
Abstract
The attempts to oral delivery of lipids can be challenging. Self-emulsifying drug delivery system (SEDDS) plays a vital role to tackle this problem. SEDDS is composed of an oil phase, surfactants, co-surfactants, emulsifying agents, and co-solvents. SEDDS can be categorized into self-nano-emulsifying agents (SNEDDS) and self-micro-emulsifying agents (SMEDDS). The characterization of SEDDS includes size, zeta potential analysis, and surface morphology via electron microscopy and phase separation methods. SEDDS can be well characterized through different techniques for size and morphology. Supersaturation is the phenomenon applied in case of SEDDS, in which polymers and copolymers are used for SEDDS preparation. A supersaturated SEDDS formulation kinetically and thermodynamically inhibits the precipitation of drug molecules by retarding nucleation and crystal growth in the aqueous medium. Self-emulsification approach has been successful in the delivery of anti-cancer agents, anti-viral drugs, anti-bacterial, immunosuppressant, and natural products such as antioxidants as well as alkaloids. At present, more than four SEDDS drug products are available in the market. SEDDS have tremendous capabilities which are yet to be explored which would be beneficial in oral lipid delivery.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Oral: The Most Preferred Administration Route
There have been numerous routes contemplated for the delivery of the drug in the systemic circulation such as oral, parenteral, nasal, topical, rectal, and cutaneous. The oral route of administration has been considered as the safer mode of administration that is often painless and non-invasive and can be self-administered (1). Drugs are delivered through an oral route compeer with the gastrointestinal physiology, which confers the “absolute” absorption site. Around 80% of dosage forms are marketed to deliver medicament through an oral route, owing to its safety, patient convenience, and cost-effectiveness. Alteration in phyiochemical properties such as partition coefficient, molecular weight, aqueous solubility, and protein binding of drug limit the bioavailability of drugs delivered orally (2). Besides being the most convenient route of administration, it has few pitfalls (3,4) including delayed onset of action, destruction of labile molecules in gastrointestinal flora (first-pass metabolism), non-specific targeting of drug molecule unless modified, difficulty in controlling the regimen release, and limitation involved in the delivery of off-putting/unsavory drug.
Lipid-Based Oral Delivery
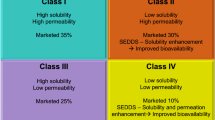
The therapeutic efficacy of an oral route of administration runs up in accordance with the contemporary factors. These factors include aqueous solubility, dissolution, and permeability (5). According to BCS (Biopharmaceutical Classification System), most of the drugs yet discovered are classified into class II (low solubility, high permeability) and class IV (low solubility, low permeability) (6,7). To combat all these issues, new technologies have been introduced in the form of novel dosage forms. It primarily targets the pathogens or its infected cells. Lipid-based formulations amplify the solubilization of drug during GI transit and fabricate the lipophilic ambiance at micro level to oblige drug transportation towards intestinal absorptive sites. This increases the absorption of the drug through numerous other concomitant mechanisms (8) namely:
-
Inhibition of drug efflux mediated by P-glycoprotein and pre-absorptive metabolism of cytochrome enzymes bounded by the gut membrane. Various reported natural, synthetic excipients including surfactants have an inhibitory action on P-gp mechanism. The high surfactant content used in self-nano-emulsifying agents (SNEDDS) can improve the intestinal drug solubilization and transcellular permeability by increasing membrane fluidity and may enhance paracellular transport by opening tight junctions. SNEDDS containing Cremophor RH40, Tween 20, and Span 80 as a dual P-gp and CYP3A4 inhibitors successfully enhanced the oral bioavailability of amiodarone by inhibiting the metabolic activity of CYP3A4 in intestinal lumen (8).
-
Bypassing the first-pass metabolism of the liver: promoting lymphatic transport as such to deliver directly to the systemic circulation.
-
Increasing permeability of the gastrointestinal membrane.
The self-convening property of lipid has turned to account for wide variety of colloidal drug carriers with diverse structure, for instance, micelles, microemulsion, emulsion, liquid crystalline nanoparticles, vesicular carriers such as liposomes, niosomes, solid lipid nanoparticles, polymer-lipid hybrid nanoparticles, and self-emulsifying/self-micro-emulsifying drug delivery system (SEDDS/SMEDDS) (Fig. 1).

Diagrammatic representation of SEDDS (self-emulsifying drug delivery system), SMEDDS (self-micro-emulsifying drug delivery system), and SNEDDS (self-nano-emulsifying drug delivery system)
Self-Emulsification in Oral Delivery
Pouton reported for the first time self-emulsifying drug delivery system, to deliver poorly water-soluble drugs via Miglyol 812 (M812, medium-chain triglyceride, MCT) and Tween 85 (T85, polyoxyethylene-20-sorbitan trioleate) incorporation to enhance both solubility and bioavailability (9). This drew the attention of researchers to work more on SEDDS. SEDDS, an isotropic mixture of oils and surfactants, intermittently accompanied by co-solvent, tends to emulsify under mild agitation mimicking the gastrointestinal flora (10,11,12). SEDDS have capacity to dissolve hydrophobic moiety, encapsulating in a single unit dosage form for oral administration. The drug moiety releases in the gastrointestinal tract lumen disperse to form the fine emulsion in contact with the gastrointestinal fluid. It equates to in situ solubilization of drug moiety, as such to be absorbed by the lymphatic system, bypassing hepatic first-pass metabolism. The droplet size of SEDDS ranging from few nanometers to several microns is further categorized as SMEDDS (self-micro-emulsifying drug delivery system) and SNEDDS (self-nano-emulsifying drug delivery system) with an emulsion of former ranging between 100 and 250 nm and later less than 100 nm (13). Enhanced drug bioavailability (14), reduced dosing frequency, more reliable time profiles of absorption, selective drug targeting considering absorption window of gastrointestinal tract (GIT), preserving of drug from hostile gut ambiance, high drug loading, availability in liquid and solid dosage forms, and flexibility in loading of sensitive drugs are the few assets of such drug delivery systems (14). Thermodynamic theory of forming microemulsion proves that entropy changes when emulsification occurs. This change in entropy that favors the dispersion is found to be greater than the energy required to increase the surface area of dispersion, furthermore to negative free energy (∆G) (15,16). This free energy asserts the requirement of energy to construct new surface between two phases, illustrated by an equation:
where ∆G denotes the free energy associated with the process N, the number of droplets, radius r and interfacial energy σ. In due course of time, the two phases separate to reduce the interfacial area. Therefore, the conventional emulsifying agent stabilizes the emulsion resulting from aqueous dilution, forming the monolayer around emulsion droplets, reducing the interfacial tension, thereby preventing coalescence. SEDDS also poses liabilities in contempt of its numerous assets namely
-
i.
chemical instability of drug
-
ii.
