
Abstract
Macrophages, as one of the most abundant tumor-infiltrating cells, play an important role in tumor development and metastasis. The frequency and polarization of tumor-associated macrophages (TAMs) correlate with disease progression, tumor metastasis, and resistance to various treatments. Pro-inflammatory M1 macrophages hold the potential to engulf tumor cells. In contrast, anti-inflammatory M2 macrophages, which are predominantly present in tumors, potentiate tumor progression and immune escape. Targeting macrophages to modulate the tumor immune microenvironment can ameliorate the tumor-associated immunosuppression and elicit an anti-tumor immune response. Strategies to repolarize TAMs, deplete TAMs, and block inhibitory signaling hold great potential in tumor therapy. Besides, biomimetic carriers based on macrophages have been extensively explored to prolong circulation, enhance tumor-targeted delivery, and reduce the immunogenicity of therapeutics to augment therapeutic efficacy. Moreover, the genetic engineering of macrophages with chimeric antigen receptor (CAR) allows them to recognize tumor antigens and perform tumor cell-specific phagocytosis. These strategies will expand the toolkit for treating tumors, especially for solid tumors, drug-resistant tumors, and metastatic tumors. Herein, we introduce the role of macrophages in tumor progression, summarize the recent advances in macrophage-centered anticancer therapy, and discuss their challenges as well as future applications.

Graphical abstract
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
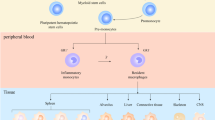
First discovered in the year of 1882, macrophages were initially described as professional phagocytes and key mediators in inflammation and innate immunity (1). They play fundamental roles in the mononuclear phagocytic system (MPS) and are pivotal orchestrators in inflammation, homeostasis, and wound healing (2). It has long been recognized that the macrophage lineage is highly heterogeneous, which is related to the specialization of macrophages in different microenvironments and locations. Multiple functional heterogeneities of macrophages have been proposed. Gordon et al. found that interleukin (IL)-4 induced alternative macrophage activation that differed from classical activation triggered by bacterial-product or pathogen-associated molecular patterns (3, 4). Soon after, Mills et al. showed that macrophages could be distinguished based on arginine catabolism, indicating that classically activated (M1) macrophages show inflammatory functions whereas alternatively activated (M2) macrophages exhibit anti-inflammatory functions (5, 6). Since then, the M1/M2 concept has been widely adopted to explain macrophage heterogeneity.
Massive infiltration of monocytes and macrophages is a common denominator of tumors. Macrophages, constituting approximately 50% of tumor mass, are the most abundant immune cells in the tumor microenvironment (TME) of many tumors (7, 8). Tissue-resident macrophages and circulating monocytes are two major sources of TAMs (9). Tumors secrete various chemoattractants to recruit these precursor cells, resulting in the vast infiltration of macrophages in the TME. Meanwhile, cytokines in the TME reprogram tissue-resident macrophages and monocyte-derived macrophages into the pro-tumor phenotype. As a class of phagocytes and antigen-presenting cells, macrophages hold the potential to directly engulf tumor cells, as well as to present tumor antigens to T cells and thus bridge innate and adaptive immunity (10). However, in the context of tumors, macrophages skew toward the M2 phenotype that exhibits immunosuppressive activities, overriding the anti-tumor function. Numerous studies have evidenced that macrophages, rather than eliminating tumor cells, adopt the pro-tumor M2 phenotype that promotes tumor progression at the primary and metastatic tumor sites (11). In a variety of types of tumors, the tumor-infiltrating macrophages maintain immunosuppression, foster tumor progression, and promote tumor recurrence. TAMs modify the tumor microenvironment and play fundamental roles in every stage of tumor progression, suppressing immune surveillance, promoting tumor angiogenesis, supporting tumor cell survival, and preparing the environment for metastatic seeding (12).
Due to their essential roles in tumor progression, TAMs have recently been in the limelight of tumor research. Various strategies have been proposed, with the aim of depleting or reprogramming macrophages to reactivate anti-tumor immunity and diminish tumor burden. Here, we summarize the current understanding of TAMs and introduce the recent progress in macrophage-centered therapies (Fig. 1). A concise overview of the functions and roles of TAMs in tumor progression is provided. We then review the emerging therapeutic strategies targeting macrophages and discuss their challenges as well as prospects.

