
Abstract
Physical properties of In0.5(ZrMg)0.75Mo3O12, including the coefficient of thermal expansion, phase stability, hygroscopicity, and decomposition temperature have been thoroughly studied by in situ x-ray powder diffraction, Raman spectroscopy and thermal methods. These investigations show that In0.5(ZrMg)0.75Mo3O12 exists in a monoclinic phase (P 21/ a) at room temperature and transforms to an orthorhombic (Pbcn) phase at ∼82 °C. In the orthorhombic form this material presents intrinsic near-zero thermal expansion (−0.16 × 10−6 K−1) in the range between 100 and 500 °C. The phase is not hygroscopic, but starts to decompose into its constituent oxides at temperatures higher than 700 °C. In comparison to the end member phase ZrMgMo3O12 in the In2Mo3O12–ZrMgMo3O12 solid solution, In0.5(ZrMg)0.75Mo3O12 is less promising for near room-temperature applications due to the phase transition from monoclinic to orthorhombic slightly above room temperature. However, the orthorhombic phase of In0.5(ZrMg)0.75Mo3O12 has potential for applications that require zero thermal expansion at temperatures higher than 100 °C.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
I. INTRODUCTION
The orthorhombic space group Pbcn (60) accommodates a large number of open-framework phases with the general chemical formula A2M3O12.1–3 In this formula M is a hexavalent cation, such as Mo6+ or W6+ (although, P5+ has been reported4 as an element capable of partial substitution for W6+ or Mo6+) forming 4-coordinated polyhedra, while A can be any small rare earth or some other trivalent cation from Al3+ to Y3+, within an octahedral environment.5–8 Tetrahedra and octahedra build an open-framework crystal structure through connected vertices. Strain screening is made possible by the open-framework structure9 and solid solutions can be synthesized with almost any combination of these cations and, therefore, there is considerable chemical and physical flexibility within the A2M3O12 family. Trivalent cations occupy one independent general 8d site, while hexavalent cations occupy two independent crystallographic positions, one general 8d and one special 4c, and oxygens adopt six independent general 8d positions. This peculiar open framework, which is related to the garnet structure by removal of the 8-coordinated cations, is responsible for the striking physical properties of these phases, such as negative or near-zero coefficients of thermal expansion (CTE), and considerable ionic conductivity.10–14 If the trivalent cation is substituted by a 1:1 mixture of tetravalent (A: Hf or Zr) and divalent cations (B: Mg) a new family (ABM3O12) is formed, assuming a lower symmetry Pna21 (33) space group. The atomic structure of these compounds was recently resolved for orthorhombic ZrMgMo3O12 by Romao et al.,15 who established that tetravalent and divalent cations occupy distinct general 4a sites, while hexavalent cations and oxygen anions occupy three and twelve independent 4a sites, respectively. The physical properties of three ABM3O12 materials (HfMgW3O12, HfMgMo3O12, and ZrMgMo3O12) have been reported, while solid solutions have been synthesized in the following systems: Al2W3O12–HfMgW3O12, Al2W3O12–ZrMgW3O12, In2Mo3O12–HfMgMo3O12, Fe2Mo3O12–ZrMgMo3O12, and Cr2Mo3O12–ZrMgMo3O12.16–23 The ABM3O12 compounds also have an open-framework crystal structure consisting of vertex-sharing coordination polyhedra. Therefore, near-zero or negative thermal expansion is expected, and has already been documented in the literature for some of these compounds (Table I). Note that orthorhombic A2M3O12 and ABM3O12 phases can transform at lower temperatures to a denser structure with the monoclinic P21/a (14) space group, which shows positive thermal expansion and, therefore, can be problematic for those applications where near-zero or negative thermal expansion is required. This phase transition is displacive24 and can be characterized as primarily a static rotation of the coordination polyhedra. In some cases, hygroscopicity can occur due to the presence of nanochannels within the cation-deficient garnet structure. If these channels are sufficiently large as to admit water molecules, the water molecules will interact with the bridging oxygen anions and hinder their transverse vibrations and, therefore, prevent negative thermal expansion.
