1). Peptidases U32 and U62 are immensely dispersed in microbes compared to other families. Peptidases U32, U57, and U69 are mainly present in the pathogenic bacteria, particularly Mycobacterium tuberculosis. The phylogenetic trees were constructed for inferring the origin and evolution of microbial unassigned peptidases (Fig. 1 and Fig.S1). Each family of this clan is formed eight major groups separately, and some members from peptidases U57, U49, U69, and U62 are distinct from the respective clusters. It revealed that the conserved domains of such families slightly diverged, despite entire protein sequences. Some members in the peptidase U49 are diverged from the members of peptidase U49 and clustered with few members in the peptidase U69 (Salmonella enterica). Peptidase U32 is clustered separately within microbial pathogens.

Dendrogram for unassigned peptidase clan in the microbial pathogens, reconstructed by Fast Minimum Evolution algorithm in the COBALT server using closely related protein sequences obtained from diverse microbial pathogens. Datasets used for the phylogenetic tree construction and trees of individual families are available in the Supplementary file. The circular view of all microbial unassigned peptidases is represented in Fig. 1a. A condensed view of respective unassigned peptidase families is represented in Fig. 1b
Estimates of genetic diversity of this clan implied that the overall transition/transversion ratio is ranged from 0.60 to 1.28, in which peptidase U57 is highly diverged by a radical nucleotide substitution (Table 2). Evolution rates (α) of peptidases U49 and U69 are relatively higher than other families, reflecting that both families may undergo rate variation among sites. Estimates of phylogenetic distance and invariant sites are closely related to one another within the family. The phylogenetic diversity of peptidases U32 and U62 is expanded with diverged functions (Fig. S2).
Estimates of population-scaled recombination pinpointed that nucleotide/amino acid diversity of peptidase U57 has expanded more than other families. Duplication/shuffling events show to impact on the functional divergence of peptidases U62 and U72. Peptidases U49, U62, and U69 are diverged for functional selection by imposing the Darwinian positive selection (Table 2). It indicated that some members of this clan widely diverge in the microbial pathogens at the species level and most of them are retained by neutral evolution within closely related species (Table S2). Conserved domains of this clan are not separately diverged but a new function might have evolved at the same evolution rate in the microbial pathogens.
Besides, recombination events were observed as evolutionary constraints for the evolution of new function or to maintain existing function in the microbial pathogenic lineage. There are no recombination events detected in the peptidases U56 and U69. The recombination/mutability rate is notably higher in the peptidases U72 and U49 compared to others (Tables S3-S4). Peptidase U62 is less conserved as the result of frequent substitution mutations (Fig. S3-S4). The primary coevolved residues are contributed to the functional evolution of the members of this clan during the coevolution process (Table S5).
Peptidase U62 contains a PmbA-TldD domain expected to undergo type-I divergence by fixing the function in both groups but variable in another group (Table 3). A conserved domain across peptidase U72 undergoes type-II divergence. The rate of gene duplication events across peptidases U62 and U72 imposed by type-I divergence is faintly higher than those events that have undergone type-II divergence. An extensive analysis of common motifs provided a clue to understanding the catalytic function and substrate-binding specificity of each family in this clan (Fig. 2). The results of our study revealed that the peptidase motif is a common conserved functional core subjected to diverge for substrate recognition.

