
Abstract
In this study, an optimized nanostructured lipid carriers (NLCs) were developed and investigated for improving the solubility and oral availability of 6-Gingerol (6G), an active and abundant component of ginger with limited applications due to its poor water solubility plus oral biological availability. The NLCs consisted of a solid lipid (glyceryl monostearate), another liquid lipid (decanoyl/octanoyl-glycerides) and mixed surfactants (Tween 80 and Poloxamer 188), and was prepared by high pressure homogenization method. The optimal 6G-NLC formulation was evaluated through physical properties such as appearance, mean particle size, zeta potential, encapsulation efficiency, and in vitro drug release, alongside techniques viz., transmission electron microscopy (TEM), differential scanning calorimetry (DSC), as well as powder X-ray diffraction (XRD). Pharmacokinetics were also evaluated in rats. The 6G-NLCs prepared with optimal formulation exhibited a homogenous spherical shape with mean particle size and zeta potential of 63.59 ± 5.54 nm and − 12.18 ± 1.06 mV. Encapsulation efficiency and drug loading were 76.71 ± 1.11 and 1.17 ± 0.35%, respectively. In vitro release profile of 6G from NLCs was sustained and fitted with Weibull equation. After oral administration of the 6G-NLCs, drug concentrations in serum, MRT, and AUC0-t were significantly higher as compared with the free 6G suspension. All these results indicated that the developed NLC formulation could be effective and promising drug carriers to improve the water solubility of 6G while sustaining the drug release as well as prolonging in vivo acting time of the drug coupled with oral bioavailability enhancement.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Ginger (Zingiber officinale Roscoe, Zingiberaceae) is known as the dried rhizome of Zingiber and is one of the important natural medicinal plants in various countries such as China, India, Mexico, and some other areas [1]. Ginger is extensively used as a food spice around the world from early on. The rhizome of ginger and its extracts have also been widely used as a kind of medicine [2]. Traditional Chinese Medicine uses ginger to treat many diseases including wind cold, cough, rheumatism, indigestion, vomiting, intoxication, infectious diseases, and so on [3,4,5]. The oleoresins extracted from rhizome of ginger involve pungent ingredients known as gingerol, shogaol, and zingerone [6] and have been proved to have all kinds of pharmacological activities [7]. Among the various ingredients in ginger, 6-Gingerol (6G) (Fig. 1) is one of the major pungent and the most abundant components which has been found to exhibit multiple pharmacological effects namely antioxidant, anti-inflammation, antitumor, antibacterial, liver and kidney protection, hypoglycemic as well as hypolipidemic activities, etc. [8,9,10,11,12,13,14,15]. However, studies in healthy rats and mice showed that after oral administration, 6G was rapidly absorbed and eliminated with a short half-life [16]. Previous report by Jiang et al. [17] showed that the 6G absorbed into plasma could be eliminated rapidly with a short half-life, indicated that the oral bioavailability of 6G was very low, and this was possibly because of the liver first-pass effect. Although 6G possesses many pharmacological activities, the difficulty in extraction and purification and the low water solubility plus bioavailability have limited its further clinical applications [17].

The molecular structure of 6-Gingerol
During the past few decades, lipid-based drug delivery systems have become promising nano-scale drug carriers for improving solubility and bioavailability of water-insoluble drugs [18,19]. Among them, nanostructured lipid carriers (NLCs) are novel second-generation lipid nanoparticle drug delivery system for sustaining drug release that is developed from solid lipid nanoparticles (SLNs) [20]. By using mixture of two lipid matrices (solid and liquid) instead of one single solid lipid material of SLNs, the NLCs could significantly overcome the shortcomings of SLNs namely poor stability, low drug encapsulation efficiency, drug leakage, and excessive water retention in the system. Meanwhile, NLCs share the advantages of SLNs, which include but not limited to desirable biocompatibility, sustained drug release, and the possibility of large-scale industrial production [21,22,23,24,25]. Traditional SLNs contain only one single lipid, which would easily form a compact crystal structure. Drugs are mostly dispersed on the surface of the lipid sphere structure and these often lead to low encapsulation efficiency, drug leakage, and burst release [26]. As for NLCs, with mixed solid lipid and another liquid lipid in the formulation prescription, cavity structure with more imperfections in the lipid core could be formed to pack the lipid-soluble drugs, therefore, obtaining a higher drug loading capacity as well as reducing drug leakage during storage [21,27]. Moreover, the combination of liquid and solid lipids, along with relatively small particle size (usually smaller than 200 nm), could effectively improve the absorption of the drug and lead to a passive targeting to certain tissues [21]. NLCs have currently been confirmed to incorporate many bioactive components to improve solubility, chemical stability, and bioavailability [23,26]. Therefore, NLCs are promising drug carriers for improving water solubility and bioavailability of poorly water-soluble drugs such as 6G and also serve as an alternative drug delivery system to bypass the first-pass metabolism, culminating in prolonging circulation time of the drug [28].