Schematic illustration of strategies that target macrophages for tumor therapy
The Role of Macrophages in Tumor Progression
Although macrophages are regarded as professional phagocytes that hold the potential to engulf abnormal, senescent, and apoptotic cells, they have been found to promote the growth and spread of tumors. TAMs promote tumor growth and metastasis through both the direct impact on the intrinsic properties of tumor cells and the indirect impact on the TME. TAMs can generate various growth factors, such as the epidermal growth factor (EGF), which stimulates the proliferation of many types of tumor cells (13, 14). Macrophages have been reported to accelerate tumor proliferation by activating signal transducer and activator of transcription 3 (STAT3) in several cancer types (15, 16). Constitutive activation of STAT3 in tumors is a key event that contributes to tumor cell survival, metastasis, and therapeutic resistance. Activation of STAT3 and NF-kB signals contributes to the self-renewal and tumorigenic activities of cancer stem-like cells (CSCs) (17). In addition, TAM-induced STAT3 activation has been reported to mediate the resistance of tumor cells against chemotherapy or radiotherapy (18, 19). TAMs induce the genetic instability of tumor cells by producing nitric oxide and reactive oxygen intermediates, which impedes the therapeutic outcome of targeted therapies (20, 21).
Macrophage heterogeneity is often explained utilizing an M1/M2 concept. Although this dichotomy of M1/M2 macrophage heterogeneity has been argued as oversimplified, it is widely adopted by most of the current research and can reflect some shared features of macrophage subgroups (9). In established tumors, TAMs are often present in an M2-like phenotype that possesses anti-inflammatory functions. They shape the tumor microenvironment by secreting various immunosuppressive cytokines and regulating cytotoxic T lymphocyte (CTL) activity. TAMs can deplete metabolites that are essential for T cell proliferation and activities. Arginase-1-expressing TAMs in murine models have been reported to metabolize L-arginine, which is critical for T cell fitness and activity (22, 23). In addition, TAMs can induce the inhibition of T cells by upregulating inhibitory signaling. Immunosuppressive TAMs also overexpress immune-checkpoint molecules, such as PD-L1, which contributes to T cell exhaustion (24, 25). Besides, TAMs produce CCL22, a ligand for CCR4, to recruit regulatory T cells (Tregs), inhibiting the antitumor activities of CTLs (26, 27).
Due to the multichannel inhibitory functions of TAMs on T cells, TAMs greatly impact the outcome of T cell-based immunotherapy (28). Studies have shown that macrophages play a key role in mediating resistance to immune checkpoint blockade therapy in triple-negative breast cancer and clear cell renal cell carcinoma (29, 30). In addition, monocyte-derived macrophages in liver metastases could establish an immune desert and diminish the responsiveness to systemic immunotherapy (31). Radiotherapy that blunted the macrophage liberated the T cell activities and improved the outcome of anti-PD-L1 therapy. Besides, murine or human Tim-4+ macrophages could sequester CTLs, and the blockade of Tim-4 boosted the efficacy of anti-PD-1 therapy in mouse peritoneal carcinomatosis (32). These studies validated that targeting macrophages to abolish their inhibitory functions on T cells can improve the outcome of immunotherapies.
TAMs can promote tumor invasiveness and metastasis by expressing enzymes that dissolve the extracellular matrix, upregulating cytokines to enhance tumor cell stemness, and releasing exosomes that transfer miRNAs into cancer cells (33,34,35). Macrophages secrete molecules such as matrix metalloproteinases 9 (MMP 9), TGF-β and growth factors that promote epithelial-mesenchymal transition and the invasiveness of tumor cells. TAMs abundantly produce signaling C-C motif ligand 18 (CCL18) that induces the migration of tumor cells (36). TAMs also prepare for the arrival and seeding of disseminated tumor cells. They play a critical role in tumor cell survival and colonization during cancer metastasis, both in primary tumors and premetastatic lesions. TAMs facilitate the extravasation of tumor cells to distant metastatic sites and improve their survival by stimulating pro-survival signaling in tumor cells and inhibiting the immune response in the metastatic lesions (37). These functions of macrophages can contribute to the spread of cancer cells and the formation of metastatic niches.