irritancy caused in GIT by a large amount of surfactant (30–60%) used in the formulation
-
iii.
incorporation of volatile co-solvent and hard gelatin capsule results in precipitation of lipophilic drugs
Pseudo-Ternary Phase Diagram: Testament of Self-Emulsification
Foremost authentication of lipid-based drug delivery systems, such as SEDDS, is by the construction of pseudo-ternary phase diagram. These diagrams demonstrate self-dispersing potential of SEDDS as thermodynamically stable nano-/microcarrier to deliver in GI lumen, providing information on phase behavior in relation to different formulation aspects. A very low free energy forms nano-sized oil droplets, for the “thermodynamically spontaneous” formulations developed. The higher the water content penetrates into oil droplet, the more easily it undergoes “self-emulsification” process. This prevents coalescence through the mechanical barrier. This diagram is constructed to optimize the quantities of different components employed in the preparation of SEDDS, with efficient self-emulsification potential, which is optically isotropic and thermodynamically stable (Fig. 2). It illustrates robustness to dilution in GI lumen apart from self-emulsification performance and ratio of co-surfactant used (16).

The pseudo-ternary phase diagram of SEDDS
Pseudo-ternary phase diagram can be constructed by performing an aqueous titration or spontaneous emulsification method, considering an oil phase, surfactant, and aqueous phase (17). Firstly, optimized surfactant can be dissolved in oil phase in the ratio 9:1, 8:1, 7:1, 6:1, 5:1, 4:1, 3:1, 2:1, and 1:1, in a glass test tube. Each ratio of surfactant and oil phase should then be titrated against aqueous phase. Turbidity is referred to as an endpoint. The percentage composition of the component in each ternary system has to be determined following up with the plotting of observed results on triangular coordinates to construct the phase diagram. The areas enclosed inside the plotted triangular coordinates are referred to as a biphasic system (heterogeneous) and the unenclosed areas are referred to as a monophasic system (homogeneous). In the dilution method, different ratios of surfactant, co-surfactant, and oil as per requirement are incorporated in preparing a ternary mixture. Formation of nano-emulsion is evaluated by diluting the ternary mixture with a suitable volume of double distilled water. The size of the globule formed by resulted dispersion is determined using spectroscopy. The ternary phase diagram is plotted for the nano-emulsion formed. The phase diagram determines the area of nano-emulsion formed with the desired size of globule (17).
SOLUBILITY, PERMEABILITY, AND BIOAVAILABILITY
Solubility, dissolution, and permeability equate to the therapeutic efficacy in formulating SEDDS (14,18). For the oral route of administration, aqueous solubility and dissolution rate are prime limiting factors in the absorption of a moiety to the site of action. SEDDS is remarked as an alternative approach to deliver a poorly aqueous soluble drug to the systemic circulation through the oral route. This novel lipid-based approach forms oil in water type of emulsion, when in contact with the gastrointestinal fluid. It spontaneously solubilizes the lipophilic drug. The fine droplets of the solubilized drugs formed exhibit a large interfacial surface area for drug absorption. Enhancement in intestinal lymphatic transport reducing the first-pass metabolism and amplification of intestinal permeability minimized the metabolism by cytochrome enzymes and inhibition of P-glycoprotein (P-gp)–mediated efflux by surfactant incorporation. P-gp-mediated efflux occurs due to consecutive driving conformational changes of the transporter. It includes nucleotide-binding domains being dimerized, efflux of drug, hydrolysis of ATP, and return to the initial state. Non-ionic surfactant, or surfactant above critical micellar concentration (CMC), plays an important role in inhibiting p-gp efflux (18). Supersaturation method and self-emulsification phenomenon are applied in drug delivery system to improve the oral bioavailability of drugs to elucidate maximal therapeutic action.
An Approach: Solidification Self-Emulsified Formulations
Encapsulation of Liquid and Semi-Solid Self-Emulsified Formulation: Capsule Filling
This is one of the modest, most extensively used method and is suitable for loading of low dose of highly potent drugs. Liquid self-emulsified preparations are filled in capsule through micro-spraying and banding process. For encapsulation of semi-solid self-emulsified preparation, the excipients are heated at temperature 20°C or above their melting point. The therapeutic agent is loaded by adding to the molten mixture. This drug-molten mixture is then filled in a capsule shell and cooled at room temperature. The filled capsule is sealed by micro spray or banding process (19).
Spray Drying
In this method, the liquid component is mixed with solid component using solvent followed by solubilization. The solubilized mixture is then atomized into a spray of fine droplets. The fine droplets are then subjected to drying chamber to evaporate the solvent. Dried particles under controlled condition and air flow are prepared. These particles can then be formulated as tablet. Preparations of solid dispersion of polyvinyl pyrrolidone (PVP) with nifedipine (20–50%) (20) and anti-HIV efavirenz with soluplus (19) are few prepared formulations to enhance dissolution and stability.
Adsorption on to Solid Carriers
The free-flowing powders possess large surface area and are capable of adsorbing oil material. The adsorbents have a capability of adsorbing 70% of liquid SEDDS. The liquid self-emulsifying formulations are adsorbed onto this free-flowing powder by simply blending the mixture (20).
Melt Granulation
This method is applied for preparing powder agglomeration by the addition of a binder. The binders used are melted or softened at relatively low temperatures. It offers numerous advantages in comparison to conventional wet granulation, as it is a one-step process, in which the addition of liquid component and the subsequent drying phases are excluded. Impeller speed, time of mixing, particle size, and viscosity of binder are the few variables to be controlled while processing (21).
Spheronization on Extrusion/Melt Extrusion
The liquid self-emulsifying formulation is first mixed with extrusion aid. The mixture is then added to water. It extrudes from a die on applying force under controlled temperature, product flow, and pressure. The maximum drug loading (approx. 60%), content uniformity, and solvent-free process offer an advantage (Fig. 3) over other methods (22).

Different methods of SEDDS solidification
Commonly Used Excipients for SEDDS Preparation
Drug
Hydrophobicity/lipophilicity of a drug is considered to be the prime factor for SEDDS formulation. Ideally, a drug should have a log p ≥ 2. A low dose of drug is formulated and should not undergo extensive first-pass metabolism (23).
Oil
Oil plays a vital role in solubilization of lipophilic drug. It enhances the availability of drug for efficient absorption in GIT through intestinal lymphatic system. The physical, melting, and the hydrophilic-lipophilic balance (HLB) characteristics of glycerides depend on the type of fatty acids and degree of etherification with respect to glycerol to yield mono or diglycerides. Both medium-chain triglycerides (MCTs) and long-chain triglycerides (LCTs) with different degrees of saturation can be used. MCTs, with 6–12 carbon chains, are transported through portal blood into the systemic circulation. LCTs with more than 12 carbon chains are transported through intestinal lymphatic. As MCT has increased fluidity, solubility properties, and prevention of oxidation, it is most widely used lipid formulation (21,24).