Unlike the abundant studies focused on its pharmacokinetics and pharmacological effects, reports related to the preparation of 6G to overcome the poorly water solubility and enhance the oral bioavailability are limited. Previous studies by Zhang et al. [29] isolated and characterized ginger-derived exosome-like nanoparticles and examined their potential use for treating inflammatory bowel diseases. Manatunga and colleagues [30] also synthesized a novel pH sensitive sodium alginate, hydroxyapatite bilayer-coated iron oxide nanoparticle composite, and loaded two anticancer drugs, Curcumin and 6-Gingerol. Although 6G has been previously processed into other formulations such as self-microemulsifying drug delivery system [31] and proliposomes [32], there still exist minuses such as easily drug leakage and complex preparation processes. Therefore, studies based on different drug delivery systems and preparation methods are worth exploring. As novel lipid-based nano-scale drug delivery systems, NLCs possess relatively small and uniform particle size distribution, better storage stability, and preparation techniques that can be adaptive for production on a large scale. In a previous study by Rosli and co-experimenters [33], ginger oil was loaded on to a NLC formulation, and the particle size, polydispersity index, and encapsulation efficiency were measured. Nonetheless, the ability of NLCs to increase the bioavailability of 6G has not been studied further. Therefore, in the present study, we formulated and assessed NLC drug delivery system for oral delivery of 6G. The formulation of 6G-NLCs was optimized, while the physicochemical properties were characterized. Subsequently, the in vitro drug release profiles and in vivo oral bioavailability of 6G-NLCs were evaluated.
MATERIALS AND METHODS
Materials
6-Gingerol (98% purity, standard product) was bought from Aladdin Corporation (Shanghai, China). 6-Gingerol (98.15% purity) was extracted and purified from a Ginger extractive (contains 10% gingerol) by a previous study [32]. Glyceryl monostearate (GMS), curcumin, and Tween 80 were bought from Sinopharm Chemical Reagent Corporation (Shanghai, China). Decanoyl/octanoyl-glycerides (MCT) was obtained from Youchuang Corporation (Shanghai, China). Poloxamer 188 was obtained from Yuanye Chemical Factory (Shanghai, China). Other related chemicals and reagents were all of analytical grade. Healthy male Sprague-Dawley rats were obtained from Experimental Animal Center of Jiangsu University (Zhenjiang, China), and the in vivo experimental procedures all abided by the ethics and regulations of animal experiments approved by the Ethic Committee of Jiangsu University.
HPLC Analysis
Contents of 6G in different samples were measured using a reverse-phase HPLC method. The HPLC system consisted of a LC-20A pump, a SIL-20A autosampler, and a LC-20AD detector (Shimadzu, Japan). Chromatographic separations were performed on a Symmetry C18 column (5 μm, 4.6 × 150 mm; Waters, Ireland), with mobile phase that consisted of methanol and water (65/35, v/v) at 1.0 mL min−1. The detection wavelength was 280 nm and the column temperature was 30°C. The HPLC condition was applied for the in vitro detection of 6G with validation of specificity, accuracy, and sensitivity. The standard curve showed a good linear within the scope of 1–200 μg mL−1, and the linear equation was: Y = 10,571X–2326.6 (R2 = 0.9999). The average recovery and relative standard deviation (RSD) were 99.64 and 3.99%, respectively (n = 3). The limit of detection (LOD) was 0.5 μg mL−1 and the limit of quantity (LOQ) was 0.2 μg mL−1, while the intra-plus inter-day variations of RSD were less than 5%.
Optimization of 6G-NLCs
The preparation process of 6G-NLCs was conducted using high pressure homogenization (HPH) method as described earlier [34] with some slight modifications. In brief, an aqueous phase composed of surfactants (Tween 80 and Poloxamer 188, 1:1, w/w) and double distilled water was mixed in a conical flask and stirred at 75–78°C. Simultaneously, a homogeneous melting lipid phase was obtained by mixing solid lipid (glycerin monostearate, GMS), liquid lipid (decanoyl/octanoyl-glycerides, MCT), and lipophilic drug (6G) alongside stirring in another flask at the same temperature. Subsequently, the hot water phase was slowly added into lipid phase, followed by constant stirring to form an emulsion solution. The final step was to homogenize the formed emulsion by a high-pressure homogenizer (APV-2000, APV, Germany) for 10 cycles at 1000 bar and the resultant NLCs were cooled to room temperature.
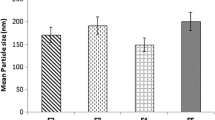
In the present study, orthogonal design was applied to screen the optimal formulation of 6G-NLCs. Based on previous studies [35,36,37], the main parameters that influence the characteristics of NLCs were the ratio of lipids and surfactants and the content of drug. Therefore, the following items were used as investigation factors: percentage of GMS (2.5–7.5%, w/v) (A), percentage of MCT (2.5–7.5%, w/v) (B), percentage of mixed surfactants (half Tween 80 and half Poloxamer 188, 2–6%, w/w) (C), and ratio of drug to lipid (1:10–1:30, w/w) (D). Through pre-experiment design, we chose the mean particle size (PS) of NLCs as the evaluation index due to its greater difference than other indexes such as zeta potential and drug encapsulation efficiency, while relatively smaller PS could efficiently improve the oral absorption of drug after administration [38]. According to the factors and levels listed in Table 1, a L9(34) orthogonal experimental design listed in Table 2 was obtained, and nine experimental conditions were conducted.
Characterization of 6G-NLCs
Particle Size and Zeta Potential
The determination of average particle size (PS), polydispersity index (PDI), as well as zeta potential (ZP) of 6G-NLCs (diluted to 1 mg mL−1) were conducted using a 90 Plus PALS instrument (Brookhaven, USA). Both measurements were conducted at 25°C for three times (n = 3).