TAMs play a crucial role in regulating angiogenesis. They produce various pro-angiogenic molecules, including tumor necrosis factor-α (TNF-α), thymidine phosphorylase, vascular endothelial growth factor (VEGF), and Wnt proteins, which are substances that promote the growth of new blood vessels (38, 39). These molecules can activate endothelial cells of blood vessels and stimulate the formation of new blood vessels. Another way that macrophages impact angiogenesis is by enhancing tumor hypoxia and glycolysis (40). The tumor microenvironment is usually characterized by hypoxia, which refers to a lack of oxygen. Tumor cells break down glucose to produce energy in the absence of oxygen, which is known as glycolysis. These conditions are both important causes of angiogenesis, as they can stimulate the production of pro-angiogenic molecules and promote the growth of new blood vessels. TAMs can help to shape the structure of blood vessels and interact with the sprouting vasculature (the sprouting of endothelial cells) to benefit the formation of complex vascular networks. In addition, a subpopulation of macrophages expresses tyrosine kinase with immunoglobulin-like and EGF-like domain 2 (TIE2), a receptor for angiopoietins that is involved in regulating the formation and maintenance of blood vessels (41). These TIE2-expressing macrophages have enhanced pro-angiogenic activity and lower pro-inflammatory activity. Targeting TIE2 or its ligand ANG2 can inhibit angiogenesis in certain tumor models, such as breast and pancreatic cancers (42).
Despite the pro-tumoral functions of TAMs introduced above, TAMs display a high degree of plasticity and hold the potential to eliminate tumor cells (43). Macrophages are professional phagocytes that can remove both foreign particles such as bacteria and altered-self particles such as apoptotic and necrotic cells. Typically, phagocytosis involves four phases: (i) identification of the target particles, (ii) membrane extending to internalize the particles, (iii) formation of phagosomes for particle degradation, and (iv) maturation of the phagosomes (44). The identification of foreign particles is usually mediated by the recognition of pathogen-associated molecular patterns (PAMPs) by the pattern-recognition receptors on macrophages (44). For the identification of dying cells, the release of some “find-me” chemoattractants such as ATP recruits distant phagocytes to the target cells to initiate the process (45). The phagocytosis of tumor cells by macrophages is regulated by “eat-me” signals such as calreticulin, signaling lymphocytic activation molecule family member 7 (SLAMF7), phosphatidylserine (PtdSer), and “don’t eat-me” signals such as CD47, PD-L1, and major histocompatibility complex (MHC) I (10). These ligands are expressed on the surface of tumor cells and interact with the corresponding receptors on macrophages. Besides, monoclonal antibodies that target tumor-specific antigens can interact with the Fcγ receptors (FcγR) on macrophages to trigger antibody-dependent cellular phagocytosis (ADCP). Studies have shown that ADCP contributes to the therapeutic efficacy of antibodies such as trastuzumab (an anti-HER2 monoclonal antibody) and rituximab (an anti-CD20 monoclonal antibody) (46).
Normalization of TAMs and blockade of inhibitory signals such as CD47-signal regulatory protein α (SIRPα) axis to activate their phagocytosis activity have emerged as an anti-tumor strategy. Pro-inflammatory M1 signatures on the macrophages can possibly convert surrounding M2 macrophages to the M1 phenotype and remodel the entire tumor microenvironment (47). In addition, after the engulfment of tumor cells, they can present tumor-associated antigens to T cells, which consequently facilitates CTL priming and killing (10, 12). Given the anti-tumor potential of macrophages, strategies targeting macrophages have emerged for tumor treatment, including depleting macrophages or limiting monocyte recruitment, reprogramming TAMs into anti-tumor phenotypes, abolishing inhibitory signals, and utilizing engineered macrophages for drug delivery and therapy.