WebLogo representations of the conserved functional motifs identified across the unassigned peptidase clan in the microbial pathogens. The residue frequencies are represented by their relative height and the site-specific probabilities as total column height, reflecting informative value for conservation and function
Homology models were generated for understudied target peptidases as shown in Fig. 3a. It has been shown that all modeled proteins have exhibited a 21–61% sequence identity with dissimilar molecular functions (Table 4). All modeled proteins were validated for their structural quality and accuracy using the Ramachandran plot. It shows that modeled residues of each target proteins are in the allowed region and few residues are in disallowed regions. We predicted above 90% residues are in allowed regions in the plot. However, structural motifs in the peptidases are analogs to those present in the structural templates. It pointed out that a conserved fold and spatial structural arrangement might have evolved separately at different evolution rates. Results of fold rate prediction show the structures of peptidases U32 and U49 consisted of all-alpha classes with the fast-folding rate (Fig. 3b). Peptidase U56 structure composes unknown structural classes at different folding rates. Peptidase U57 structure has a mixed structural class with a slow folding rate. The structures of the peptidases U62, U69, and U72 have mixed structural classes with a fast-folding rate. The results of this study are considerably better to understand the entanglement of folding imprints in the arrangement of structural elements and associated molecular functions.

Graphical representation of protein models (a) and protein folding imprints (b) of unassigned peptidase clan in the microbial pathogens based on their fold rates
Discussion
Unassigned peptidase U32 family
Peptidase U32 degrades soluble fibrillar type I collagen and does not have a zinc-binding motif similar to bacterial collagenases belonging to the peptidase M9 family (Kato et al. 1992). Peptidase U32 is essential for initial penetration of the host, facilitating the prompt establishment of infection with the concomitant progression of chronic periodontitis (Han et al. 2008; Grenier and La 2011; Figaj et al. 2019). Collagenolysis mechanism of this family remains speculative due to the existence of different functional regions and domain organization. In this study, peptidase U62 from C. botulinum type A was closely related to the C. botulinum neurotoxins peptidase, as described earlier (Doxey et al. 2008). It is also found that the existence of invariant sites in different functional regions and domains has influenced the phylogenetic divergence and functional expansion of this family within microbial pathogens. Microbial peptidase U32 might have evolved by gene duplication and divergence, as previously hypothesized for clostridial collagenases (Bond and Wart 1984; Matsushita et al. 1999). The members of peptidase U32 had a distinct structure and common functional analogy to the collagenases as a result of the functional convergence (Galperin et al. 2012).
Unassigned peptidase U49 family
Lit is a constitutively expressed peptidase that hydrolyzes a single peptide bond within the universally conserved switch region (R58GV-ITI motif) of the host translation factor EF-Tu1. A putative Zinc-binding catalytic motif H160EXXHX67H232 and His169 are important for Lit peptidase activity that mediates exclusion in Escherichia coli K-12 (Copeland and Kleanthous 2005). EF-Tu1 forms a weak complex with a major capsid protein gp23 (Gol peptide) that serves as a signal for viral infection, ultimately leading to the arrest of protein synthesis and cell death before phage maturation (Georgiou et al. 1998; Bingham et al. 2000). In our study, Lit peptidase shared very little global similarity with other peptidases. Even if most of the family members in this family are related, few of them were diverged from the same family and clustered to the peptidase U69. It revealed that functional purification of this family in the microbial pathogens can be imposed by strain-specific neutral evolution at a slow non-synonymous substitution rate.
Unassigned peptidase U56 family
Homomultimeric peptidase (linocin M18) is an antilisterial bacteriocin that belongs to peptidase U56, which can hydrolyze chymotrypsin, trypsin, and casein. There is no amino acid sequence homology to any other peptidases (Boucabeille et al. 1997). Linocin M18 from T. maritima is a perspective for its antimicrobial ecological strategies in a hyperthermophilic niche (Hicks et al. 1998, 2001). As shown by our analysis, the members of this family have evolved by several evolutionary forces. We observed the coevolution process as an evolutionary force to bring functional divergence across the peptidase U56 in the microbial pathogens. It was agreed to the previous hypothesis on the coevolution of functionally constrained characters described by Wagner (1984).