Transmission Electron Microscopy
6G-NLCs of 50 μL (2.5 mg mL−1) was diluted to optimal concentration with water and put on a copper grid prior to analysis. After negatively stained with phosphotungstic acid, the copper grid was dried in the air and subjected to transmission electron microscopy (TEM; Tecnai 12, Philips, Holland) for physical appearance observation.
Encapsulation Efficiency and Drug Loading
A HPLC method as mentioned in a previous work [39] was conducted to determine the encapsulation efficiency (EE) of 6G in our study. Briefly, the 6G-NLCs were dissolved in methanol to disrupt its structure and then injected to RP-HPLC to measure the total concentration of 6G in NLCs. Meanwhile, 1 mL of the prepared 6G-NLCs was filtered through a 0.22-μm filter in order to remove unencapsulated 6G. The obtained filtrate was diluted with methanol and then analyzed by RP-HPLC to obtain the concentration of 6G encapsulated in NLCs. The drug loading (DL) was calculated by dividing the content of encapsulated drug to total vehicle (w/w). The equation for the EE (1) and DL (2) was as follows:
where Cencapsulated means the concentration of 6G incorporated in the NLCs, Ctotal means the theoretical concentration of 6G in the NLCs recipe. V represents the volume of formulated 6G-NLCs in one recipe, and Wlipids represents the weight of the vehicle.
X-Ray Diffraction Analysis
The 6G-NLC dispersion was lyophilized with a cryoprotectant of glucose by a FreeZone 6 plus freeze dryer (Labconco, USA) prior to scanning. X-ray diffraction analysis (XRD) of the samples (GMS, glucose (cryoprotectant), freeze-dried blank-NLCs, mixture of 6G and free-dried blank-NLCs, and 6G-NLCs powder) was performed by a D8 Advance X-ray diffractometer (Bruker, Germany) equipped with a Cu-Kα radiation. In brief, the samples were scanned over a range of 5° to 80° and the scanning rate was 5° min−1. The diffraction curves were recorded.
Differential Scanning Calorimetry
Differential scanning calorimetry (DSC) analysis was conducted by a STA 449 C thermal analysis (Netzsch, Germany). Samples (6G, solid lipid (GMS), blank-NLCs, and 6G-NLCs) were weighted accurately then placed in aluminum pans and sealed. DSC thermograms of different samples were measured at 25–300°C in a nitrogen environment, and the heating speed was 10°C min−1.
In Vitro Drug Release
In vitro drug release studies were conducted using dialysis method to simulate the in vivo biological conditions in different release media, including HCl (pH 1.2), double distilled water (pH 7.0), and PBS (pH 7.4). Briefly, 6G solution (5 mg mL−1, 1 mL) and 6G-NLCs (the same 6G content) were put into dialysis bags separately, then placed in conical flask, and added with 100-mL different release medium, respectively. The test was performed on a 37 ± 1°C digital water-bath shaker with a speed of 100 rpm. An aliquot of 1 mL was sampled at intervals of 0.25, 0.5, 1, 2, 4, 6, 8, 10, 12, 24, 36, 48 h and promptly replaced with 1 mL of fresh 37 ± 1°C medium to maintain constant volume. The samples were centrifuged (10,000 rpm for 10 min) and then injected into RP-HPLC to determine the amount of 6G released.
Pharmacokinetic Studies in Rats
Eight male Sprague-Dawley rats (200 ± 20 g) were fed individually under normal laboratory environments and divided randomly into two groups before experiment. The two groups were separately administered 6G-NLCs alongside 6G suspension (6G dispersed in 4% sodium carboxymethylcellulose (CMC-Na) solution) through oral gavage at a dose of 200 mg kg−1. At 0.083, 0.25, 0.5, 0.75, 1, 1.5, 2, 3, 4, 6, 8, 10, and 12 h after administration, 0.5 mL of blood samples was drawn from the suborbital vein using capillaries, then centrifuged at 3700 rpm for 10 min to separate the serums. To extract the drug in the serum, 50 μL of Curcumin (10 μg mL−1; internal standard) and 500 μL of ethyl acetate were added to 200 μL of serum and vortexed, then centrifuged at 3000 rpm for 5 min. The ethyl acetate layer was carefully transferred to a new tube and volatilized under a stream of nitrogen at 37°C. The residue was re-dissolved with 400 μL of 65% methanol-water (v/v) and centrifuged at 10,000 rpm for 10 min after vortexing. The supernatant was injected into RP-HPLC for determination of 6G in serum. The HPLC apparatus included a LC-20A High Performance Liquid Chromatography (Shimadzu, Japan) with a LC-20A pump (Shimadzu, Japan) and an UV detector (Shimadzu, Japan). A column of Symmetric C18 (5 μm, 4.6 mm × 150 mm; Waters, Ireland) was used for chromatographic separation at a detection wavelength of 230 nm. The mobile phase contained 65% methanol and 35% water with a flow rate of 1 mL min−1, and temperature was at 37°C. Meanwhile, the main pharmacokinetic parameters were determined. The peak concentration of serum drug (Cmax) and time to achieve the peak concentration (Tmax) could be directly derived from the serum drug concentration-time curve, while the area under the concentration-time curve (AUC) as well as mean residence time (MRT) and other related indices were calculated by a Drug and Statistics (DAS) 2.0 software.