Therapy Strategies
Depletion of TAM
Given the negative role of TAMs in tumor treatment, reducing TAM numbers by direct depletion of TAMs or indirect inhibition of the recruitment of their precursor cells has great potential in tumor management. One attractive target for TAM and monocyte depletion is the colony-stimulating factor 1–colony-stimulating factor 1 receptor (CSF-1–CSF-1R) axis. CSF-1R is exclusively expressed by macrophages and monocytes and plays an important role in their survival (48). Antibodies, siRNAs, and small molecule compounds have been investigated to inhibit the CSF-1–CSF-1R axis (49,50,51). Immunosuppressive cells such as TAMs can promote tumor cell immune escape and hinder T cell function, thus restricting the efficacy of immune checkpoint blockade therapy. Li et al. developed a hydrogel loaded with Pexidartinib (PLX) and anti-PD-1-conjugated platelets (P-aPD-1) for the prevention of tumor recurrence after surgery (Fig. 2) (52). PLX-loaded dextran nanoparticles (PLX-NPs) were harbored in the hydrogel, which was implanted in the tumor surgical cavity, allowing the gradual release of PLX to block CSF-1R for the local depletion of TAMs. P-aPD-1 could be either injected systemically or loaded in the hydrogel to activate the infiltrating T cells for anti-tumor immunity. PLX-NP-loaded hydrogel contributed 2.3-fold and 1.8-fold decreases in intratumoral TAM densities compared to PLX and PLX-NP. The results suggested that local TAM depletion boosted the anti-tumor efficacy of both local and systemic immune checkpoint inhibitor treatment. The hydrogel served as a depot that released PLX locally in a controlled manner, which could maximize TAM depletion efficacy while minimizing the potential side effects on normal tissues.

Schematic illustration of the hydrogel loaded with Pexidartinib (PLX) and anti-PD-1-conjugated platelets (P-aPD-1) for the prevention of tumor recurrence after surgery. Reproduced with permission. (52) Copyright 2022, Springer Nature
Bisphosphonates, such as clodronate, have a higher cytotoxicity on TAMs than macrophages in bones, especially when the drugs are delivered by liposomes (53, 54). Besides, TAM-targeted delivery of several antitumor chemotherapeutics such as DOX also results in effective TAM depletion. Some membrane receptors highly expressed on macrophages, such as mannose receptor (MMR/CD206), can be targeted by using delivery systems modified with mannose or their analogues (55). However, mannose moieties can bind to other receptors, such as dendritic cell-specific intracellular adhesion molecules (ICAM)-3 grabbing non-integrin (DC-SIGN), liver/lymph node-specific ICAM-3 grabbing non-integrin (L-SIGN), and mannose-binding lectins. This can be addressed by using ligands with higher selectivity or by designing stimuli-responsive systems. The CSPGAKVRC (UNO) peptide showed a better selectivity that preferentially homes to tumor tissues and binds to mannose receptors on TAMs (56). In addition, dual-targeting nanoparticles that are equipped with fusion peptides containing two targeting units showed high affinity to M2-like TAM and improved the targeted delivery of anti-CSF-1R siRNA to TAMs (49). The application of delivery systems that shield mannose in circulation and expose it can further improve the efficacy of TAM depletion. For instance, calcium zoledronate-loaded nanoparticles with mannose as the targeting motif were further modified with a pH-responsive sheddable PEG layer, leading to efficient TAM depletion (Fig. 3) (57).

Schematic illustration of macrophage-targeted delivery of calcium zoledronate for the depletion of TAM. Zol, zoledronate. Reproduced with permission. (57) Copyright 2019, American Chemical Society
Chemotactic signals present in the tumor microenvironment mediate the mobilization of the precursor monocytes to primary and metastatic tumor sites. The mechanism for the infiltration of monocytes into the tumor area is complicated, involving the participation of cytokines (such as CSF-1), complement components (such as C5a), and chemokines (such as CCL2) (58,59,60,61,62). Based on the chemoattractant involved in monocyte recruitments, one strategy for depleting monocytes in the tumor microenvironment is to target the signaling pathways and reduce monocyte-attracting molecules that regulate their recruitments and survivals. Inhibitors targeting the CSF-1–CSF-1R axis can deplete monocytes and TAMs from the tumor microenvironment. Targeting CCR2, which is expressed on Ly6C+/CCR2+ inflammatory monocytes, may effectively deplete this subset of monocytes and reduce TAM accumulation in tumors (62). Similarly, blocking the Notch signaling pathway, which is involved in the differentiation of inflammatory monocytes into TAMs, has been shown to reduce TAM accumulation and tumor growth (63, 64).