Surfactant and Co-surfactant
Surfactants create an interfacial film and reduce the interfacial tension, thereby exhibiting dispersion. HLB value needs to be monitored during formulating SEDDS. In order to achieve higher emulsification, the surfactant with HLB value greater than 12 is selected. It forms small oil in water (O/W) droplets and assists in rapid spreading of the meant formulation. Mostly, non-ionic surfactants are chosen for its non-toxic nature to formulate SEDDS although they may cause moderate irreversible alteration in permeability of wall of GIT. Approximately 30–60% w/w of the formulation of surface-active agents yields better self-emulsification in GIT. Large concentration of surfactant may irritate the wall of GIT (25).
Co-surfactant reduces the interfacial tension to an even smaller transient negative value. It imparts flexibility to the interfacial film in order to achieve different curvatures for the formation of different concentrations of microemulsion. The larger concentration of surfactant (nearly 30%) can be counterfeited by the introduction of co-surfactant. At this point, the expansion of interface leads to the formation of finely dispersed droplets. It will adsorb more surfactant or surfactant/co-surfactant ratio, until the film gets depleted enough to make the interfacial tension to be positive again. It yields to the formation of “spontaneous emulsion.” Medium-chain length alcohols (C3–C8) are mostly employed as co-surfactant (26).
Size, Surface Morphology, and Phase Separation
The characterization methods of SEDDS include size, zeta potential analysis, surface morphology via electron microscopy, and phase separation methods. The size and zeta potential greatly affect the physical and chemical properties of SEDDS. Size and polydispersity index (PDI) can affect the melting properties of lipid formulation as well as penetration and dissolution rate. It implies the release of drug encapsulated. In the literature, the size of oil droplets of SEDDS formulation was reported (27,28). Size can be determined by some routine techniques like small angle X-ray or neutron scattering. Zeta potential provides information about the charge present on the droplets that will decide its entry across the plasma membrane to reach the target site. Size can be determined by some routine techniques like a small angle X-ray or neutron scattering and dynamic light scattering (DLS) (29). Electron microscopy (SEM and TEM) and atomic force microscopy (AFM) including X-ray spectroscopy can be used to evaluate the surface morphology of SEDDS. These techniques give information about the uniformity and homogeneity of the prepared emulsion and stability of formulation. Measuring the small phase angle during polarization is however challenging. Spectroscopic methods such as Raman, IR, and NMR may be used for the characterization and evaluating drug-carrier compatibilities of SEDDS (30,31,32). Phase separation evaluation is used to decipher the stability of prepared SEDDS. Sample is diluted with distilled water and centrifuged. The cloud point was determined to investigate the stability of SEDDS in gastrointestinal tract. The cloud point of SEDDS should be more than 37°C; otherwise, absorption of emulsion can be interrupted as the cloudy emulsion affects the absorption by dehydration of ingredients used in SEDDS preparation (33).
Supersaturable SEDDS and Pharmacokinetic Parameters
Supersaturation is the phenomenon of maintaining the solubility of compound above the equilibrium state without precipitation that results in the crossing of a biological membrane by different drug molecules. Nearly 70% of the potential drug molecule failed to achieve their therapeutic activity up to the highest level due to poor solubility. It leads to a slower absorption rate. There are many alternative techniques such as cyclodextrin complexation, solid dispersion, and lipid-based drugs, but SEDDS-based formulation achieves much attention to resist this solubility problem. Numerous SEDDS-based formulations have been reported for the different categories of drugs such as anti-cancer, anti-diabetic, anti-viral, anti-bacterial, and some natural products. Some of them are summarized in the subsequent sections of this review. A supersaturated (S) solution consists of high drug content that can increase the driving force for flux across the GI membrane and enhanced absorption in substantial period of time (34).
Polymers used in the preparation of S-SEDDS formulation kinetically and thermodynamically inhibit the precipitation of drug molecules by retardation of nucleation and crystal growth in the aqueous medium. A number of S-SEDDS formulations such as silybin and celecoxib are explored for poorly water-soluble drugs which provided a clear vision that polymers used in these formulations prevent the precipitation and come out as a better option to improve the oral bioavailability and absorption. Polyvinyl pyrrolidone (PVP) polymer has been widely used to improve the water solubility of various poorly soluble drugs for, e.g., sulfathiazole (35), phenytoin (36), chlorothiazide (37), estradiol (38), norethindrone acetate (39), fluocinonide (40), and hydrocortisone acetate (41,42). Badische Anilin-und-Soda-Fabrik (BASF), a named company, introduced an amphiphilic copolymer, polyvinyl caprolactam-polyvinyl acetate-polyethylene glycol (soluplus) graft copolymer to produce solid dispersions. It is widely explored to prepare S-SEDDS with excellent solubilization, oral bioavailability, and higher absorption rate. This copolymer is utilized as a suitable stabilizer for preparation of S-SEDDS. Recently, Quan et al. prepared S-SEDDS using soluplus with fenofibrate, a sparingly soluble drug using solvent evaporation method. The authors constructed a super saturable drug delivery system to improve the oral bioavailability and solubility of the drug via precipitation inhibitor. Sometimes, the supersaturation may lead to rapid precipitation of drug. This problem can be overcome by the use of precipitation inhibitors. A supersaturation assay was performed to evaluate the precipitation inhibitory effect of prepared drug delivery system. The results revealed that the soluplus formulation of fenofibrate showed more retardation towards precipitation than the free drug effectively. It released the drugs in a sustained manner up to 2 h. The AUC for S-SEDDS formulation was 1.4 times more in the presence of soluplus than the free drug. The in vitro studies exhibited the results with a statement that all the excipients used in the preparation of S-SEDDS are biocompatible. It showed excellent results against Caco-2 cell lines in MTT assay. Based on the in vivo pharmacokinetic studies performed in beagle dogs, it was concluded that the super saturable preparation of fenofibrate with soluplus precipitation inhibitor provided an increase of 40% relative bioavailability and can work as a potent candidature to increase the oral bioavailability of drug (43).
Gao and colleagues applied supersaturation phenomenal approach to improve the water solubility of PNU-9153, an agent that can enhance the activity of insulin (44). In 1960, Higuchi was the first scientist who used the supersaturation process in transdermal drug delivery to enhance the drug efflux through the skin (45). Different water-soluble cellulose-based polymers such as methylcellulose, hydroxypropyl methylcellulose phthalate, and sodium carboxymethyl cellulose are reported for precipitation inhibition in supersaturation method by inhibiting the crystal growth. This study described the supersaturation process used to develop a formulation of PNU-9135 with improved solubility and oral bioavailability. Hydroxypropyl methylcellulose (HPMC) was incorporated in the formulation to inhibit the precipitation. The formulation was developed with different polymers like PEG-400 and propylene glycol (PG) and compared. Cremophor as a surfactant, PEG 400, Pluronic L-44, HPMC, and other minor components were used for the preparation of S-SEDDS. It showed an oral bioavailability of 76% that is equivalent to a formulation composed of tween (bioavailability 68%). An in vivo study of PG composed S-SEDDS conducted on dogs elucidated a data with fivefold greater bioavailability than the formulation of PEG-400. The significant results were obtained with HPMC for PNU-9135 in the in vivo studies (46).