Statistical Analysis
In the present paper, data were expressed as means ± standard deviation (SD). Statistical significance was assessed using Student’s t test by SPSS 22.0 software and p < 0.05 was considered as significant.
RESULTS AND DISCUSSION
Optimized Formulation
In this research, the formulation of 6G-NLCs was optimized through an orthogonal design and the calculation was conducted using the software SPSS 22.0. The orthogonal results are shown in Table 2, while the variance analysis result is indicated in Table 3. Intuitively, the ranged analysis in Table 2, the values represented the influence order of the factors. Therefore, according to the range values, influence order of four factors was A > B > C > D. As shown in Table 3, through the variance analysis, only factor A had an influence (p < 0.05) on the mean PS of the prepared NLCs. Meanwhile, the smallest mean PS was obtained with factor A in level 1 (Table 2); thus, the mean PS ranges from 73.21 to 92.43 nm. However, the other three factors had no statistical influence on the mean PS of the prepared NLCs (p > 0.05). According to the results of visual and variance analyses via the orthogonal experimental design, the optimal formulation of 6G-NLCs was obtained as follows: GMS 2.5% (w/w), MCT 2.5% (w/w), mixed Tween 80 and Poloxamer 188 6% (w/w), with the drug to lipid ratio being 1:10 (w/w).
Morphology, Particle Size, and Zeta Potential of 6G-NLCs
The 6G-NLCs prepared showed a transparent appearance with slightly light blue opalescence. The average PS of 6G-NLCs was 63.59 ± 5.54 nm (Fig. 2, right) with PDI of 0.243 ± 0.039, and the ZP was − 12.18 ± 1.06 mV. The PS showed a small value and a normal distribution with a relatively low PDI, which could affect the uptake by reticuloendothelial system, and thereby increase the oral absorption alongside bioavailability of the drug, leading to passive accumulation in tissues [40]. The value of ZP was often used to evaluate the physical stability of nano-formulations during storage [41]. In this study, the ZP value of 6G-NLCs was − 12.18 mV, which could be considered as a stable system. The TEM imaging (Fig. 2, left) showed homogeneous spherical shapes with smooth surface, as well as no alignment of particles. The mean size of particles through the TEM figure was in the range of 50–70 nm, which was consistent with the mean PS result. These results indicated that the optimized formulation was suitable for preparing 6G-NLCs, as the prepared formulation was homogeneous and stable.

TEM image (left) and mean particle size distribution (right) of optimized 6G-NLCs
Encapsulation Efficiency and Drug Loading
The NLCs often possess higher drug EE as compared with SLNs due to the addition of liquid lipid which could change the inner structure of particles [42]. In this study, the samples were measured by RP-HPLC method as mentioned before. The average EE and DL of optimized 6G-NLCs were 76.71 ± 1.11 and 1.17 ± 0.35%, respectively. The possible reason for high EE of 6G in NLCs could be ascribed to the hydrophobic character of 6G, which led to high solubility of 6G in the lipids and thus could evenly disperse into the matrix of particles [43].
X-Ray Diffraction Analysis
Phase identification of a crystalline material can be obtained through rapid analytical technique, XRD which could subsequently provide information on unit cell dimensions. In order to assess the crystal form of solid lipid and the drug after formulation into NLCs, the XRD technique was employed. As displayed in Fig. 3, the diffraction peaks of GMS appeared around the 2θ values of 5.38°, 19.49°, 20.48°, and 23.38°. However, the characteristic peaks of GMS were vanished or reduced in the curve of NLCs, which indicated a less ordered crystalline state of lipid in NLCs. In addition, no more peaks appeared in the NLCs curves other than the peaks that appeared in GMS and glucose curves, which suggested that the drug was successfully incorporated in the formulation. The diffraction curve of mixture of 6G and freeze-dried blank-NLCs showed more peaks as compared with the freeze-dried blank NLC curve, which could be attributed to the diffraction peaks of 6G. However, most of these peaks disappeared in the curve of freeze-dried 6G-NLCs, suggesting that the drug was incorporated into the NLCs. These results demonstrated that the mixture of solid and liquid lipids led to a disordered crystal structure in NLCs. This result could be illustrated further by DSC analysis.

X-ray diffraction (XRD) patterns of GMS (a), glucose (b), blank-NLCs (c), physical mixture of 6G and blank-NLCs (d), and 6G-NLCs (e)
Differential Scanning Calorimetry
In this study, DSC analysis was conducted to analyze the melting and crystallizing process of drug and lipid in the NLCs. Figure 4 shows the DSC curves of GMS, 6G, physical mixture of 6G and GMS, and 6G-NLCs under setting conditions. The curve of 6G exhibited a melting peak at about 242.74°C, while for GMS, the melting peak appeared at 62.98°C. The individual peaks of 6G and GMS all appeared in the thermogram of physical mixture with nearly same values. However, in the thermogram of 6G-NLCs, the melting peak of 6G around 240°C disappeared, and the peak of GMS shifted to a low temperature. The change of peaks between raw materials and NLCs suggested that the drug was uniformly dispersed in the NLCs in an amorphous phase.