The thermal expansion, phase transitions, and hygroscopicity of In2Mo3O12–ZrMgMo3O12 solid solutions have not previously been reported, although these properties have been described for the end-members.15,25 As reported elsewhere25 In2Mo3O12 is monoclinic (P21/a) at room temperature and transforms to an orthorhombic (Pbcn) phase at a temperature of 335 °C; the latter presents a negative coefficient of thermal expansion (−1.85 × 10−6 K−1) until at least 760 °C. The first report on ZrMgMo3O12 by Song et al.,20 indicated that it exhibited an orthorhombic phase with a negative CTE (−3.8 × 10−6 K−1) from −150 °C to 927 °C. However, a more recent, thorough study,15 showed that ZrMgMo3O12 is in the orthorhombic phase between −126 °C and 700 °C with a very near-zero coefficient of thermal expansion (0.16 × 10−6 K−1) from 25 °C to 450 °C, increasing by about an order of magnitude to 1.0 × 10−6 K−1 above 450 °C. Below −126 °C the orthorhombic phase transforms to monoclinic.
Therefore, the rule of mixtures predicts that a compound with the chemical formula In2x(ZrMg)1−xMo3O12, with x = 0.25 (In:(Zr + Mg) = 1:3) would present a negative CTE of about −3.3 × 10−6 K−1 if the CTE determined by Song et al.,20 were correct. The same approach predicts that the CTE of In2x(ZrMg)1−xMo3O12 (x = 0.25) should be an order of magnitude lower and, therefore, near zero (−0.3 × 10−6 K−1), if the study carried out by Romao et al.15 gave a more accurate CTE of ZrMgMo3O12. It is, therefore, possible that In0.5(ZrMg)0.75Mo3O12 could have a near-zero CTE over a large range of superambient temperatures. The phase transition temperature in this new material is also of concern since the addition of In, a more electronegative atom in comparison to Zr and Mg, would be expected to increase the temperature of the monoclinic (positive CTE) to orthorhombic (negative or near-zero CTE) phase transition. Also, the insertion of a larger cation (In3+) into the crystal structure could potentially cause In0.5(ZrMg)0.75Mo3O12 to become hygroscopic.
Therefore, it is scientifically and technologically relevant to study the thermal expansion, phase transition, and hygroscopicity of In0.5(ZrMg)0.75Mo3O12; these three main properties are indispensable for the evaluation of the potential of such a material as a filler in composites or as a consolidated ceramic object. Also, the temperature at which the molybdate phase starts to decompose into constituent oxides, followed by MoO3 sublimation, is relevant, considering the sintering processes necessary to produce dense polycrystalline objects.
II. EXPERIMENTAL METHODS
A. Synthesis and consolidation of In0.5(ZrMg)0.75Mo3O12
In0.5(ZrMg)0.75Mo3O12 was synthesized through a total evaporation route from a stoichiometric mixture of 0.1 M aqueous solutions of Mg(NO3)2 (Sigma Aldrich, St. Louis, Missouri, 98%), In(NO3)2 (Sigma Aldrich, 99.9%), (NH4)6Mo7O24·4H2O (Isofar, 99%), and ZrO(NO3)2 (Johnson Matthey Company, New York, New York, 99.99%). Since 0.1 M aqueous solution of ZrO(NO3)2 is not soluble in water, it was necessary to add 100 mL of HNO3 (Sigma Aldrich, 69%) until complete dissolution. The acidic solution obtained by the mixture of the four reagents (pH = 0.35) was continuously stirred and heated at 70 °C, until complete water evaporation (∼12 h). Afterward, the as-prepared precursor material was oven dried at 100 °C for 8 h without any further post treatments. It was analyzed by differential scanning calorimetry (DSC) in a Perkin–Elmer (Seer Green, United Kingdom) Simultaneous Thermal Analyzer (STA-6000), using a heating rate of 5 K/min in air flux (20 mL/min), for determination of the crystallization temperature. The DSC curve (see Supplementary Materials) showed an exothermic peak at 718 °C, ascribed to the crystallization of In0.5(ZrMg)0.75Mo3O12 from an amorphous precursor.
Therefore, the precursor powder was calcined at 750 °C (heating rate of 20 K/min) for 5 h in a Tubular Maitec–INTI FET 1600/H (Maitec, São Carlos, Brazil) furnace in air atmosphere, followed by an additional heating (with the same heating rate) to 800 °C with dwell time of 1 min.
Monolithic ceramics were prepared using the as-synthesized crystalline powder with the addition of 5 mass% of polyethylene glycol (98%; Vetec, Rio de Janeiro, Brazil) as binder, mixed with a small amount of distilled water until tacky and then pressed at 100 MPa of uniaxial pressure for 1 min. The as-prepared green cylindrical specimens were presintered at 800 °C for 1 h.