CAR-M
The CAR is a synthetic surface receptor that recognizes specific antigens and bypasses the MHC and TCR binding for direct T cell activation (65). CARs can be transduced into various immune cells, operating as a critical recognition device for tumor cells by the immune system. Current immunotherapies harness CAR to better recognize target antigens that are uniformly expressed on tumor cells, but not on normal tissues. As surface CARs bind to target tumor cell surface antigens, cytotoxic immune cells are redirected to tumor cells that express that antigen (66). Revolutionary therapies have utilized the integration of CAR in the cell membrane of T cells (CAR-T) to vastly improve hematological tumor cell recognition and elimination by the immune system (67). However, many trials that have attempted to utilize CAR-T cells specific to solid tumors have not shown an effective antitumor response, largely due to the inefficient recruitment of T cells to the tumor site and immunosuppressive tumor microenvironment that halts CAR-T cell function (65).
Macrophages can also be transduced with CAR, in a similar fashion to CAR-T cells to create CAR-Macrophages (CAR-Ms). CARs for macrophage engineering are typically composed of an extracellular antigen-recognition domain, a hinge domain, a transmembrane domain, and intracellular signaling domains (68). The antigen recognition can be directed by a single-chain variable fragment (scFv) against targets, such as CD19 and HER2 (69). Intracellular domains such as CD3ζ and macrophage-native Fc receptor common gamma chain (FcRγ) can then activate the phagocytosis functions of macrophages. Other motifs can be added to intracellular domains to endow the engineered macrophage with improved abilities to engulf large targets, secrete cytokines, present antigens, and activate T cells (68, 70). As a result, the CARs could redirect the phagocytic functions of macrophages toward a specific target and hold the potential to evoke an adaptive immune response. CAR-M treatment is encouraging due to the tumor’s recruitment ability of myeloid cells and thus the ability of macrophages transduced with CAR to enter the solid tumor (71, 72). CAR-M cells, once in the solid tumor microenvironment, directly kill antigen-expressing cells via phagocytosis. In addition, they promote a pro-inflammatory tumor microenvironment utilizing cytokine and chemokine secretion. The pro-inflammatory state of the TME is thus more accepting of the entry of other immune cells like tumor-specific CTLs which recognize tumor-specific antigens presented by engineered CAR-Ms to destroy the target cells (69). By counteracting the immunosuppressive TME, CAR-Ms can further boost the adaptive immune response. In this regard, CAR-M cell therapy may outweigh current CAR-T cell therapy for the treatment of solid tumors.
Mounting evidence has implied that CAR-Ms directly kill tumor cells and indirectly sculpt the TME, stimulate T cells, and induce a vaccinal effect. Klichinsky et al. utilized a replication-incompetent adenoviral vector (Ad5f35) for CAR delivery to macrophages and generated anti-HER2 CAR-MΦs (73). Primary human macrophages were generated from CD14+ peripheral blood monocytes with granulocyte macrophage stimulating colony factor (GM-CSF). It was demonstrated that the Ad5f35-transduced human macrophages expressed anti-HER2 effectively. The resulting anti-HER2 CAR-MΦ eradicated a HER2-positive ovarian cancer cell line (SKOV3) in a solid tumor xenograft mouse model and also demonstrated antigen-specific phagocytosis in HER2+ beads and tumor cells. Additionally, the adenovirally transduced macrophages, regardless of their CAR expression, inducted the expression of interferon-associated response genes like IFIT1, ISG15, and IFITM1, consistent with an anti-tumoral M1 phenotype. Moreover, the surrounding M2 macrophages were converted to the M1 phenotype, which decreased the secretion of immunosuppressive cytokines while enhancing the antigen presentation to T cells. This phenotype was maintained at least 40 days after transduction. This study demonstrated that CAR gene manipulation in human macrophages can alter their phagocytic activity against tumors. An anti-HER2 CAR-M product (CT-0508) developed based on this study has entered the phase 1 clinical trial for the treatment of HER2-overexpressing solid tumors and demonstrated good feasibility (74).