Differential scanning calorimetric (DSC) thermograms of 6G (a), GMS (b), physical mixture of 6G and GMS (c), and 6G-NLCs (d)
In Vitro Release
The dialysis method was the most common method for measuring the in vitro release of nano-formulations. In the present study, the release profiles of free 6G from 6G-NLCs in different media at 37 ± 1°C are shown in Fig. 5. The release curves of free 6G were only displayed within 12 h because there was no more free drug release after that, while the unload of 6G from NLCs was sustained and continued till 48 h. The release of 6G from 6G-NLCs in different media were fitted to Weibull kinetics model and the fitted equations were as follows: lnln(1/(1-Q/100)) = 0.6313lnt-1.73543, r = 0.9652 (water), lnln(1/(1-Q)) = 0.586lnt-1.931, r = 0.9517 (pH 1.2), lnln(1/(1-Q/100)) = 0.7064lnt-1.96717, r = 0.9929 (pH 7.4). The accumulated release of free 6G in different media reached the maximum at the first 12 h, which was about 60–80%. In contrast, the release of free 6G from 6G-NLCs showed a controlled and continuous release, the release ratio of 6G-NLCs was observed in water, pH 1.2 HCl and pH 7.4 PBS with about 65, 55, and 60% at first 12 h, followed by a relatively slower release over 48 h. The accumulated drug release rates after 48 h were about 75.35, 60.21, and 83.69% in water, pH 1.2 HCl, and pH 7.4 PBS, respectively. Meanwhile, the cumulative release of 6G in pH 7.4 PBS was higher than the other two media. This might be ascribed to the higher solubility of 6G in alkaline condition due to the phenolic hydroxyl groups in the molecular structure [10]. The solubility of 6G in three different release medium was measured in a pre-experiment and the solubility values were 252.31 μg mL−1 (water), 226.24 μg mL−1 (pH 1.2 HCl), and 258.87 μg mL−1 (pH 7.4 PBS), respectively. The maximum solubility was in pH 7.4 PBS, which was in agreement with the accumulate release rate. The differences between the release profile of pH 1.2 HCl and pH 7.4 PBS might suggest that the successful incorporation of 6G in NLCs could protect the drug from the strong acid environment of the gastric, which seems to suggest that the 6G-NLCs reached the small intestinal and subsequently undergoes intestinal absorption process. There was no burst release pattern observed in any of the medium (as shown in Fig. 5), which indicated that the formulation prepared was homogenous and the drug was successfully loaded into the inner of NLCs. In addition, the sustained release might be due to the loading of 6G into the cavities of two different mixed lipids in the lipid core of NLCs, as this was considered to provide more space for drug incorporation in NLCs [36]. Thus, the sustained in vitro release property might lead to an effective and continuous treatment in vivo and enhance the bioavailability.

In vitro release profiles of free 6G and 6G-NLCs in pH 1.2 HCl solution (a), pH 7.4 PBS (b), and water (c) at 37 ± 1°C (mean ± SD, n = 3)
In Vivo Pharmacokinetics Study
The chromatographic conditions mentioned before was confirmed as a feasible tool for accurate quantitative analysis of 6G in serum. The endogenous substances in serum did not interfere with the determination of 6G and IS under the condition. The regression equation in the range of 0.125–25 μg mL−1 was as follows: A = 0.3346C + 0.0521, with r2 = 0.9995. The serum concentration-time curves of free 6G suspension and 6G-NLCs in rats following oral gavage are shown in Fig. 6, and the relevant pharmacokinetic parameters are listed in Table 4. As shown in Fig. 6, the serum concentrations of 6G were higher in 6G-NLC group as compared with free 6G suspension group at every time point. The Cmax value of 6G in NLCs (7.467 ± 1.079 μg mL−1) was significantly higher (p < 0.05) than 6G suspension (1.869 ± 0.176 μg mL−1). Moreover, T1/2 and MRT values were also delayed in the NLC group from 1.971 and 1.510 h to 5.156 and 4.478 h. Twelve hours after oral administration of 6G-NLCs, the 6G serum concentration was still more than 0.5 μg mL−1, whereas the drug concentration was under the limit of detection after 4 h in rats treated with 6G suspension. These results indicated that the formulated NLCs had sustained release characteristics, which correlated well with the in vitro release results. The AUC(0 → t) of 6G after administration of 6G suspension was 2.742 ± 0.318 h μg mL−1, while that of 6G-NLCs was 5.86 times greater (16.059 ± 1.519 h μg mL−1). The bioavailability of 6G was low probably due to its poor water solubility and the excessive elimination in the liver [17]. As for NLCs, the encapsulation of drug into the lipid core could largely avoid the first-pass effect [44], and the use of surfactants in the formulations might have increased the epithelial permeability and resulted in an improvement of absorption [45]. On the other hand, the small PS (less than 100 nm) could also increase the surface area of the formulation particles, which coupled with the high dispersity could result in sufficient and steady intestinal absorption after administration [46]. There was no difference between the Tmax value of 6G and 6G-NLCs group as other parameters were significantly increased. This might be due to the readily absorption and widely distribution of 6G influenced by the increased permeability and small particle size of the NLCs. The smaller particle size could improve drug permeation across the intestinal membrane due to enhanced drug solubility as well as enlarged surface area of drug contacting [47]. These results suggested that the formulation of NLCs could significantly improve the oral absorption of 6G. The prepared 6G-NLCs showed enormous potential for increasing the water solubility and oral bioavailability of the drug.