Unassigned peptidase U57 family
A sporulation-specific peptidase (YabG) has a relatively complex role for proteolysis in the maturation of spore cortex and coat assembly proteins such as cotF, cotT, yrbA, spoIVA, yeeK, and yxeE (Takamatsu et al. 2000a, b). The protein modification process involved in the spore germination is mediated by comprehensive interactions among yabG, Tgl, and their substrates in the mature spores (Henriques and Moran 2000; Kuwana et al. 2006). Our study described that the evolution of such a sporulation-specific family is quite complex encompassing multiple tandem duplications (Galperin et al. 2012). We observed phylogenetic proximity between peptidase U57 and peptidase U72, suggestive of the inter-transitory evolutionary ancestry of these families. Bacillus cytotoxicus sporulation-specific peptidase has shown to diverge from the same family as a result of nucleotide/amino acid diversity during the evolution process. Interestingly, the members of this family were remarkably diverged compared to others due to amino acid substitutions and transition/transversion processes. Moreover, the protein structure of the members of this family might have evolved with mixed structural classes with a slow folding rate.
Unassigned peptidase U62 family
Microcin B17 is a ribosomally synthesized peptide produced by diverse strains of Gram-negative bacteria carrying a pMCCB17 plasmid (Allali et al. 2002). Microcin-processing peptidase 1 cleaves the microcin B17 precursor at the Gly26-Val27 in the peptide clan. Consequently, it participates in the process of controlling DNA gyrase activity by controlling cell death (ccdA) of sex factor F, which ultimately leads to cell death (Murayama et al. 1996). Phylogenetic inference of our study indicated that the members of peptidase U62 were evolutionarily distinct from distantly related organisms. A conserved domain PmbA_TldD diverged within the family members, which could be resulted due to the type-I divergence. Results of this study also suggested that the members in the peptidase U62 might have evolved via recombination process and amino acid substitutions in accordance to the earlier work (Bedau and Packard 2003; Clark et al. 2012).
Unassigned peptidase U69 family
Auto-transporters are secreted proteins that are assembled in the outer membrane of bacterial cells. AIDA-I self-cleaving autotransporter protein serves as a virulence protein in different toxigenic E. coli strains (Henderson and Nataro 2001). During the proteolytic cleavage, its processed mature passenger domain is stabilized by non-covalent interaction with the 30 kDa β-domain. It has been reported to involve in the bacterial pathogenesis (Benz and Schmidt 1992, 1993). As a result of our study, the members of this family might undergo a rate variation among sites during the evolutionary process since the evolution rate was relatively different from others (Bedau and Packard 2003). There were no recombination events and low evolutionary change for functional divergence. Interestingly, a selective strength for protein function was rather than gene function. Some members of this family might have evolved with a specific and distinct function, which is adaptive to the particular niche occupied by the microbial pathogens. As shown by our analysis, structural domains of this family member could be evolutionarily optimized during the colonization of new niches owing to the overall passenger domain scaffold being widely divergent (Kostakioti and Stathopoulos 2006; Celik et al. 2012).
Unassigned peptidase U72 family
Deamidase of Pup (prokaryotic ubiquitin-like protein) (Dop) converts the C-terminal glutamine of Pup to glutamate, which forms an isopeptide bond with an ε-amino group of lysine residues of target proteins by the enzyme PafA (proteasome accessory factor A) (Özcelik et al. 2012; Ofer et al. 2013; Prathiviraj and Chellapandi 2020). It is an evolutionary derivative of glutamine synthetases consisting of GhExE as a distinctive signature (Iyer et al. 2009). Pup depupylase/deamidase belongs to the carboxylate-amine ligase family (Barandun et al. 2012). Dop and PafA are similar in overall structure and fold with differences in loop regions. The active site is located in a broad β-sheet cradle accessible at one end (Özcelik et al. 2012). Pup-proteasome system is evaluated as a promising drug target for emerging multi-drug-resistant M. tuberculosis strains (Ofer et al. 2013; Prathiviraj and Chellapandi 2020). In the present study, the Pup protein has a conserved motif with a G[EQ] signature at the C-terminus. It was structurally unrelated to the ubiquitin fold, and the function of the protein modifier was converged. It also found that a phyletic pattern of mycobacterial PafA was closely related to the Pup and both of these proteins were genomic neighbors in all bacterial lineages. We observed a functional linkage between Pup/PafA and the archaeal-type proteasome (Pearce et al. 2008). The rate of gene duplication events across the peptidase U72 by type-I divergence was faintly more than those resulted from type-II divergence. A new molecular function of this family member might have evolved due to radical changes in the segregating sites with a high recombination rate, as described by Clark et al. (2012).