To simplify CAR-MΦ preparation, Chen et al. developed a nanoporter-loaded hydrogel nanoparticles for in situ induction of CAR-MΦs (Fig. 4) (75). CD133 is a pentaspan transmembrane glycoprotein expressed on cancer stem cells that drives glioblastoma (GBM) relapse and chemoradioresistance. CD133-targeted CAR gene was laden in nanoporter via electrostatic interaction and was then loaded in the hydrogel superstructure. The hydrogel was injected into a post-surgical cavity following GBM tumor debulking, and the nanoporter with the targeting ability for the CD206 receptor to deliver the CARs to engineer M2 macrophages. The results suggested that the macrophages engineered by the nanoparticles had high phagocytotic activity against CD133+ glioma stem cells in vitro and aided in macrophage reprogramming in vivo. In an orthotopic patient-derived glioma humanized mouse model, it was determined that intracranial codelivery of NP-pCAR MΦs and anti-CD47 antibodies by the nanoporter-hydrogel structure achieved the most robust antitumor efficacy. This combination therapy greatly increased the phagocytotic ability of the NP-pCAR-MΦs against glioma stem cells by simultaneously targeting CD47 “don’t eat me” signaling. This treatment generated a robust anti-tumor immune response in the post-surgery tumor cavity and inhibited GBM recurrence post-operation, contributing to an 83% survival rate in 120 days. A typical manufacturing workflow of CAR-M cells requires time-intensive isolation, genetic modification, and selective expansion before infusion back into a patient. The development of novel in situ macrophage engineering approaches may expedite the preparation and lower the cost of CAR-M cell therapies.

Schematic showing the in situ induction of CAR-MΦs by a hydrogel loaded with nanoporters and the working mechanism of resultant CAR-MΦs. GSCs, glioma stem cells. Reproduced with permission. (75) Copyright 2022, AAAS
Macrophage-Based Drug Delivery
Cell-based delivery systems can disguise cargo as self-components, thus helping it escape the clearance of MPS during circulation. Specifically, macrophages are a main component of MPS and have a long lifespan ranging from several months to years (76). The surface receptors and other molecules displayed on macrophages allow them to react to inflammatory signals and migrate to the diseased site (77). The tumor microenvironment is often associated with chronic inflammations that are prone to recruit macrophages (48). Therefore, efforts have been made to exploit macrophage-based delivery carriers, principally through backpacking drugs onto macrophages, in situ macrophage hitchhiking, as well as loading drugs inside macrophages.
Nanoparticles deemed “backpacks” have been designed to adhere to mobile circulatory cells such as macrophages and modulate the functions of these cells. For example, the discoidal backpacks comprised of a polyvinyl alcohol (PVA) layer sandwiched between two polylactic-co-glycolic acid (PLGA) layers have been developed using microcontact printing (78). PVA’s hydrophilicity allowed for the incorporation of interferon-γ (IFN-γ), a cytokine that functions as the primary activator of macrophages, into the middle layer of the backpack (Fig. 5). Backpacks also contain a cell adhesive layer composed via layer-by-layer assembly of polyallylamine and aldehyde-modified hyaluronic acid. Backpacks with this cell-adhesive layer bound to 86.9% of bone marrow-derived macrophages, and the anisotropic shape of backpacks helped to avoid phagocytosis by disrupting the formation of actin structure necessary to complete phagocytosis. Macrophages carrying IFN-γ backpacks not only shifted polarization of TAMs toward M1 phenotypes but also maintained their M1 phenotypes in the immunosuppressive solid tumor environment as supported by increased expression of inducible iNOS, MHCII, and CD80, relative to unpolarized macrophages. Macrophages, when left unpolarized or polarized from MΦs in free IFN-γ ex vivo, lost the M1 phenotype in the tumor microenvironment without the presence of the cellular backpack.