Mechanistic Fate of Drug Delivery
Gastrointestinal tract is profoundly supplied with lymph and blood vessels. SEDDS/SMEDDS target the intestinal lymphatic system to deliver therapeutic agent. Higher the lipophilicity of a drug, higher is the extent of lymphatic transport. It enhances the bioavailability directly or indirectly through reduction in first-pass metabolism. It helps in effective luminal drug solubility. Chylomicrons (CML), predominantly composed of triacylglycerol, are stimulated to carry self-emulsified formulation via lymphatic transport. The transport pathways include paracellular, fusion, and transcytosis followed by a drug from a self-emulsification drug delivery system (Fig. 4). CML are low-density lipoproteins. The absorption rate constant (Ka) and permeability coefficient (Papp) affect the intestinal lymphatic absorption of drug (47). Incorporation of drug into CML either directly (if polar) or digested results in sailing of chylomicrons. It further increases its solubilization capacity in GI tract. These drugs carrying CML are subjected to secretion from enterocyte to initiate lymphatic transport, rather than portal circulation. The therapeutic agent in intestinal lymph gets into the systemic circulation via junction of left internal jugular and subclavian veins. This leads to avoidance of first-pass metabolism (48). Uptake of self-emulsified formulation is depicted in Fig. 4.

Schematic representation of uptake of SEDDS with different pathways
Drug Delivery Applications
Since the evolution of the earth, plants are a big source of medicinal values. These naturally occurring compounds are facing many problems in their delivery to the body such as low solubility, low bioavailability, and rapid release. A self-emulsification method, as a lipid-based drug delivery system, has been used exhaustively to improve their physio-chemical parameters. Some of the antioxidants and alkaloids are discussed below and summarized in Table I.
Solubility and Bioavailability Enhancement of Natural and Other Bioactives
Antioxidants play a vital role in body defense system against ROS (reactive oxygen species) system (62). SEDDS are one of the drug delivery vehicles, which resulted in improved delivery of antioxidants.
Quercetin
Quercetin is an antioxidant and dietary flavonoid found in vegetables, fruits, nuts, and red wine. The poor aqueous solubility and bioavailability is the major hurdle of its intestinal absorption. Self-nano-emulsifying drug delivery systems (SNEDDS) have gained much attention to improve the solubility and the bioavailability of hydrophobic drugs. Several marketed SEDDS-based products are available in the market such as Sandimmune, Neoral (cyclosporine), Norvir (ritonavir), and Fortovase (saquinavir) with improved effectivity (63).
Jain and coworkers enhanced the oral bioavailability and antioxidant potential of quercetin (QN) through SEDDS formulation. Capmul MCM acting as the oil phase, ethanol as co-surfactant, and Tween 80 as surfactants were used. DPPH scavenging assay was performed to compare the activity of antioxidants in QN-SEDDS and pure quercetin. The stability testing was done according to the ICH guidelines for 6 months in accelerated conditions. QN-SEDDS showed a significant increase in the cellular uptake by 23.75-fold than free QN. QN-SEDDS oral bioavailability was higher than the free QN. QN-SEDDS exhibited significantly higher in vivo antioxidant potential than free QN against cardiotoxicity and nephrotoxicity induced by doxorubicin and cyclosporin A, respectively (49).
Resveratrol
Resveratrol or 3,5,4′-trihydroxy trans-stilbene is a natural antioxidant. It has a wide range of biomedical applications (64,65). However, chemical instability and the non-aqueous solubility limit its application. Several SEDDS formulation were prepared to improve its solubility and absorption through the intestine. Mamadou and coworkers developed resveratrol formulation via SEDDS to improve the solubility and absorption of this natural product. For the preparation of semi-solid and semi-liquid nano-emulsions Labrasol, Gelucire, Miglyol, and Montanox surfactants were used by this group, respectively. The chamber was used for analysis of the absorption profile in the jejunum area of the intestine. The results suggested that RSV-SEDDS may be evaluated further and emerged as a suitable candidate for RSV delivery via the oral route (50).
Alkaloids
Alkaloids are important natural products found in the plants and showed different pharmacological activities such as the anti-cancer activity of vinblastine and taxanes and analgesic activities of morphine (66). Naturally, occurring alkaloids such as noscapine (N) exhibited anti-tumor activity. It is potentiated by the induction of cell death. It is a lipophilic compound with poor aqueous solubility, therefore, suffering from the oral bioavailability issue. To tackle this problem, Andey et al. developed and characterized a mannose-based formulation of noscapine using self-emulsification method to improve the oral bioavailability of this natural compound. The SMEDDS and SEDDS formulations with mannosamine and without mannosamine were prepared by a spray drying method and compared. Labrasol, Tween 80, and HPMC (hydroxypropyl methylcellulose), a semi-synthetic polymer, were used as co-surfactant, surfactant, and an emulsifying agent to prepare the formulations. SMEDDS size range was found in nanometer while SEDDS formulation was 5–6 μm in size. The mannosamine SEDDS formulation was released for 6 h with controlled release effect during the in vitro release studies. The cell cytotoxicity studies were performed on a Caco-2 cell line against H1650 SP cells and showed effective results (51).
Anti-cancer Drugs
Docetaxel
Docetaxel (DCT) is a taxane derivative and act as a potent chemotherapeutic agent in the cancer treatment battle. DCT is a poorly soluble drug with aqueous solubility (< 5 μg/mL) and low bioavailability (5%) (67). The bulky polycyclic structure of this drug is the reason of low solubility. SEDDS can be used as a better option to enhance oral bioavailability. Seo et al. prepared a SNEDDS formulation to improve the bioavailability and overall efficacy. DCT-loaded SNEDDS were prepared by rational blending of Capryol 90, Labrasol, and Transcutol HP using nano-emulsion method. Anti-tumor activities of prepared SNEDDS were greater than the free drug. The in vivo studies confirmed that the D-SNEDDS increased the oral bioavailability by 17%. The reduced toxicity profile of D-SNEDDS was observed when compared with Taxotere® in the in vivo studies. All the results claimed that the D-SNEDDS may be used as a potent formulation through oral dosage with enhanced anti-tumor activity and reduced toxicity (52).