B. Characterization of In0.5(ZrMg)0.75Mo3O12 powder
X-ray powder diffraction of the calcined powder was carried out in a D8 Discovery diffractometer (Bruker, Karlsruhe, Germany), using Cu Kα radiation in steps of 0.02° (1 s per step) from 10° to 80° (2θ). In situ XRPD was carried out in a D8 Advance diffractometer using Cu Kα radiation, equipped with an XRK 900 reactor chamber (Anton Paar GmbH, Graz, Austria), at different temperatures (100, 200, 300, and 500 °C) in air atmosphere. The XRPD patterns were acquired in the range of 10°−80° (2θ) with a step size of 0.02° (1 s per step). The analysis of raw data was performed using the Le Bail method with Topas 4.2 Software (Bruker) for determination of the intrinsic CTE and evaluation of the phase transition temperature.
DSC was carried out at a Perkin–Elmer Simultaneous Thermal Analyzer (STA 8000) in air flux (20 mL/min) in the temperature range between −10 and 300 °C, at a heating rate of 10 K/min. The DSC was calibrated with In and Ag.
Thermogravimetry was performed in a Perkin–Elmer Simultaneous Thermal Analyzer (STA-6000) between 25 and 900 °C using a heating rate of 10 K/min in air flux (20 mL/min).
The morphology of the calcined powder was determined by a field-emission scanning electron microscope (FE-SEM) (JSM-6701F; JEOL Ltd., Tokyo, Japan) in secondary electron mode operating at 1 kV. The chemical composition was examined by energy-dispersive x-ray spectroscopy (EDS).
Raman spectroscopy was carried out in situ from room temperature to 250 °C using a Jobin Yvon T64000 spectrometer (Horiba Scientific, Longjumeau, France), which was equipped with a cooled charge-coupled device detector. The backscattering geometry was used to analyze the scattered light and the spectral excitation was provided by an Ar+ laser using the 514.5 nm laser line. The slits were set for a spectral resolution of 2 cm−1.
Additional Raman spectra of materials in the A2M3O12 and ABM3O12 families (see Sec. III. B.) were acquired at room temperature using a Nicolet NXR 9650 FT-Raman spectrometer (Thermo Fisher Scientific, Waltham, Massachusetts) with a 1064 nm excitation laser, with spectral resolution of 1 cm−1.
C. Characterization of In0.5(ZrMg)0.75Mo3O12 monolithic ceramic
Relative density measurements of green and bulk specimens were carried out using Archimedes’ principle. Wax was applied to the surface to prevent water entrance. Distilled water (30 °C, density 0.9957 g/cm3) was used as the liquid. The accuracy of the pellet mass measurement on the analytical balance was ±0.001 g. As will be demonstrated in the Results section, crystalline In0.5(ZrMg)0.75Mo3O12 powder was considered monophasic and monoclinic (P21/a) at room temperature for calculation of the theoretical density (3.44 g/cm3). The density of the green specimen was 62% of theoretical density (TD), while 80% of TD was determined for the pre-sintered samples.
Dilatometry of a bulk sample was performed in air using a NETZSCH dilatometer DIL 402C (NETZSCH Group, Selb, Germany) in the temperature range between 25 and 800 °C, with a heating rate of 10 K/min; the cooling rate was not controlled.
III. RESULTS AND DISCUSSION
A. Crystal system at room temperature and hygroscopicity of In0.5(ZrMg)0.75Mo3O12
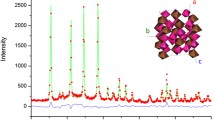
FE-SEM analysis [Fig. 1(a)] showed that the calcined powder was constituted of faceted sub-micron to micron-sized particles, in the general range 0.7–1.6 µm. EDS analysis [Fig. 1(b)] indicated that the stoichiometric ratio of the chemical elements is in agreement with the proposed formula In0.5(ZrMg)0.75Mo3O12 (Table SI in Supplementary Material).

(a) FEG-SEM image of In0.5(ZrMg)0.75Mo3O12 powder, (b) EDS spectrum, and (c) room-temperature XRPD pattern, illustrating the Le Bail fit to the monoclinic P21/a space group (a = 16.2694 Å, b = 9.5610 Å, c = 18.9782 Å and β = 125.56°); the experimental pattern is black, the calculated pattern is red and the difference plot is green [inset: magnified view of XRPD in the range between 21° and 27° (2θ), with two weak diffraction lines at 25.8° and 23.7° (2θ) related to the monoclinic system marked as *].