Conclusions
We conducted a detailed proteomic survey to understand molecular and functional diversity of unassigned peptidase clan in the microbial pathogens. The members of this clan are evolutionarily related, but the sequence and functional diversity are specific to the individual strains. A conserved domain (peptidase) in the respective families is conserved across the genera and evolved as distinct substrate specificity for harboring the full virulence of microbial pathogens. Genetic drift, amino acid substitutions, and coevolution are major evolutionary constraints imposing on the functional divergence of this clan in the microbial pathogens occupying a different pathological niche. The Darwinian positive selection is an important evolutionary process that determines the neofunctionalization of highly diverged family members from a conserved functional core of this clan. The present study describes the apparent involvement and associations of members of this clan in the molecular pathogenesis and virulence of clinically important pathogens. As a result, the evolution of history and origin of this clan provides a new idea for the development of antimicrobial agents targeting the understudied proteins. The targeted proteins of this study are promising targets for antibiotic drug discovery and the development of resistance for many classes of clinically used antibiotics. Nevertheless, experimental evaluations of the target proteins are very important to know their structural, functional, and biochemical characteristics during the infection process.
References
Allali N, Afif H, Couturier M, Van Melderen L (2002) The highly conserved TldD and TldE proteins of Escherichia coli are involved in microcin B17 processing and in CcdA degradation. J Bacteriol 184:3224–3231. https://doi.org/10.1128/jb.184.12.3224-3231.2002
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402. https://doi.org/10.1093/nar/25.17.3389
Bailey TL, Johnson J, Grant CE, Noble WS (2015) The MEME Suite. Nucleic Acids Res 43:W39–W49. https://doi.org/10.1093/nar/gkv416
Barandun J, Delley CL, Weber-Ban E (2012) The pupylation pathway and its role in mycobacteria. BMC Biol 10:95. https://doi.org/10.1186/1741-7007-10-95
Barrett AJ, Rawlings ND (2007) Species’ of peptidases. Biol Chem 388:1151–1157. https://doi.org/10.1515/BC.2007.151
Bedau MA, Packard NH (2003) Evolution of evolvability via adaptation of mutation rates. Biosystems 69:143–162. https://doi.org/10.1016/s0303-2647(02)00137-5
Benz I, Schmidt MA (1992) AIDA-I, the adhesin involved in diffuse adherence of the diarrhoeagenic Escherichia coli strain 2787 (O126:H27), is synthesized via a precursor molecule. Mol Microbiol 6:1539–1546. https://doi.org/10.1111/j.1365-2958.1992.tb00875.x
Benz I, Schmidt MA (1993) Diffuse adherence of enteropathogenic Escherichia coli strains-processing of AIDA-I. Zentralbl Bakteriol 278:197–208. https://doi.org/10.1016/s0934-8840(11)80837-6
Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, Kiefer F, Gallo Cassarino T, Bertoni M, Bordoli L, Schwede T (2014) SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res 42:W252–W258. https://doi.org/10.1093/nar/gku340
Bingham R, Ekunwe SI, Falk S, Snyder L, Kleanthous C (2000) The major head protein of bacteriophage T4 binds specifically to elongation factor Tu. J Biol Chem 275:23219–23226. https://doi.org/10.1074/jbc.M002546200