GLM-7
GLM-7 is a novel acrylamide drug under preclinical phase but showed poor water solubility in the pre-formulation studies (68,69). Wang and research group prepared a SEDDS formulation of GLM-7 and enhanced the oral bioavailability of the drug. The mean size of the prepared emulsion was observed in the range of 400 nm with spherical shape and uniformity. The dissolution study results explained that more than 80% in the case of GLM-7 SEDDS in acidic medium. In the plasma of beagle dogs, prepared SEDDS concentration was ninefold higher than the reference or control used in the in vivo studies. In addition, an area under the curve results also suggested that the SEDDS enhanced the oral bioavailability of GLM-7 (53).
DIM-14
DIM (3,3-diindolylmethane) is an active anti-cancer molecule with low solubility. Patela et al. employed the cancer stem cells (CSC) targeting approach with DIM-14 to potentiate the tumor treatment. The poor water solubility of DIM-14 compound restricts its benefits in tumor therapy. Self-emulsifying preparation of DIM-14 showed greater solubility and oral bioavailability than the pure DIM-14 compound. The cytotoxicity study was performed against H1650 stem cell lines and exhibited improved activity with SED system. The particle size of DIM-14 SED system was observed in the range of 250 nm with negative zeta potential. Pharmacokinetic study results suggested that the prepared DIM-41 SED system improved the absorption rate by threefold. The tumor volume was reduced up to 60% by the prepared formulation against tumor models (54).
Immunosuppressant
Tacrolimus (TC) is an immunosuppressive drug and exhibited variant oral bioavailability with a range of 4 to 89%. The main reason for this variability include (i) poor aqueous solubility, (ii) extensive metabolism by cytochrome P450 3A4 (CYP3A4), and (iii) P-glycoprotein (P-gp) efflux transport. Wang et al. reported a preparation of TC based SMEDDS formulation to improve the solubility and oral bioavailability and optimize the formulation using a pseudo-ternary phase diagram. Tocopheryl polyethylene glycol succinate (TPGS) and Cremophor EL40 were selected as excipients for the SEDDS formulation P-glycoprotein (P-gp) inhibitors. Miglyol 840 was chosen as the oil phase and Transcutol P as co-surfactant for the preparation of TC-SEDDS in the ratio of 1:2. As per solubility test results, solubility has been reported to be higher with the use of Miglyol 840. The pseudo-ternary phase testing was performed to optimize the concentration of oil, surfactants, and co-surfactants. The average sizes of oil droplets were observed to be 20 nm in size. Bioavailability and pharmacokinetic studies stated that Cmax and area under the curve of TC-SEDDS were higher than the free TC. A significant increase in oral bioavailability of TC with SMEDDS was noticed despite of no significant results seen for inhibition of P-gp efflux (55).
Anti-bacterial
Satranidazole (SZ) is an anti-protozoal drug belonging to the category of nitroimidazoles. It is a potent and active drug used for parasitic diseases (intestinal and hepatic amebiasis, giardiasis, trichomoniasis, and anaerobic infections). The low bioavailability and poor aqueous solubility limit the wide spectrum application of this drug. Several alteration techniques such as cyclodextrin complexation, micronization, lipid drugs, and micelles were applied to overcome this problem. Out of these delivery systems, SEDDS provided better results to tackle the challenges faced by SZ. SEDDS provided more stable and efficient transport across the cell membrane. Gurav et al. prepared microemulsion for SZ using oleic acid as the oil phase, Tween 20 as the surfactant, and PEG400 as co-surfactant. SZ-SEDDS formulation stability and dispersibility were analyzed by different methods. A total of nine formulations were prepared from F1–F9. The microemulsion region was estimated by constructing a pseudo-ternary phase diagram and optimized the formulation. In the in vitro release studies, only three formulations showed sustained release effect up to 70% in 45 min. The size of oil droplets was found to be in the range of 750 nm for all nine formulations. The three best ones showed a size of 18.4 ± 9.5, 122.5 ± 21.9, and 85.4 ± 5.42 nm. The ideal size range for microemulsion is up to 250 nm. Therefore, SEDDS formulations can be a potential alternative method to improve the solubility of SZ (56).
Amphotericin B (AMB)
Bioavailability and aqueous solubility of amphotericin B (AMB) are very low. Recently, the lipid-based drug delivery system has gained attention in terms to overcome this problem. Wasan and research group developed a formulation of AMB-SEDDS and evaluated its effectivity against Aspergillus fumigatus– or Candida albicans–infected rats. They developed SEDDS formulation using PEG phospholipids and medium-chain fatty acid triglycerides and glyceryl mono-oleate via the solvent evaporation method. Phase stability of formulation was determined by atomic force microscopy (AFM). The mean size of SEDDS was found to be 200–400 nm. The formulation of triglycerides was prepared with different grades of PEG 350, 550, 750, and 1000 and showed an excellent solubilization range. Glyceryl mono-oleate PEG2000–based oral AMB treatment significantly decreased total fungal CFU (colony functional unit) concentrations in the A. fumigatus–infected rats. This formulation at the doses of 5 mg/kg and 10 mg/kg drastically reduced fungal CFU concentrations in the kidney by > 75% and > 95%, respectively, in comparison to non-treated controls in the Candida albicans–infected rats (57).
Anti-viral Drugs
Tipranavir (TPV) is a drug act by inhibition of non-peptide protease used for the treatment of HIV-AIDS (70). The self-emulsification method is utilized to increase the solubility and bioavailability of TPV. The TPV-SEDDS formulation in soft gelatin capsule enhanced the bioavailability twice than the TPV filled in hard gelatin capsules without self-emulsification approach (58).
Saquinavir (SQV) is another active and powerful anti-HIV drug which falls in the category of protease inhibitor (PI) and used as a regimen for highly active antiretroviral therapy (HAART). Presently, SQV is available in hard and gelatin capsule form in the market. SQV is water-insoluble drug; hence, to dissolve this drug in the formulation, Capmul is used as an excipient. The high concentration of Capmul causes rapid release of the drug and diarrhea as side effects in patients. To overcome this side effect, a new approach has been applied. SEDDS formulations of SQV along with ritonavir reduce the side effects at some instant and enhance the bioavailability as well (59).
Anti-hypertensive and Cardiovascular Drugs
Felodipine (FP) is an anti-hypertensive agent and acts by blocking the calcium channel during signaling pathway. Despite its potent action, poor water solubility, fast liver metabolism, and low bioavailability limit its pharmacological benefits. In recent years, some approaches have been used to enhance the dissolution of felodipine like mesoporous silica nanoparticles and wet-milling (71,72).
Jing et al. successfully enhanced the bioavailability of FP by preparing self-emulsifying tablets. They prepared solid and liquid self-emulsifying formulations and compared them. The size of solid tablets was found to be equivalent to SMEDDS liquid formulations. X-ray diffractometry (XRD) data suggested the polymorphism state of prepared tablets. In vivo studies were performed on fasted beagle dogs, and solid FP tablet bioavailability was twofold higher than the oral dosage form available in the market. However, no significant differences were observed between solid and liquid dosage forms of FP. Dissolution studies, stability studies, and bioavailability assay results revealed that the solid emulsifying formulation is an interesting and effective approach for FP solubility improvement (60).