In vivo serum concentration-time curves of 6G after oral administration of 6G suspension and 6G-NLC at a dosage of 200 mg kg−1 to rats (mean ± SD, n = 4)
CONCLUSIONS
In the present study, 6-Gingerol-loaded nanostructured lipid carriers (6G-NLCs) were successfully developed through high pressure homogenization method and optimized by a four-factor three-level orthogonal design. The optimized NLCs had a homogeneous spherical shape with small mean PS and negative ZP. The XRD and DSC results displayed amorphous or less ordered forms of 6G and lipids in NLCs. In vitro release study revealed that 6G-NLCs possessed a sustained release profile, and in vivo pharmacokinetic study showed that the AUC0-t of 6G-NLCs formulation was 5.86-fold as compared to 6G suspensions after oral administration. All the results indicated that the optimized NLC formulation could availably improve the water solubility and enhance the oral bioavailability of 6G. Therefore, this study provides useful references for further research and applications of 6G, as well as numerous other water-insoluble pharmacological compounds.
References
Ghosh AK, Banerjee S, Mullick HI, Banerjee J. Zingiber officinale: a natural gold. Int J Pharma Bio Sci. 2011;2:283–94.
Zick SM, Ruffin MT, Djuric Z, Normolle D, Brenner DE. Quantitation of 6-, 8- and 10-Gingerols and 6-Shogaol in human plasma by high-performance liquid chromatography with electrochemical detection. Int J Biomed Sci. 2010;6:233–40.
Kim JS, Lee SI, Park HW, Yang JH, Shin TY, Kim YC, et al. Cytotoxic components from the dried rhizomes of Zingiber officinale roscoe. Arch Pharm Res. 2008;31:415–8. https://doi.org/10.1007/s12272-001-1172-y.
An KJ, Zhao DD, Wang ZF, Wu JJ, Xu YJ, Xiao GS. Comparison of different drying methods on Chinese ginger (Zingiber officinale roscoe): changes in volatiles, chemical profile, antioxidant properties, and microstructure. Food Chem. 2016;197:1292–300. https://doi.org/10.1016/j.foodchem.2015.11.033.
Wu H, Horng C, Tsai S, Lee Y, Hsu S, Tsai Y, et al. Relaxant and vasoprotective effects of ginger extracts on porcine coronary arteries. Int J of Mol Med. 2018;41(4):2420–8. https://doi.org/10.3892/ijmm.2018.3380.
Semwal RB, Semwal DK, Combrinck S, Viljoen AM. Gingerols and Shogaols: important nutraceutical principles from ginger. Phytochemistry. 2015;117:554–68. https://doi.org/10.1016/j.phytochem.2015.07.012.
Feng X, Kong W, Wei J, Ou-Yang Z, Yang M. HPLC fingerprint analysis combined with chemometrics for pattern recognition of ginger. Pharm Biol. 2014;52:362–7. https://doi.org/10.3109/13880209.2013.837493.
Nile S, Park S. Chromatographic analysis, antioxidant, anti-inflammatory, and xanthine oxidase inhibitory activities of ginger extracts and its reference compounds. Ind Crop Prod. 2015;70:238–44. https://doi.org/10.1016/j.indcrop.2015.03.033.
Yang MQ, Ye LL, Liu XL, Qi XM, Lv JD, Wang G, et al. Gingerol activates noxious cold ion channel TRPA1 in gastrointestinal tract. Chin J Nat Med. 2016;14:434–40. https://doi.org/10.1016/s1875-5364(16)30040-1.
Singh AB, Akanksha, Singh N, Maurya R, Srivastava AK. Anti-hyperglycaemic, lipid lowering and anti-oxidant properties of [6]-gingerol in db/db mice. Int J Med Med Sci. 2009;1:536–44.
Tzeng TF, Chang CJ, Liu IM. 6-Gingerol inhibits rosiglitazone-induced adipogenesis in 3T3-L1 adipocytes. Phytother Res. 2014;28:187–92. https://doi.org/10.1002/ptr.4976.
Young HY, Luo YL, Cheng HY, Hsieh WC, Liao JC, Peng WH. Analgesic and anti-inflammatory activities of [6]-gingerol. J Ethnopharmacol. 2005;96:207–10. https://doi.org/10.1016/j.jep.2004.09.009.
Almada da Silva J, Becceneri AB, Sanches Mutti H, Martin A, Silva M, Fernandes J, et al. Purification and differential biological effects of ginger-derived substances on normal and tumor cell lines. J Chromatogr B Analyt Technol Biomed Life Sci. 2012;903:157–62. https://doi.org/10.1016/j.jchromb.2012.07.013.
Rodrigues F, Prata M, Oliveira I, Alves N, Freitas R, Monteiro H, et al. Gingerol fraction from Zingiber officinale protects against gentamicin-induced nephrotoxicity. Antimicrob Agents Chemother. 2014;58:1872–8. https://doi.org/10.1128/aac.02431-13.
Sabina E, Pragasam S, Kumar S, Rasool M. 6-Gingerol an active ingredient of ginger, protects acetaminophen-induced hepatotoxicity in mice. J Chin Integr Med. 2011;9:1264–9. https://doi.org/10.3736/jcim20111116.
Bhattarai S, Tran V, Duke C. Stability of [6]-gingerol and [6]-shogaol in simulated gastric and intestinal fluids. J Pharm Biomed Anal. 2007;45:648–53. https://doi.org/10.1016/j.jpba.2007.07.006.