Bond MD, Wart HEV (1984) Relationship between the individual collagenases of Clostridium histolyticum: evidence for evolution by gene duplication. Biochemistry 23:3092–3099. https://doi.org/10.1021/bi00308a037
Boratyn GM, Schäffer AA, Agarwala R, Altschul SF, Lipman DJ, Madden TL (2012) Domain enhanced lookup time accelerated BLAST. Biol Direct 7:12. https://doi.org/10.1186/1745-6150-7-12
Boucabeille C, Mengin-Lecreulx D, Henckes G, Simonet JM, van Heijenoort J (1997) Antibacterial and hemolytic activities of linenscin OC2, a hydrophobic substance produced by Brevibacterium linens OC2. FEMS Microbiol Lett 153:295–301. https://doi.org/10.1111/j.1574-6968.1997.tb12588.x
Celik N, Webb CT, Leyton DL, Holt KE, Heinz E, Gorrell R, Kwok T, Naderer T, Strugnell RA, Speed TP, Teasdale RD, Likić VA, Lithgow T (2012) A bioinformatic strategy for the detection, classification and analysis of bacterial autotransporters. PLoS One 7:e43245. https://doi.org/10.1371/journal.pone.0043245
Chellapandi P, Prisilla A (2017) Structure, function and evolution of Clostridium botulinum C2 and C3 toxins: Insight to poultry and veterinary vaccines. Curr Protein Pept Sci 18:412–424. https://doi.org/10.2174/1389203717666161201203311
Chellapandi P, Sakthi Shree S, Bharathi M (2013) Phylogenetic approach for inferring the origin and functional evolution of bacterial ADP-ribosylation superfamily. Protein Pept Lett 20:1054–1065. https://doi.org/10.2174/0929866511320090012
Chellapandi P, Prathiviraj R, Prisilla A (2018) Molecular evolution and functional divergence of IspD homologs in malarial parasites. Infect Genet Evol 65:340–349. https://doi.org/10.1016/j.meegid.2018.08.013
Chellapandi P, Prathiviraj R, Prisilla A (2019) Deciphering structure, function and mechanism of Plasmodium IspD homologs from their evolutionary imprints. J Comput Aided Mol Des 33:419–436. https://doi.org/10.1007/s10822-019-00191-2
Clark BK, Wabick KJ, Weidner JG (2012) Inversion and crossover recombination contributions to the spacing between two functionally linked genes. Biosystems 109:169–178. https://doi.org/10.1016/j.biosystems.2012.04.013
Copeland NA, Kleanthous C (2005) The role of an activating peptide in protease-mediated suicide of Escherichia coli K12. J Biol Chem 280:112–117. https://doi.org/10.1074/jbc.M411280200
de Castro E, Sigrist CJ, Gattiker A, Bulliard V, Langendijk-Genevaux PS, Gasteiger E, Bairoch A, Hulo N (2006) ScanProsite: detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res 34:W362–W365. https://doi.org/10.1093/nar/gkl124
Doxey AC, Lynch MD, Muller KM, Meiering EM, McConkey BJ (2008) Insights into the evolutionary origins of clostridial neurotoxins from analysis of the Clostridium botulinum strain A neurotoxin gene cluster. BMC Evol Biol 8:316. https://doi.org/10.1186/1471-2148-8-316
Duarte AS, Correia A, Esteves AC (2016) Bacterial collagenases - A review. Crit Rev Microbiol 42:106–126. https://doi.org/10.3109/1040841X.2014.904270
Figaj D, Ambroziak P, Przepiora T, Skorko-Glonek J (2019) The role of proteases in the virulence of plant pathogenic bacteria. Int J Mol Sci 20:E672. https://doi.org/10.3390/ijms20030672
Galperin MY, Mekhedov SL, Puigbo P, Smirnov S, Wolf YI, Rigden DJ (2012) Genomic determinants of sporulation in Bacilli and Clostridia: towards the minimal set of sporulation-specific genes. Environ Microbiol 14:2870–2890. https://doi.org/10.1111/j.1462-2920.2012.02841.x