The room-temperature XRPD pattern [Fig. 1(c)] indicated that the calcined powder was monophasic and monoclinic (P21/a space group), not orthorhombic. Two weak diffraction peaks at 25.8° and 23.7° (2θ), marked in the inset of Fig. 1(c), can be associated with a lower symmetry monoclinic space group (P21/a),10 and are absent in the orthorhombic space groups (Pbcn and Pna21) adopted by members of the A2M3O12 and ABM3O12 families. Therefore, the substitution of 1/4 of the low electronegativity Zr (1.4) and Mg (1.2) atoms by In with higher electronegativity (1.7) significantly increases the thermal stability range of the monoclinic phase in comparison to the end-member compound ZrMgMo3O12 (transition temperature of −126 °C).
Note that In0.5(ZrMg)0.75Mo3O12 is not hygroscopic: its mass loss on heating to 600 °C was negligible (<0.3 mass%) (Fig. 2). However, above 700 °C significant mass loss occurs because of partial decomposition of In0.5(ZrMg)0.75Mo3O12 into its constituent oxides followed by sublimation of MoO3.15 The absence of hygroscopicity is not unexpected since the end-members ZrMgMo3O12 and In2Mo3O12 are not hygroscopic,15 and hygroscopicity in the A2M3O12 family is generally reported for the orthorhombic phases with large trivalent cations, such as rare earths from Y to Ho.24 The evidence of partial molybdate decomposition above 700 °C (Fig. 2) might be an obstacle to attaining high densities during sintering.

TGA curve of In0.5(ZrMg)0.75Mo3O12 powder.
B. Phase transition in In0.5(ZrMg)0.75Mo3O12
The DSC thermogram of In0.5(ZrMg)0.75Mo3O12 [Fig. 3(a)] shows an endothermic event on heating, associated with a monoclinic to orthorhombic phase transition, with an enthalpy change of 2.5 kJ/mol and an onset temperature of 82 °C. This kind of phase transition for the compounds belonging to the A2M3O12 family is reported to be structurally subtle and comprises very low enthalpy changes (ΔH ≤ 2 kJ/mol),8 i.e., on the same order of magnitude as observed here for In0.5(ZrMg)0.75Mo3O12. This is a displacive phase transition due to the breakage of secondary oxygen–oxygen bonds (the same ones responsible for the existence of molecular solid tetraoxides, such as RuO4 and OsO4) between neighboring polyhedra,1,26 resulting in more empty space within the orthorhombic crystal structure. No primary bonds are broken during this event. Therefore, the phase transition is thermodynamically driven by the increase of vibrational entropy in the transition to a less dense structure (orthorhombic), causing more disorder (i.e., increase of distinctive configurations) in distribution of thermal energy over harmonic oscillators (atoms). The dilatometric curve of a pre-sintered specimen confirms this transition through an abrupt increase of sample length (thermal deformation) between 70 and 90 °C [Fig. 3(b)], characteristic of a phase transition from a dense to a more open crystal structure, such as in the case of the monoclinic (P21/a) to orthorhombic (Pbcn or Pna21) transition, inherent to the A2M3O12 and ABM3O12 families. This abrupt change of specimen length is not associated with water loss since the latter is insignificant for In0.5(ZrMg)0.75Mo3O12.

(a) DSC curve of In0.5(ZrMg)0.75Mo3O12 powder and (b) dilatometric curve of a pre-sintered specimen of In0.5(ZrMg)0.75Mo3O12.
In the A2M3O12 family, the subtle structural differences that exist between Pbcn, Pna21, and P21/a phases can be difficult to resolve by x-ray diffraction alone.15,18 Raman spectroscopy provides an important complementary technique because it is sensitive to different structural features. Specifically, there exist stretching modes of the AO6 and MO4 coordination polyhedra that are sensitive to the identity of the A and M cations.27 Unlike the low-energy bending and librational modes, which involve coordinated motions of atoms throughout the unit cell, the energies of the stretching modes show significant differences for the Pbcn, Pna21, and P21/a phases.15,27 However, the number of Raman-active vibrational modes in A2M3O12 materials is large due to their large unit cells and the absence of atoms at special positions.15,27 Therefore, to quantify differences in the vibrational spectra between phases, a deconvolution approach has been used, using Fityk software.28 This approach fits the spectra to peaks that represent bands rather than individual vibrations, an approach which is supported by previous computational and Raman studies.15,27,29–33
In Fig. 4 the fitted stretching regions of the Raman spectra of In0.5(ZrMg)0.75Mo3O12 at 40 and 250 °C are shown. The fits were performed with pseudo-Voigt profile functions, with the goal of finding the minimum number of peaks required for a satisfactory fit. The criterion used was to deem the fit satisfactory when the addition of a new peak resulted in a reduction in the residual which was larger than the residual resulting from the error in fitting the shape of the largest stretching peak. Eight peaks were needed to fit the spectrum acquired at 40 °C [Fig. 4(a)], whereas the spectrum acquired at 250 °C [Fig. 4(b)] required only five peaks, indicating an increase in symmetry upon heating. This result is consistent with previous reports of transitions between monoclinic and orthorhombic phases in the A2M3O12 family.29,32,33 However, peak deconvolution has not previously been applied to these materials. Therefore, we have additionally determined and deconvoluted the room-temperature Raman spectra of A2M3O12 and ABM3O12 materials such as In2Mo3O12, Al2Mo3O12, ZrMgMo3O12, and Sc2W3O12; all these phases synthesized as described elsewhere,25 are known and were used here to make comparisons with In0.5(ZrMg)0.75Mo3O12.