Schematic illustration of IFN-γ-loaded cellular backpacks for maintaining pro-inflammatory M1 phenotypes of adoptively transferred MΦ. a MΦs polarized with IFN-γ rapidly lose their M1 phenotypes following a traditional adoptive macrophage transfer. b MΦs armed with IFN-γ backpacks maintain their proinflammatory phenotypes and repolarize endogenous TAMs. Reproduced with permission. (78) Copyright 2020, AAAS
Due to their phagocytic nature, macrophages can actively phagocytose bacteria or bacteria-mimetic nanosystems. Gao et al. developed phagocytic immune cell-hitchhiking gold nanoparticles (GNPs) for anti-tumor photothermal treatment (PTT) (Fig. 6) (79). GNPs were modified with β-cyclodextrin (β-CD) or adamantane (ADA), which formed the host-guest pair that could drive the supramolecular self-assembly of GNPs. The modified GNPs were further coated with Escherichia coli outer membrane vesicles (OMVs), which facilitate the phagocytosis of GNPs by phagocytic immune cells, including macrophages. Following the phagocytosis, the OMVs were degraded, and GNPs without the photothermal effect could self-assemble into GNP aggregates with the photothermal effect. The formation of intracellular GNP aggregates with large sizes also reduced their leakage from immune cells. Phagocytic cells carry the loaded GNPs to melanoma sites through inflammatory tropism-mediated targeting. When a portion of phagocytic immune cell-hitchhiking GNPs reached the tumor sites, an initial irradiation was performed that induced damage and inflammation at tumor sites, facilitating the recruitment of more phagocytic cells together with more GNPs. A secondary irradiation is then conducted to trigger a potent PTT effect. The positive feedback between macrophage-mediated delivery and PTT treatment contributed to improved therapeutic effects.

Schematic illustration of phagocytic immune cell-hitchhiking gold nanoparticles (GNPs) for enhanced PTT. a The construction of OMV-coated nanoparticles. b The phagocytosis by phagocytic immune cells triggered intracellular aggregation of GNPs, forming GNP aggregates with the photothermal property. c The positive feedback between macrophage-mediated delivery and PTT treatment contributed to improved therapeutic effects. Reproduced with permission. (79) Copyright 2022, AAAS
In addition, macrophage membrane-coated nanoparticles and macrophage-derived exosomes have also been explored for drug delivery. Cao et al. coated liposomes with macrophage membranes for the delivery of emtansine, displaying enhanced inhibition activity against the lung metastasis of breast cancer (80). In addition, the presence of the blood−brain barrier (BBB) and blood−brain−tumor barrier (BBTB) restricts the accumulation of therapeutics in glioblastoma. Due to their ability to cross the BBB, macrophages and their exosomes have been employed as carriers for brain drug delivery. The exosomes can be further decorated with motifs, such as AS1411 aptamer or cRGD peptide, to improve their tumor-targeting capability (81, 82). Combined with different therapeutics, the macrophage exosome-based delivery system improved the efficacy of sonodynamic therapy, gene therapy, and chemotherapy on brain tumor models. In addition, exosomes derived from M1 macrophages display the ability to foster M1 polarization of macrophages, establish a local immunostimulatory microenvironment, and promote anti-tumor immunity (83).
Macrophage-based drug delivery systems have been developed for the treatment of a variety of diseases, such as rheumatoid arthritis, sepsis, and atherosclerosis (84,85,86). For example, Hou et al. adoptively transferred macrophages loaded with antimicrobial peptide and cathepsin B (AMP-CatB) mRNA for the treatment of sepsis induced by multidrug-resistant bacteria (84). AMP-CatB mRNA encoding an AMP, a CatB, and a CatB-responsive linker was loaded in vitamin C-derived lipids (VCLNPs). The VCLNPs were internalized by macrophages via caveolae-mediated endocytosis, and then the CatB–an endogenous lysosomal protein could carry the payload into lysosomes. The VCLNPs-loaded macrophage displayed a strong bactericidal activity in the sepsis mice. These systems may also provide inspiration for macrophage-based tumor therapy.