Cilnidipine is a calcium channel blocker anti-hypertensive drug with poor solubility and low bioavailability issues. Bakhle and Avari reported the solid SEDDS of cilnidipine (CDP) to enhance solubility and bioavailability of CDP for oral delivery. Capryol 90, Tween, and Neusilin US2 80 act as the oil phase, surfactant, and adsorbent for the preparation of microemulsion. The area of microemulsion was identified with the help of pseudo-ternary phase diagram. Dissolution study results remarked the prepared CDP-SEDDS formulation to be more soluble than the CDP marketed formulations. The crystalline form of CDP in SEDDS preparation was changed into the amorphous form that can be observed in the X-Ray powder diffraction studies. CDP-SEDDS absorption rate was higher than the pure drugs (61).
Atorvastatin (AVS) is a drug used for the treatment of hyperlipidemia and cardiovascular attacks. It is insoluble in acidic pH and sparingly soluble in water. High pre-systemic clearance is the main reason for AVS poor solubility and low oral bioavailability. Therefore, the lipid carrier-based approach is explored to combat aqueous solubility and bioavailability issues. Kosnik et al. developed solid and liquid lipid-based self-emulsifying system for AVS to improve its solubility. Liquid SEDDS have some disadvantages; hence, the author also prepared solid SEDDS formulation and compared both of them. Solubility test, pseudo-ternary phase diagram, stability test, and size analysis were performed for both formulations. Solid SEDDS was prepared by spray drying technique and method was optimized. The in vitro dissolution assay revealed that the solid self-emulsifying formulation exhibited better solubilization properties than liquid SEDDS. Solid SEDDS showed less turbidity than the liquid ones in the phase diagram assay. Hence, the authors successfully prepared a solid SEDDS of AVS. It increased the solubility of this BCS II class drug and improve its absorption rate (10).
CHALLENGES AND FUTURE PROSPECTS
Certain biopharmaceutical issues involved with SEDDS are such as amount of drug used for preparation, risk of precipitation, surfactants, and excipients used in SEDDS and the effect of oil droplet size. The optimization of the formulation is based on the number of factors such as the dose encapsulation efficiency, adequate solvent capacity to dissolve the drug, and the fate of drug after absorption from intestinal mucosa. Several times the choice of drug formulation depends on the solvent capacity. The drugs having a log P value about 2 is more devoted to this problem. Hydrophobic drugs cross the biological membrane fastly but the dissolution rate of these drugs is very low in gut or mucosa. It is a common problem for practitioners working on lipid soluble drugs (23). Many drugs failed in preclinical studies due to this reason. At this time, it is wise to use co-solvent and some other strategies to improve the drug bioavailability. In some cases, lipidic drugs may produce a pharmacological response at a lower concentration. If the drug is active and potent and sufficiently dissolves in the solvent system, then one should go with different SEDDS preparation methods but the precipitation rate must be considered with keeping in mind the adequate absorption rate for formulation in the gut. In 1989, Tarr and Yalkowsky successfully evaluated the effect of droplet size on absorption via gut perfusion experiment in case of cyclosporin A (73). The fine particles of microemulsion showed a better absorption rate than larger particles. The ideal size of particles of the microemulsion is considered within the range of 250 nm. A formulation with high content of solubilizing agents and fine dispersion provides particles of size 100 nm. The size of particles also affected the bioavailability; a significant bioavailability range difference was noticed between the particle sizes of 100 nm and 250 nm but more evidence are awaited to prove this statement until now (74). The risk of precipitation is involved with the number of polymers, surfactants, and triglycerides used in the preparation of SEDDS. At present, there are no established methods available to determine the risk of precipitation. Excessive use of co-solvent, copolymers, polymers, and other excipients may affect the supersaturation process in self-emulsification of drug candidates (75). No doubt SEDDS has been successfully used to enhance the solubility and bioavailability of poorly aqueous soluble drugs and many publications are the shreds of evidence for this statement. Self-emulsification approach has been established as an important tool to improve the absorption rate and overall therapeutic efficacy of drugs explained in the literature. Many anti-cancer, anti-diabetic, anti-bacterial, and anti-viral drugs solubility parameters are improved via self-emulsification by different methods of preparation. In conclusion, there is a need to pay attention to the characteristic analysis of SEDDS. The priority of future research should be based on bioavailability studies and the mechanism of action of a different formulation.
References
Mahmooda A, Bernkop-Schnurch AM. SEDDS: a game changing approach for the oral administration of hydrophilic macromolecular drugs. Adv Drug Del Rev. 2018. https://doi.org/10.1016/j.addr.2018.07.001.
Viswanathan P, Muralidaran Y, Ragavan G. Challenges in oral drug delivery: a nano-based strategy to overcome nanostructures for oral medicine in EA Grumezescu. Nanostruct Oral Med. 2017:173–20.
Gupta H, Bhandari D, Sharma. A recent trends in oral drug delivery: a review. Recent Pat Drug Deliv Formul. 2009;3:162–73.
Verma P, Thakur AS, Deshmukh K, Jha AK, Verma S. Routes of drug administration. Int J Pharm Res. 2010;1:54–9.
Feeney OM, Crum MF, McEvoy CL, Trevaskis NL, Pouton CW, Charman WN, et al. 50 years of oral lipid-based formulations: provenance, progress and future perspectives. Adv Drug Del Rev. 2016;101:167–94.
Kalepun S, Manthina M, Padavala V. Oral lipid-based drug delivery systems: an overview. Acta Pharm Sin B. 2013;3:361–72.
Gursoy RN, Benita S. Self-emulsifying drug delivery systems (SEDDS) for improved oral delivery of lipophilic drugs. Biomed Pharmacother. 2004;58:173–82.
Karan M, Rajashree CM, Arti RT. Challenges in oral delivery: role of P-gp efflux pump. Curr Drug Ther. 2014;9:47–55.
Pouton CW. Formulation of self-emulsifying drug delivery systems. Adv Drug Del Rev. 1997;25:47–58.
Kosnik AC, Szekalska M, Amelian A, Szymanska E. Development and evaluation of liquid and solid self-emulsifying drug delivery system for atorvastatin. Molecules. 2015;20:21010–22.
Charman SA, Charman WN, Rogge MC, Wilson TD, Dutko FJ, Pouton CW. Self-emulsifying drug delivery systems: formulation and biopharmaceutic evaluation of an investigational lipophilic compound. Pharm Res. 1992;9:87–93.
Craig DQM, Lievens HSR, Pitt KG, Storey DE. An investigation into the physicochemical properties of self-emulsifying systems using low frequency dielectric spectroscopy, surface tension measurement and particle size analysis. Int J Pharm. 1993;96:147–55.