Stretching regions of the Raman spectra of In0.5(ZrMg)0.75Mo3O12 at (a) 40 °C and (b) 250 °C, showing experimental data (black circles), the fit to the data (blue line), and the peaks composing the fit (orange lines).
Figure 5 shows the stretching regions of the Raman spectra of In2Mo3O12 and Al2Mo3O12, two A2M3O12 materials that adopt a monoclinic P21/a phase at room temperature.25,34 In both cases, the deconvolution procedure revealed eight peaks. The significant differences in the positions and intensities of the peaks between the two materials can be attributed to the Al3+ cation participating in the stretches to a greater extent than Sc3+ due to the greater rigidity of the AlO6 polyhedron.15,27 The identification of eight peaks in the spectra shown in Fig. 5 corroborates the P21/a phase at room temperature for In0.5(ZrMg)0.75Mo3O12.

In Fig. 6, the stretching regions of the Raman spectra of ZrMgMo3O12 and Sc2W3O12, two materials whose orthorhombic phases are stable at room temperature,15,35 are shown. Sc2W3O12 adopts a Pbcn structure typical for A2M3O12 materials,35 whereas the related Pna21 phase of ZrMgMo3O12 has ordered Zr4+ and Mg2+ cations.15 A′2x(A″Mg)1−xM3O12 materials such as In0.5(ZrMg)0.75Mo3O12 can adopt Pna21 phases where the A″(Zr or Hf) and Mg atoms are ordered and the A′ atom is disordered,23 or Pbcn phases where all three A site atoms are disordered.23 Deconvolution of the stretching regions of the Raman spectra revealed 11 peaks for ZrMgMo3O12 and five for Sc2W3O12. The large differences between the two can be attributed to the decrease in symmetry between Pbcn and Pna21, and the differences in chemical bonding between the ZrO6 and MgO6 polyhedra.15 The presence of five peaks in the stretching region of the Raman spectrum of In0.5(ZrMg)0.75Mo3O12 at 250 °C therefore corroborates its assignment to the Pbcn phase, while the presence of eight peaks in the room temperature spectrum confirms the assignment of this phase to the monoclinic P21/a space group as previously ascribed through XRPD [Fig. 1(a)]. The increased breadth of the peaks in Fig. 4 can be attributed to disorder at the A site.

Stretching regions of the Raman spectra of (a) ZrMgMo3O12 and (b) Sc2W3O12 at room temperature, showing experimental data (black circles), the fit to the data (blue line), and the peaks composing the fit (orange lines). ZrMgMo3O12 is in the Pna21 phase while Sc2W3O12 is in the Pbcn phase at this temperature.15,35
C. Thermal expansion of orthorhombic In0.5(ZrMg)0.75Mo3O12
Suzuki and Omote23 showed for Al2x(HfMg)1−xW3O12 that when x increases from 0 to ∼0.3, the space group changes to Pbcn from a lower symmetry orthorhombic space group (originally thought to be Pnma but now known to be Pna21).15 This agrees with our analysis of the Raman spectra of In0.5(ZrMg)0.75Mo3O12 (x = 0.25), which attributed the spectrum at 250 °C, i.e., above the phase transition temperature, to the orthorhombic space group Pbcn[see Supplementary Materials for all Raman spectra of In0.5(ZrMg)0.75Mo3O12 from room temperature to 250 °C]. In situ XRPD (Fig. 7) additionally corroborates that the high temperature phase adopts the Pbcn space group due to the absence of two closely spaced diffraction lines at around 13.5° (2θ), present in the case of Pna21 space group in accordance to the findings of Suzuki and Omote.23Figure 8 presents the dependence of the natural logarithm of unit-cell volume of In0.5(ZrMg)0.75Mo3O12 in the orthorhombic (Pbcn) phase with temperature. The intrinsic linear CTE, calculated from the slope of this curve, gives a value very close to zero (−0.16 ± 0.07) × 10−6 K−1 between 100 and 500 °C, in reasonable agreement to the value estimated by the rule of mixtures (−0.3 × 10−6 K−1), using the CTE of the end-member ZrMgMo3O12 reported by Romao et al.,15 The CTE of In0.5(ZrMg)0.75Mo3O12 is in the same range as ZrMgMo3O12 (0.16 × 10−6 K−1), which is considered a near-zero thermal expansion material with a CTE similar to that of fused quartz and Invar.