Georgiou T, Yu YN, Ekunwe S, Buttner MJ, Zuurmond A, Kraal B, Kleanthous C, Snyder L (1998) Specific peptide-activated proteolytic cleavage of Escherichia coli elongation factor Tu. Proc Natl Acad Sci USA 95:2891–2895. https://doi.org/10.1073/pnas.95.6.2891
Grenier D, La VD (2011) Proteases of Porphyromonas gingivalis as important virulence factors in periodontal disease and potential targets for plant-derived compounds: a review article. Curr Drug Targets 12:322–331. https://doi.org/10.2174/138945011794815310
Gromiha MM, Thangakani AM, Selvaraj S (2006) FOLD-RATE: prediction of protein folding rates from amino acid sequence. Nucleic Acids Res 34:W70–W74. https://doi.org/10.1093/nar/gkl043
Gu X, Zou Y, Su Z, Huang W, Zhou Z, Arendsee Z, Zeng Y (2013) An update of DIVERGE software for functional divergence analysis of protein family. Mol Biol Evol 30:1713–1719. https://doi.org/10.1093/molbev/mst069
Han HJ, Taki T, Kondo H, Hirono I, Aoki T (2008) Pathogenic potential of a collagenase gene from Aeromonas veronii. Can J Microbiol 54:1–10. https://doi.org/10.1139/w07-109
Henderson IR, Nataro JP (2001) Virulence functions of autotransporter proteins. Infect Immun 69:1231–1243. https://doi.org/10.1128/IAI.69.3.1231-1243.2001
Henriques AO, Moran CP (2000) Structure and assembly of the bacterial endospore coat. Methods 20:95–110. https://doi.org/10.1006/meth.1999.0909
Hicks PM, Rinker KD, Baker JR, Kelly RM (1998) Homomultimeric protease in the hyperthermophilic bacterium Thermotoga maritima has structural and amino acid sequence homology to bacteriocins in mesophilic bacteria. FEBS Lett 440:393–398. https://doi.org/10.1016/s0014-5793(98)01451-3
Hicks PM, Chang LS, Kelly RM (2001) Homomultimeric protease and putative bacteriocin homolog from Thermotoga maritima. Methods Enzymol 330:455–460. https://doi.org/10.1016/s0076-6879(01)30397-x
Huson DH, Bryant D (2006) Application of phylogenetic networks in evolutionary studies. Mol Biol Evol 23:254–267. https://doi.org/10.1093/molbev/msj030
Iyer LM, Abhiman S, Maxwell Burroughs A, Aravind L (2009) Amidoligases with ATP-grasp, glutamine synthetase-like and acetyltransferase-like domains: synthesis of novel metabolites and peptide modifications of proteins. Mol Biosyst 5:1636–1660. https://doi.org/10.1039/b917682a
Kato T, Takahashi N, Kuramitsu HK (1992) Sequence analysis and characterization of the Porphyromonas gingivalis prtC gene, which expresses a novel collagenase activity. J Bacteriol 174:3889–3895. https://doi.org/10.1128/jb.174.12.3889-3895.1992
Kostakioti M, Stathopoulos C (2006) Role of the alpha-helical linker of the C-terminal translocator in the biogenesis of the serine protease subfamily of autotransporters. Infect Immun 74:4961–4969. https://doi.org/10.1128/IAI.00103-06
Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018) MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35:1547–1549. https://doi.org/10.1093/molbev/msy096
Kuwana R, Okuda N, Takamatsu H, Watabe K (2006) Modification of GerQ reveals a functional relationship between Tgl and YabG in the coat of Bacillus subtilis spores. J Biochem 139:887–901. https://doi.org/10.1093/jb/mvj096
Letunic I, Bork P (2018) 20 years of the SMART protein domain annotation resource. Nucleic Acids Res 46:D493–D496. https://doi.org/10.1093/nar/gkx922