Jiang SZ, Wang NS, Mi SQ. Plasma pharmacokinetics and tissue distribution of [6]-gingerol in rats. Biopharm Drug Dispos. 2008;29:529–37. https://doi.org/10.1002/bdd.638.
Beloqui A, Solinís MÁ, Rodríguez-Gascón A, Rodríguez-Gascón A, Almeida AJ, Préat V. Nanostructured lipid carriers: promising drug delivery systems for future clinics. Nanomedicine. 2015;12:143–61. https://doi.org/10.1016/j.nano.2015.09.004.
Shi F, Wei Z, Zhao Y, Xu X. Nanostructured lipid carriers loaded with Baicalin: an efficient carrier for enhanced antidiabetic effects. Pharmacogn Mag. 2016;12(47):198–202. https://doi.org/10.4103/0973-1296.186347.
Souto E, Almeida A, Müller R. Lipid nanoparticles (SLN®, NLC®) for cutaneous drug delivery: structure, protection and skin effects. J Biomed Nanotechnol. 2007;3:317–31. https://doi.org/10.1166/jbn.2007.049.
Fang CL, Al-Suwayeh SA, Fang JY. Nanostructured lipid carriers (NLCs) for drug delivery and targeting. Recent Pat Nanotechnol. 2013;7:41–55. https://doi.org/10.2174/187221013804484827.
Aditya N, Macedo A, Doktorovova S, Souto E, Kim S, Chang P, et al. Development and evaluation of lipid nanocarriers for quercetin delivery: a comparative study of solid lipid nanoparticles (SLN), nanostructured lipid carriers (NLC), and lipid nanoemulsions (LNE). LWT-Food Sci Technol. 2014;59:115–21. https://doi.org/10.1016/j.lwt.2014.04.058.
Liu Y, Wang L, Zhao Y, He M, Zhang X, Niu M. Nanostructured lipid carriers versus microemulsions for delivery of the poorly water-soluble drug luteolin. Int J Pharm. 2014;476:169–77. https://doi.org/10.1016/j.ijpharm.2014.09.052.
Yuan L, Liu C, Chen Y, Zhang Z, Zhou L, Qu D. Antitumor activity of tripterine via cell-penetrating peptide-coated nanostructured lipid carriers in a prostate cancer model. Int J Nanomedicine. 2013;8:4339. https://doi.org/10.2147/ijn.s51621.
Wang F, Chen L, Zhang D, Jiang S, Shi K, Huang Y, et al. Methazolamide-loaded solid lipid nanoparticles modified with low-molecular weight chitosan for the treatment of glaucoma: vitro and vivo study. J Drug Target. 2014;22(9):849–58. https://doi.org/10.3109/1061186x.2014.939983.
Weiss J, Decker EA, Mcclements DJ, Kristbergsson K, Helgason T, Awad T. Solid lipid nanoparticles as delivery systems for bioactive food components. Food Biophys. 2008;3:146–54. https://doi.org/10.1007/s11483-008-9065-8.
Karn-orachai K, Smith SM, Phunpee S, Treethong A, Puttipipatkhachorn S, Pratontep S, et al. The effect of surfactant composition on the chemical and structural properties of nanostructured lipid carriers. J Microencapsul. 2014;31:609–18. https://doi.org/10.3109/02652048.2014.911374.
Ferreira M, Chaves LL, Lima SA, Reis S. Optimization of nanostructured lipid carriers loaded with methotrexate: a tool for inflammatory and cancer therapy. Int J Pharm. 2015;492:65–72. https://doi.org/10.1016/j.ijpharm.2015.07.013.
Zhang M, Viennois E, Prasad M, Zhang Y, Wang L, Zhang Z, et al. Edible ginger-derived nanoparticles: a novel therapeutic approach for the prevention and treatment of inflammatory bowel disease and colitis-associated cancer. Biomaterials. 2016;101:321–40. https://doi.org/10.1016/j.biomaterials.2016.06.018.
Manatunga D, Silva R, Silva K, Silva N, Bhandari S, Yap Y, et al. pH responsive controlled release of anti-cancer hydrophobic drugs from sodium alginate and hydroxyapatite bi-coated iron oxide nanoparticles. Eur J Pharm Biopharm. 2017;117:29–38. https://doi.org/10.1016/j.ejpb.2017.03.014.
Xu Y, Wang QL, Feng YS, Firempong CK, Zhu Y, Omari-Siaw E, et al. Enhanced oral bioavailability of [6]-Gingerol-SMEDDS: preparation, in vitro and in vivo evaluation. J Funct Foods. 2016;27:703–10. https://doi.org/10.1016/j.jff.2016.10.007.
Wang QL, Wei QY, Yang QX, Cao X, Li Q, Shi F, et al. A novel formulation of [6]-gingerol: Proliposomes with enhanced oral bioavailability and antitumor effect. Int J Pharm. 2018;535:308–15. https://doi.org/10.1016/j.ijpharm.2017.11.006.
Rosli NA, Hasham R, Aziz AA, Noor NM, & Aziz RA Formulation and characterization of [6]-Gingerol loaded nanostructured lipid carrier (NLC). International Science Postgraduate Conference 2014.
Shi F, Zhao YY, Firempong CK, Xu XM. Preparation, characterization and pharmacokinetic studies of linalool-loaded nanostructured lipid carriers. Pharm Biol. 2016;54:2320–8. https://doi.org/10.3109/13880209.2016.1155630.