In situ XRPD patterns of In0.5(ZrMg)0.75Mo3O12 powder in the temperature range between 25 and 500 °C.

Natural logarithm of the unit-cell volume of orthorhombic (Pbcn space group) In0.5(ZrMg)0.75Mo3O12 as a function of temperature.
It is worth noting that In0.5(ZrMg)0.75Mo3O12 shows anisotropic expansion along its three crystallographic axes, as the a-axis expands and b- and c-axes contract on heating (Supplementary Material, Fig. S3). Thermal expansion anisotropy can be quantitatively expressed as the maximum difference between the axial thermal expansion coefficients (Δαmax) as proposed by Srikanth36 and it was determined here to be Δαmax = 9.2 × 10−6 K−1 (from 100 to 500 °C). This value is almost the same as previously determined for In(HfMg)0.5Mo3O1216 and is significantly lower than the values generally estimated for the phases from A2M3O12 and ABM3O12 families.16 The CTE can also be obtained from the dilatometric curve, however, pronounced hysteresis [Fig. 3(b)] indicates microcrack healing within bulk specimens. Thus, the bulk CTE (1.5 × 10−6 K−1) determined dilatometrically on heating from 100 to 500 °C is biased due to the microcracking contribution increasing the CTE compared with the intrinsic CTE, a phenomenon previously documented that can occur on heating and cooling of bulk specimens.37 It is possible that sintered bodies of In0.5(ZrMg)0.75Mo3O12 with nanometric grains could exhibit a bulk CTE close to the intrinsic value due to reduction of microcracks in accordance with Griffith theory.38
IV. CONCLUSIONS
In0.5(ZrMg)0.75Mo3O12 is a novel non-hygroscopic and near-zero thermal expansion phase from the A′2x(A″Mg)1−xM3O12 family. Nevertheless, distinct from the closer solid solution end-member, ZrMgMo3O12, it is monoclinic (P21/a) at room temperature and transforms to an orthorhombic (Pbcn) crystal system at ∼82 °C. Above this temperature to 500 °C the orthorhombic phase shows a CTE of (−0.16 ± 0.07) × 10−6 K−1 and, therefore, is considered a near-zero thermal expansion material. At the temperatures higher than 700 °C this phase starts to decompose into its constituent oxides accompanied by MoO3 volatilization. Therefore, special precautions should be used during sintering of this phase to achieve high densities and protect In0.5(ZrMg)0.75Mo3O12 from decomposition. In comparison to the end-member phase ZrMgMo3O12 of the In2Mo3O12–ZrMgMo3O12 solid solution series, In0.5(ZrMg)0.75Mo3O12 is generally less promising for application purposes due to the phase transition from monoclinic to orthorhombic crystal system slightly above room temperature. Nevertheless, In0.5(ZrMg)0.75Mo3O12 is more isotropic in its thermal expansion along the three principal axes than generally reported for the phases from A2M3O12 and ABM3O12 families, which can lead to reduced thermally induced stresses within bulk polycrystals.
References
J.S.O. Evans, T.A. Mary, and A.W. Sleight: Negative thermal expansion in a large molybdate and tungstate family. J. Solid State Chem. 133, 580 (1997).
C.P. Romao, K.J. Miller, C.A. Whitman, M.A. White, and B.A. Marinkovic: Negative thermal expansion (thermomiotic) materials. In Comprehensive Inorganic Chemistry II, Vol. 4, J. Reedijk and K. Poeppelmeier, eds. (Elsevier: Oxford, 2013); p. 128–151.
C. Lind: Two decades of negative thermal expansion research: Where do we stand? Materials 5 (6), 1125 (2012).
J.S.O. Evans: Negative thermal expansion materials. J. Chem. Soc., Dalton Trans. 19, 3317 (1999).
J.S.O. Evans, T.A. Mary, and A.W. Sleight: Negative thermal expansion materials. Phys. B Condens. Matter. 241–243, 311 (1997).