Mahapatra AK, Murthy PN. Self-emulsifying drug delivery systems (SEDDS): an update from formulation development to therapeutic strategies. Int J Pharm Tech Res. 2014;6:545–68.
Sebastain G, Rajasree PH, George J, Gowda DV. Self-micron emulsifying drug delivery systems (SMEEDS) as a potential drug delivery system-novel applications and future perspectives: a review. Int J Pharm. 2016;6:105–10.
Patel SN, Patel DN, Patel TD, Prajapati TH, Parikh BN. Self-emulsifying drug delivery system. J Glob Pharm Tech. 2010;2:29–37.
Xu X, Cao M, Ren L, Qian Y, Chen G. Preparation and optimization of rivaroxaban by self-nanoemulsifying drug delivery system (SNEDDS) for enhanced oral bioavailability and no food effect. AAPS PharmSciTech. 2018;19:1847–59.
Khedekar K, Mittal S. Self-emulsifying drug delivery system: a review. Int J Pharm Sci Res. 2013;4:4494–507.
Fotouh KA, Allam AA, El-Badry M, El-Sayed AM. Self-emulsifying drug-delivery systems modulate P-glycoprotein activity: role of excipients and formulation aspects. Nanomedicine. 2018;13. https://doi.org/10.2217/nnm-2017-0354.
Lavra ZMM, Santana DPD, Re M.I. Solubility and dissolution performances of spray dried solid dispersion of efavirenz in soluplus. Drug Dev Ind Pharm 2017;43: 42–54.
Ikasari ED, Fudholi A, Martono S, Marchaban. Investigation of nifedipine solid dispersion with solvent PVP K-30. Int J Pharm Pharm Sci. 2015;7:389–92.
Hauss DJ, Fogal SE, Ficorilli JV, Price CA, Roy T, Jayaraj AA, et al. Lipid-based delivery systems for improving the bioavailability and lymphatic transport of poorly water-soluble LTB4 inhibitor. J Pharm Sci. 1998;87:164–9.
Pillay V, Fassihi R. Unconventional dissolution methodologies. J Pharm Sci. 1999;88:843–51.
Pouton CW. Lipid formulations for oral administration of drugs: non-emulsifying, self-emulsifying and ‘self-microemulsifying’ drug delivery systems. Eur J Pharm Sci. 2000;2:S93–8.
Kimura M, Shizuki M, Miyoshi K, Sakai T, Hidaka H, Takamura H, et al. Relationship between the molecular structures and emulsification properties of edible oils. Biosci Biotechnol Biochem. 1994;58:1258–61.
Gershanik T, Benita S. Positively charged self-emulsifying oil formulation for improving oral bioavailability of progesterone. Pharm Dev Technol. 1996;1:147–57.
Reiss H. Entropy-induced dispersion of bulk liquids. J Colloid Interface Sci. 1975;53:61–70.
Boltri L, Canal T, Esposito PA, Carli F. Lipid nanoparticles: evaluation of some critical formulation parameters. Proc Intern Symp Control Rel Bioact Mater. 1993;20:346–7.
Xu R. Progress in nanoparticles characterization: sizing and zeta potential measurement. Particuology. 2008;6:112–5.
Pecora R. Dynamic light scattering measurement of nanometer particles in liquids. J Nanopart Res. 2009;2:123–31.
Goddeeris C, Goderis B, van den Mooter G. Lyotropic, liquid crystalline nanostructures of aqueous dilutions of SMEDDS revealed by small-angle X-ray scattering: impact on solubility and drug release. Eur J Pharm Sci. 2010;40:110–7.
Gradzielski M. Recent developments in the characterization of microemulsions. Curr Opin Cold Int Sci. 2008;13:263–9.
Vogt FG. Solid-state characterization of amorphous dispersions. In: Newman A, editor. Amorph solid dispersions: Pharm; 2015. p. 117–78.
Elnaggar YSR, El-Massik MA, Abdallah OY. Self-nano-emulsifying drug delivery systems of tamoxifen citrate: design and optimization. Int J Pharm. 2009;380:133–41.
Kataoka M, Sugano K, da Costa Mathews C. Application of dissolution/permeation system for evaluation of formulation effect on oral absorption of poorly water-soluble drugs in drug development. Pharm Res. 2012;29:1485–94.
Simonelli AP, Mehta SC, Higuchi WI. Inhibition of sulfathiazole crystal growth by polyvinyl pyrrolidone. J Pharm Sci. 1970;59:633–8.
Sekikawa H, Fujiwara J, Naganuma T, Nakano M, Arita T. Dissolution behaviors and gastrointestinal absorption of phenytoin in phenytoin-polyvinyl pyrrolidone coprecipitate. Chem Pharm Bull. 1978;26:3033–9.
O’Driscoll KM, Corrigan OI. Chlorothiazidepolyvinyl pyrrolidone (PVP) interactions: influence on membrane permeation (everted rat intestine) and dissolution. Drug Dev and Ind Pharm. 1982;8:547–64.
Megrab NA, Williams AC, Barry BW. Oestradiol permeation through human skin silastic membrane: effects of propylene glycol and supersaturation. J Control Release. 1995;36:277–94.
Ma X, Taw J, Chiang C. Control of drug crystallization in transdermal matrix system. Int J Pharm. 1996;142:115–9.
Schwarb FP, Imanidis G, Smith EW, Haigh JM, Surber C. Effect of concentration and degree of saturation of topical fluocinonide formulations availability on human skin. Pharm Res. 1997;16:909–15.
Raghavan SL, Trividic A, Davis AF, Hadgraft J. Crystallization of hydrocortisone acetate: influence of polymers. Int J Pharm. 2001a;212:213–21.
Raghavan RL, Kiepfer B, Davis AF, Kazarian SG, Hadgraft J. Membrane transport of hydrocortisone acetate from supersaturated solutions; the role of polymers. Int J Pharm. 2001b;221:95–105.
Quan G, Niu B, Singh V, Zhou Y, Wu CY, Pan X, et al. Supersaturable solid self-micro emulsifying drug delivery system: precipitation inhibition and bioavailability enhancement. Int J Nanomed. 2017;12:8801–11.
Gao P, Guyton ME, Huang T, Bauer J, Stefanski KJ, Lu Q. Enhanced oral bioavailability of a poorly water-soluble drug pnu-91325 by super saturatable formulations. Drug Dev Ind Pharm. 2004;30:221–9.
Higuchi T. Physical chemical analysis of percutaneous absorption process. J Soc Cosmet Chem. 1960;11:85–97.
Halliwel B, Gutteridge JMC. The definition and measurement of antioxidants in biological systems. Free Radic Biol Med. 1995;18:125–6.