Martin DP, Lemey P, Lott M, Moulton V, Posada D, Lefeuvre P (2010) RDP3: a flexible and fast computer program for analyzing recombination. Bioinformatics 26:2462–2463. https://doi.org/10.1093/bioinformatics/btq467
Matsushita O, Jung CM, Katayama S, Minami J, Takahashi Y, Okabe A (1999) Gene duplication and multiplicity of collagenases in Clostridium histolyticum. J Bacteriol 181:923–933. https://doi.org/10.1128/JB.181.3.923-933.1999
Mayrose I, Graur D, Ben-Tal N, Pupko T (2004) Comparison of site-specific rate-inference methods: Bayesian methods are superior. Mol Biol Evol 21:1781–1791. https://doi.org/10.1093/molbev/msh194
Mittal M, Siddiqui MR, Tran K, Reddy SP, Malik AB (2014) Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Signal 20:1126–1167. https://doi.org/10.1089/ars.2012.5149
Murayama N, Shimizu H, Takiguchi S, Baba Y, Amino H, Horiuchi T, Sekimizu K, Miki T (1996) Evidence for involvement of Escherichia coli genes pmbA, csrA and a previously unrecognized gene tldD, in the control of DNA gyrase by letD (ccdB) of sex factor F. J Mol Biol 256:483–502. https://doi.org/10.1006/jmbi.1996.0103
Ofer N, Forer N, Korman M, Vishkautzan M, Khalaila I, Gur E (2013) Allosteric transitions direct protein tagging by PafA, the prokaryotic ubiquitin-like protein (Pup) ligase. J Biol Chem 288:11287–11293. https://doi.org/10.1074/jbc.M112.435842
Özcelik D, Barandun J, Schmitz N, Sutter M, Guth E, Damberger FF, Allain F, Ban N, Weber-Ban E (2012) Structures of Pup ligase PafA and depupylase Dop from the prokaryotic ubiquitin-like modification pathway. Nat Commun 3:1014. https://doi.org/10.1038/ncomms2009
Papadopoulos JS, Agarwala R (2007) COBALT: constraint-based alignment tool for multiple protein sequences. Bioinformatics 23:1073–1079. https://doi.org/10.1093/bioinformatics/btm076
Pearce MJ, Mintseris J, Ferreyra J, Gygi SP, Darwin KH (2008) Ubiquitin-like protein involved in the proteasome pathway of Mycobacterium tuberculosis. Science 322:1104–1107. https://doi.org/10.1126/science.1163885
Pond SL, Frost SD, Muse SV (2005) HyPhy: hypothesis testing using phylogenies. Bioinformatics 21:676–679. https://doi.org/10.1093/bioinformatics/bti079
Popoff MR, Bouvet P (2009) Clostridial toxins. Future Microbiol 4:1021–1064. https://doi.org/10.2217/fmb.09.72
Prathiviraj R, Chellapandi P (2020) Deciphering molecular virulence mechanism of Mycobacterium tuberculosis dop isopeptidase based on its sequence-structure-function linkage. Protein J 39:33–45. https://doi.org/10.1007/s10930-019-09876-x
Prathiviraj R, Prisilla A, Chellapandi P (2016) Structure-function discrepancy in Clostridium botulinum C3 toxin for its rational prioritization as a subunit vaccine. J Biomol Struct Dyn 34:1317–1329. https://doi.org/10.1080/07391102.2015.1078745
Prisilla A, Prathiviraj R, Sasikala R, Chellapandi P (2016) Structural constraints-based evaluation of immunogenic avirulent toxins from Clostridium botulinum C2 and C3 toxins as subunit vaccines. Infect Genet Evol 44:17–27. https://doi.org/10.1016/j.meegid.2016.06.029
Prisilla A, Prathiviraj R, Chellapandi P (2017) Molecular evolutionary constraints that determine the avirulence state of Clostridium botulinum C2 toxin. J Mol Evol 86:174–186. https://doi.org/10.1007/s00239-017-9791-y