S. Sumithra and A.M. Umarji: Negative thermal expansion in rare earth molybdates. Solid State Sci. 8 (12), 1453 (2006).
M. Ari, P.M. Jardim, B.A. Marinkovic, F. Rizzo, and F.F. Ferreira: Thermal expansion of Cr2x Fe2−2 xMo3O12, Al2 xFe2−2 xMo3O12 and Al2 xCr2−2 xMo3O12 solid solutions. J. Solid State Chem. 181 (6), 1472 (2008).
T. Varga, J.L. Moats, S.V. Ushakov, and A. Navrotsky: Thermochemistry of A2M3O12 negative thermal expansion materials. J. Mater. Res. 22 (9), 2512 (2007).
A.L. Goodwin, S.A. Wells, and M.T. Dove: Cation substitution and strain screening in framework structures: The role of rigid unit modes. Chem. Geol. 225 (3–4), 213 (2006).
K.J. Miller, M.B. Johnson, M.A. White, and B.A. Marinkovic: Low-temperature investigations of the open-framework material HfMgMo3O12. Solid State Commun. 152 (18), 1748 (2012).
J.S.O. Evans and T.A. Mary: Structural phase transitions and negative thermal expansion in Sc2(MoO4)3. Int. J. Inorg. Mater. 2 (1), 143 (2000).
B.A. Marinkovic, P.M. Jardim, R.R. de Avillez, and F. Rizzo: Negative thermal expansion in Y2Mo3O12. Solid State Sci. 7 (11), 1377 (2005).
B.A. Marinkovic, M. Ari, R.R. de Avillez, F. Rizzo, F.F. Ferreira, K.J. Miller, M.B. Johnson, and M.A. White: Correlation between AO6 polyhedral distortion and negative thermal expansion in orthorhombic Y2Mo3O12 and related materials. Chem. Mater. 21 (13), 2886 (2009).
M.M. Wu, Y. Zu, J. Peng, R.D. Liu, Z.B. Hu, Y.T. Liu, and D.F. Chen: Controllable thermal expansion properties of In2−xCrxMo3O12. Cryst. Res. Technol. 47 (7), 793 (2012).
C.P. Romao, F.A. Perras, U. Werner-Zwanziger, J.A. Lussier, K.J. Miller, C.M. Calahoo, J.W. Zwanziger, M. Bieringer, B.A. Marinkovic, D.L. Bryce, and M.A. White: Zero thermal expansion in ZrMgMo3O12: NMR crystallography reveals origins of thermoelastic properties. Chem. Mater. 27 (7), 2633 (2015).
K.J. Miller, C.P. Romao, M. Bieringer, B.A. Marinkovic, L.P. Prisco, and M.A. White: Near-zero thermal expansion in In(HfMg)0.5Mo3O12. J. Am. Ceram. Soc. 96 (2), 561 (2012).
W. Song, B. Yuan, X. Liu, Z. Li, J. Wang, and E. Liang: Tuning the monoclinic-to-orthorhombic phase transition temperature of Fe2Mo3O12 by substitutional co-incorporation of Zr4+ and Mg2+. J. Mater. Res. 29 (7), 849 (2014).
A.M. Gindhart, C. Lind, and M. Green: Polymorphism in the negative thermal expansion material magnesium hafnium tungstate. J. Mater. Res. 23 (1), 210 (2008).
B.A. Marinkovic, P.M. Jardim, M. Ari, R.R. De Avillez, F. Rizzo, and F.F. Ferreira: Low positive thermal expansion in HfMgMo3O12. Phys. Status Solidi 245 (11), 2514 (2008).
W.B. Song, E.J. Liang, X.S. Liu, Z.Y. Li, B.H. Yuan, and J.Q. Wang: A negative thermal expansion material of ZrMgMo3O12. Chin. Phys. Lett. 30 (12), 126502 (2013).
W.B. Song, J.Q. Wang, Z.Y. Li, X.S. Liu, B.H. Yuan, and E.J. Liang: Phase transition and thermal expansion property of Cr2−xZr0.5 xMg0.5 xMo3O12 solid solution. Chin. Phys. B 23 (6), 066501 (2014).
F. Li, X. Liu, W. Song, B. Yuan, Y. Cheng, H. Yuan, F. Cheng, M. Chao, and E. Liang: Phase transition, crystal water and low thermal expansion behavior of Al2−2 x(ZrMg)xW3O12· n (H2O). J. Solid State Chem. 218, 15 (2014).