Zhuang CY, Li N, Wang M, Zhang XN, Pan WS, Peng JJ, et al. Preparation and characterization of vinpocetine loaded nanostructured lipid carriers (NLC) for improved oral bioavailability. Int J Pharm. 2010;394:179–85. https://doi.org/10.1016/j.ijpharm.2010.05.005.
Liu D, Liu Z, Wang L, Zhang C, Zhang N. Nanostructured lipid carriers as novel carrier for parenteral delivery of docetaxel. Colloids Surf B Biointerfaces. 2011;85:262–9. https://doi.org/10.1016/j.colsurfb.2011.02.038.
Yang Y, Corona A, Schubert B, Reeder R, Henson M. The effect of oil type on the aggregation stability of nanostructured lipid carriers. J Colloid Interface Sci. 2014;418:261–72. https://doi.org/10.1016/j.jcis.2013.12.024.
Chu C, Tong S, Xu Y, Wang L, Fu M, Ge Y, et al. Proliposomes for oral delivery of dehydrosilymarin: preparation and evaluation in vitro and in vivo. Acta Pharmacol Sin. 2011;32:973–80. https://doi.org/10.1038/aps.2011.25.
Wang Y, Wang S, Firempong CK, Zhang H, Wang M, Zhang Y, et al. Enhanced solubility and bioavailability of naringenin via liposomal nanoformulation: preparation and in vitro and in vivo evaluations. AAPS PharmSciTech. 2016;18:586–94. https://doi.org/10.1208/s12249-016-0537-8.
Li H, Chen M, Su Z, Sun MJ, Ping Q. Size-exclusive effect of nanostructured lipid carriers on oral drug delivery. Int J Pharm. 2016;511:524–37. https://doi.org/10.1016/j.ijpharm.2016.07.049.
Jia L, Shen J, Zhang D, Duan C, Liu G, Zheng D, et al. In vitro and in vivo evaluation of oridonin-loaded long circulating nanostructured lipid carriers. Int J Biol Macromol. 2012;50:523–9. https://doi.org/10.1016/j.ijbiomac.2012.01.024.
Wang W, Chen L, Huang X, Shao A. Preparation and characterization of minoxidil loaded nanostructured lipid carriers. AAPS PharmSciTech. 2017;18(2):509–16. https://doi.org/10.1208/s12249-016-0519-x.
Ni S, Sun R, Zhao G, Xia Q. Quercetin loaded nanostructured lipid carrier for food fortification: preparation, characterization and in vitro study. J Food Process Eng. 2014;38:93–106. https://doi.org/10.1111/jfpe.12130.
Tran T, Ramasamy T, Truong D, Choi H, Yong C, Kim J. Preparation and characterization of Fenofibrate-loaded nanostructured lipid carriers for oral bioavailability enhancement. AAPS PharmSciTech. 2014;15(6):1509–15. https://doi.org/10.1208/s12249-014-0175-y.
Zhu Y, Wang M, Zhang J, Peng W, Firempong CK, Deng W, et al. Improved oral bioavailability of capsaicin via liposomal nanoformulation: preparation, in vitro drug release and pharmacokinetics in rats. Arch Pharm Res. 2015;38:512–21. https://doi.org/10.1007/s12272-014-0481-7.
Feng Y, Sun C, Yuan Y, Zhu Y, Wan J, Firempong CK, et al. Enhanced oral bioavailability and in vivo antioxidant activity of chlorogenic acid via liposomal formulation. Int J Pharm. 2016;501(1–2):342–9. https://doi.org/10.1016/j.ijpharm.2016.01.081.
Luan J, Zheng F, Yang X, Yu A, Zhai G. Nanostructured lipid carriers for oral delivery of baicalin: In vitro and in vivo evaluation. Colloid Surface A. 2015;466:154–9. https://doi.org/10.1016/j.colsurfa.2014.11.015.
Acknowledgements
The authors also thank the University Ethics Committee for the kind guidance in the animal experiments.
Funding
This work was supported by National Natural Science Foundation of China (81473172, 81503025, 81720108030 and 81773695), National “Twelfth Five-Year” Plan for Science & Technology Support (Grant 2013BAD16B07-1), Special Funds for 333 and 331 projects (BRA2013198), Industry-University-Research Institution Cooperation (JHB2012-37, GY2013055) in Jiangsu Province and Zhenjiang City, Program for Scientific Research Innovation Team in Colleges and Universities of Jiangsu Province (SJK-2015-4), and a project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Healthy male Sprague-Dawley rats were obtained from Experimental Animal Center of Jiangsu University (Zhenjiang, China), and the in vivo experimental procedures all abided by the ethics and regulations of animal experiments approved by the Ethic Committee of Jiangsu University.
Conflict of Interest
The authors declare that they have no conflicts of interest.
Additional information
Guest Editor: Sanyog Jain
Rights and permissions
About this article

Cite this article
Wei, Q., Yang, Q., Wang, Q. et al. Formulation, Characterization, and Pharmacokinetic Studies of 6-Gingerol-Loaded Nanostructured Lipid Carriers. AAPS PharmSciTech 19, 3661–3669 (2018). https://doi.org/10.1208/s12249-018-1165-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1208/s12249-018-1165-2