Rawlings ND (2010) Peptidase inhibitors in the MEROPS database. Biochimie 92:1463–1483. https://doi.org/10.1016/j.biochi.2010.04.013
Rawlings ND, Barrett AJ (1993) Evolutionary families of peptidases. Biochem J 290:205–218. https://doi.org/10.1042/bj2900205
Rawlings ND, Tolle DP, Barrett AJ (2004) Evolutionary families of peptidase inhibitors. Biochem J 378:705–716. https://doi.org/10.1042/BJ20031825
Rawlings ND, Morton FR, Barrett AJ (2006) MEROPS: the peptidase database. Nucleic Acids Res 34:D270–D272. https://doi.org/10.1093/nar/gkj089
Rawlings ND, Barrett AJ, Bateman A (2012) MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res 40:D343–D350. https://doi.org/10.1093/nar/gkr987
Rife C, Schwarzenbacher R, McMullan D, Abdubek P, Ambing E, Axelrod H, Biorac T, Canaves JM, Chiu HJ, Deacon AM, DiDonato M, Elsliger MA, Godzik A, Grittini C, Grzechnik SK, Hale J, Hampton E, Han GW, Haugen J, Hornsby M, Jaroszewski L, Klock HE, Koesema E, Kreusch A, Kuhn P, Lesley SA, Miller MD, Moy K, Nigoghossian E, Paulsen J, Quijano K, Reyes R, Sims E, Spraggon G, Stevens RC, van den Bedem H, Velasquez J, Vincent J, White A, Wolf G, Xu Q, Hodgson KO, Wooley J, Wilson IA (2005) Crystal structure of a putative modulator of DNA gyrase (pmbA) from Thermotoga maritima at 1.95 A resolution reveals a new fold. Proteins 61:444–448. https://doi.org/10.1002/prot.20468
Sharov AA (2014) Evolutionary constraints or opportunities? Biosystems 123:9–18. https://doi.org/10.1016/j.biosystems.2014.06.004
Takamatsu H, Imamura A, Kodama T, Asai K, Ogasawara N, Watabe K (2000a) The yabG gene of Bacillus subtilis encodes a sporulation specific protease which is involved in the processing of several spore coat proteins. FEMS Microbiol Lett 192:33–38. https://doi.org/10.1111/j.1574-6968.2000.tb09355.x
Takamatsu H, Kodama T, Imamura A, Asai K, Kobayashi K, Nakayama T, Ogasawara N, Watabe K (2000b) The Bacillus subtilis yabG gene is transcribed by SigK RNA polymerase during sporulation, and yabG mutant spores have altered coat protein composition. J Bacteriol 182:1883–1888. https://doi.org/10.1128/jb.182.7.1883-1888.2000
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25:4876–4882. https://doi.org/10.1093/nar/25.24.4876
Wagner GP (1984) Coevolution of functionally constrained characters: Prerequisites for adaptive versatility. Biosystems 17:51–55. https://doi.org/10.1016/0303-2647(84)90015-7
Wang D, Zhang Y, Zhang Z, Zhu J, Yu J (2010) KaKs_Calculator 2.0: a toolkit incorporating gamma-series methods and sliding window strategies. Genomics Proteomics Bioinformatics 8:77–80. https://doi.org/10.1016/S1672-0229(10)60008-3
Acknowledgements
Life Science Research Board-Defense Research and Development Organization (Sanction No. DLS/81/48222/LSRB-249/BTB/2012), New Delhi, India, is duly acknowledged for partial financial support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors confirm that this article’s content has no conflicts of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 1201 kb)
Rights and permissions
About this article

Cite this article
Prathiviraj, R., Chellapandi, P. Evolutionary genetic analysis of unassigned peptidase clan-associated microbial virulence and pathogenesis. Biologia 75, 2083–2092 (2020). https://doi.org/10.2478/s11756-020-00529-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.2478/s11756-020-00529-4