T. Suzuki and A. Omote: Zero thermal expansion in (Al2 x(HfMg)1−x)(WO4)3. J. Am. Ceram. Soc. 89 (2), 691 (2006).
A.W. Sleight and L.H. Brixner: A new ferroelastic transition in some A2(MO4)3 molybdates and tungstates. J. Solid State Chem. 7 (2), 172 (1973).
B.A. Marinkovic, M. Ari, P.M. Jardim, R.R. de Avillez, F. Rizzo, and F.F. Ferreira: In2Mo3O12: A low negative thermal expansion compound. Thermochim. Acta 499 (1–2), 48 (2010).
M. Pley and M.S. Wickleder: Two crystalline modifications of RuO4. J. Solid State Chem. 178 (10), 3206 (2005).
W. Paraguassu, M. Maczka, A.G.S. Filho, P.T.C. Freire, F.E.A. Melo, J.M. Filho, and J. Hanuza: A comparative study of negative thermal expansion materials Sc2(MoO4)3 and Al2(WO4)3 crystals. Vib. Spectrosc. 44 (1), 69 (2007).
M. Wojdyr: Fityk: A general-purpose peak fitting program. J. Appl. Crystallogr. 43 (5 Part 1), 1126 (2010).
V. Sivasubramanian, T.R. Ravindran, R. Nithya, and A.K. Arora: Structural phase transition in indium tungstate. J. Appl. Phys. 96 (1), 387 (2004).
A.C. Torres Dias, C. Luz Lima, W. Paraguassu, K. Pereira Da Silva, P.T.C. Freire, J. Mendes Filho, B.A. Marinkovic, K.J. Miller, M.A. White, and A.G. Souza Filho: Pressure-induced crystal-amorphous transformation in Y2Mo3O12. Vib. Spectrosc. 68, 251 (2013).
M. Maczka, W. Paraguassu, A.G. Souza Filho, P.T.C. Freire, J. Mendes Filho, F.E.A. Melo, and J. Hanuza: High-pressure Raman study of Al2(WO4)3. J. Solid State Chem. 177 (6), 2002 (2004).
Q.J. Li, B.H. Yuan, W.B. Song, E.J. Liang, and B. Yuan: The phase transition, hygroscopicity, and thermal expansion properties of Yb2−xAlxMo3O12. Chin. Phys. B 21 (4), 046501 (2012).
T.R. Ravindran, V. Sivasubramanian, and A.K. Arora: Low temperature Raman spectroscopic study of scandium molybdate. J. Phys. Condens. Matter 17 (2), 277 (2005).
W.T.A. Harrison, A.K. Cheetham, and J. Faber: The crystal structure of aluminum molybdate, Al2(MoO4)3, determined by time-of-flight powder neutron diffraction. J. Solid State Chem. 76 (2), 328 (1988).
J.S.O. Evans, T.A. Mary, and A.W. Sleight: Negative thermal expansion in Sc2(WO4)3. J. Solid State Chem. 137 (1), 148 (1998).
V. Srikanth, E.C. Subbarao, and G.V. Rao: Thermal expansion anisotropy, microcracking and acoustic emission of Nb2O5 ceramics. Ceram. Int. 18 (4), 251 (1992).
P.M. Jardim, E.S. Garcia, and B.A. Marinkovic: Young’s modulus, hardness and thermal expansion of sintered Al2W3O12 with different porosity fractions. Ceram. Int. 42 (4), 5211 (2016).
C.B. Carter and M.G. Norton: Ceramic Materials (Springer, New York, NY, 2013).
ACKNOWLEDGMENTS
B.A.M. is grateful to CNPq (National Council for Scientific and Technological Development) for a Research Productivity Grant. L.P.P. and P.I.P. are grateful to CNPq for scholarships. The authors are grateful to undergraduate student Gabriella Faro for technical assistance. This study was supported by the NSERC Canada (grants to M.A.W.), and the Canada Foundation for Innovation, the Atlantic Innovation Fund and other partners that fund the Facilities for Materials Characterization managed by the Institute for Research in Materials at Dalhousie University. Aspects of this study were sponsored by the Department of the Army, U.S. Army Research Office.
Author information
Authors and Affiliations
Corresponding author
Supplementary Material
Rights and permissions
About this article

Cite this article
Prisco, L.P., Pontón, P.I., Paraguassu, W. et al. Near-zero thermal expansion and phase transition in In0.5(ZrMg)0.75Mo3O12. Journal of Materials Research 31, 3240–3248 (2016). https://doi.org/10.1557/jmr.2016.329
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1557/jmr.2016.329