Li F, Hu R, Wang B, Gui Y, Cheng G, Gao S, et al. Self-microemulsifying drug delivery system for improving the bioavailability of huperzine A by lymphatic uptake. Acta Pharm Sin B. 2017;7(3):353–60.
Charman WN, Stella VJ. Estimating the maximal potentials for intestinal lymphatic transport of lipophilic drug molecules. Int J Pharm. 1986;34:175–8.
Jain S, Jain AK, Pohekar M, Kaushik T. Novel self-emulsifying formulation of quercetin for improved in vivo antioxidant potential: implications on drug induced cardiotoxicity and nephrotoxicity. Free Radic Biol Med. 2013;65:117–30.
Mamadou GC, Charrueau JD, Nzouzi NL, Eto B, Ponchel G. Increased intestinal permeation and modulation of pre-systemic metabolism of resveratrol formulated into self-emulsifying drug delivery systems. Int J Pharm. 2017;521:150–5.
Andey T, Patel A, Marepally S, Chougule M, Spencer SD, Rishi AK, et al. Formulation, pharmacokinetic, and efficacy studies of mannosylated self-emulsifying solid dispersions of noscapine. PLoS One. 2016;11:e0146804.
Seo YG, Kima DH, Ramasamy T, Kim JH, Marasini N, Oh YK, et al. Development of docetaxel-loaded solid self-nanoemulsifying drug delivery system (SNEDDS) for enhanced chemotherapeutic effect. Int J Pharm. 2013;452:412–20.
Wang YJ, Sun J, Zhang T, Liu H, He F, He Z. Enhanced oral bioavailability of tacrolimus in rats by self-micro emulsifying drug delivery systems. Drug Dev and Ind Pharm. 2011;37:1225–30.
Patela AR, Doddapanenia R, Andeya T, Wilson H, Safeb S, Singh M. Evaluation of self-emulsified DIM-14 in dogs for oral bioavailability and in Nu/nu mice bearing stem cell lung tumor models for anticancer activity. J Control Release. 2015;10:18–26.
Wang Y, Yu N, Guo R, Yang M, Shan L, Huang W, et al. Enhancing in vivo bioavailability in beagle dogs of GLM-7 as a novel anti-leukemia drug through a self-emulsifying drug delivery system for oral delivery. Curr Drug Deliv. 2016;13:131–42.
Gurav NP, Dandagi MP, Gadad AP, Masthiholimath VS. Solubility enhancement of satranidazole using self-emulsified drug delivery systems. Ind J Pharm Educ Res. 2015;50:3.
Wasan EK, Bartlett K, Gershkovich P, Sivak O, Banno B, Wong Z, et al. Development and characterization of oral lipid-based Amphotericin B formulations with enhanced drug solubility, stability and antifungal activity in rats infected with Aspergillus fumigatus or Candida albicans. Int J Pharm. 2009;372:76–84.
Cohen SJW, Schuurman R, Burger DM, Koopmans PP, Sprenger HG, Juttman JR, et al. Randomized trial comparing saquinavir soft gelatin capsules versus indinavir as part of triple therapy (CHEESE study). JAIDS. 1999;13:53–8.
Buss N, Snell P, Bock J, Hsu A, Jorga K. Saquinavir and ritonavir pharmacokinetics following combined ritonavir and saquinavir (soft gelatin capsules) administration. Br J Clin Pharmacol. 2001;52:255–64.
Jing B, Wang Z, Yang R, Zheng X, Zhao X, Tang S, and He Z. Enhanced oral bioavailability of felodipine by novel solid self-microemulsifying tablets. Drug Dev Ind Pharm. 2016;42:506–12.
Bakhle SS, Avari JG. Development and characterization of solid self-emulsifying drug delivery system of cilnidipine. Chem Pharm Bull. 2015;63:408–17.
Boxin OU, Dejian H, Maureen AF, Elizabeth KD. Analysis of antioxidant activities of common vegetables employing oxygen radical absorbance capacity (ORAC) and ferric reducing antioxidant power (FRAP) assays: a comparative study. J Agric Food Chem. 2002;5:223–8.
Date AA, Desai N, Dixit R, Nagarsenker M. Self-nanoemulsifying drug delivery systems: formulation insights, applications and advances. Nanomedicine. 2010;5:1595–616.
Baur JA, Sinclair DA. Therapeutic potential of resveratrol: the in vivo evidence. Nat Rev Drug Disov. 2006;5:493–506.
Chen Y, Zhang H, Yang J, Sun H. Improved antioxidant capacity of optimization of a self-microemulsifying drug delivery system for resveratrol. Molecules. 2015;20:21167–77.
Li W, Shao Y, Hu L. BM6, a new semi-synthetic vinca alkaloid, exhibits its potent in vivo anti-tumor activities via its high binding affinity for tubulin and improved pharmacokinetic profiles. Cancer Biol Ther. 2007;6:787–94.
Liu Z, Liu D, Wang L, Zhang J, Zhang N. Docetaxel-loaded pluronic P123 polymeric micelles: in vitro and in vivo evaluation. Int J Mol Sci. 2011;12:1684–96.
Sun S. Acrylamide derivative and use thereof in manufacture of medicament. 2010; Patent, US2012/0116075A1.
Sun S. Acrylamide derivative and use thereof in manufacture of medicament. 2010; Patent, CN102421754B.
Yeni P. Tipranavir: a protease inhibitor from a new class with distinct antiviral activity. JAIDS. 2003;34:S91–4.
Meng J, Li S, Yao Q, Zhang L, Weng Y, Cai C, et al. In vitro/in vivo evaluation of felodipine micropowders prepared by the wet-milling process combined with different solidification methods. Drug Dev Ind Pharm. 2014;40:929–36.
Karavas E, Ktistis G, Xenakis A, Georgarakis E. Miscibility behavior and formation mechanism of stabilized felodipine-polyvinyl pyrrolidone amorphous solid dispersions. Drug Dev Ind Pharm. 2005;31:473–89.
Tarr BD, Yalkowsky SH. Enhanced intestinal absorption of cyclosporin in rats through the reduction of emulsion droplet size. Pharm Res. 1989;6:40–3.
Constantinides PP. Lipid microemulsions for improving drug dissolution and oral absorption: physical and biopharmaceutical aspects. Pharm Res. 1995;12:1561–72.
Kauss T, Gaubert A, Tabaran L, Tonelli G, Phoeung T, Langlois MH, et al. Development of rectal self-emulsifying suspension of a moisture-labile water-soluble drug. Int J Pharm. 2018;536:283–91.
Author information
Authors and Affiliations
Corresponding author
Additional information
Guest Editor: Sanyog Jain
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Rani, S., Rana, R., Saraogi, G.K. et al. Self-Emulsifying Oral Lipid Drug Delivery Systems: Advances and Challenges. AAPS PharmSciTech 20, 129 (2019). https://doi.org/10.1208/s12249-019-1335-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1208/s12249-019-1335-x