Repolarization of Macrophages
When triggered in response to environmental cues, macrophages exhibit different functional programs that are subcategorized into classically activated (M1) or alternatively activated (M2) macrophages. M1 macrophages are polarized by lipopolysaccharides and cytokines like IFN-γ or GM-CSF to exhibit strong effector functions against cancer cells and pathogens (87, 88). In addition to their high phagocytic ability, M1-macrophages heighten pro-inflammatory cytokines like IL-12, IL-23, and TNF-α which facilitate leukocyte recruitment and activation during injury (89). Alternatively, polarization by IL-4 and IL-13 results in M2-macrophages which perform anti-inflammatory functions. M2-macrophages clear debris and release transforming growth factor-β (TGF-β), platelet-derived growth factor (PDGF), and VEGF to aid in wound healing and repair (90, 91). Furthermore, M2 macrophages contribute to inflammation resolution by producing immunosuppressive cytokines like IL-10. Of note, macrophages demonstrate high plasticity between phenotypes. M2 macrophages can shift to M1 macrophages under the induction of certain signals, making polarization a dynamic process.
Reprogramming TAMs toward the M1-like phenotype may activate their phagocytosis functions, representing a promising strategy for tumor treatment. The repolarization of TAMs toward the M1 phenotype can be achieved through some therapeutic modalities, such as chemotherapy, radiotherapy, and immunotherapy (92, 93). Low-dose local radiotherapy can reprogram macrophages toward an iNOS+/M1 phenotype that promotes infiltration and activity of CTLs, thus orchestrating effective immunotherapy (94). Many cytokines (including CSF-1), immunostimulants, transcription signal modulators, and chemical compounds can be utilized to reprogram TAMs. CpG oligodeoxynucleotides (CpG ODNs) can be recognized by toll-like receptor 9 to polarize macrophages towards the M1 phenotype. Loading CpG ODNs in macrophage-targeted nanoparticles promoted M1-type polarization and abolished tumor-associated immunosuppression (95). Besides, the upregulation of some metabolic pathways supports and accelerates cancer progression by driving M2 polarization in tumors. Sialylation is an established hallmark of tumors and is associated with immunosuppression status. Tumor-specific desialylation could be achieved by an antibody-sialidase conjugate, which resulted in a phenotype shift of TAMs toward M1 polarization (96).
Containing abundant contents from their parent cells, exosomes derived from M1 macrophages could reprogram M2 toward M1 phenotype. Nie et al. engineered exosomes from M1 macrophages with immune stimulatory antibodies for tumor therapy (Fig. 7) (97). MΦ were incubated with Manganese2+ (Mn2+), which exemplified successful hydroxyl radical generation via a Fenton-like reaction and thus induced M1 polarization. M1 exosomes were then modified with surface azide groups and were conjugated with dibenzocylclooctyne-modified immune stimulatory antibodies aCD47 and aSIRPα via click chemistry. In an acidic tumor microenvironment, pH-sensitive benzoic imine bonds are cleaved, selectively releasing the antibodies that block the inhibitory receptor SIRPα from interacting with CD47 tumor cell transmembrane proteins and thus induce improved phagocytic abilities of the M1 macrophages. The treatment switched pro-tumoral M2 macrophages to anti-tumoral M1 macrophages via M1 exosome-mediated re-education and blockage of CD47-SIRPα signaling. The fluorescence from tumor cells was dispersed into 73% of the M1 Exo-treated M2, while the signal can hardly be detected in untreated pristine M2, confirming the macrophage reprogram ability of M1 Exo. In addition, natural killer (NK) cells have been reported to promote the M1 polarization of macrophages via some NK membrane proteins, such as DNAM-1 and RANKL. Deng et al. cloaked photosensitizer-loaded nanoparticles with NK cell membranes (NK-NPs) (98). NK-NPs contributed to a 5.5-fold increase in the percentage of M1 macrophages in tumors, working synergistically with photodynamic therapy-induced immunogenic cell death to augment the anti-tumor efficacy.

Schematic illustration of engineered exosomes from M1 macrophages for M1 polarization. M1 Exo, M1 macrophage exosomes. Reproduced with permission. (97) Copyright 2020, Wiley-VCH
Blocking Inhibitory Signaling to Enhanced Phagocytosis
CD47 is a transmembrane protein widely present in normal cells, functioning as an inhibitor of phagocytosis to maintain immune homeostasis. However, to evade the phagocytosis by macrophages, tumor cells upregulate their expression of CD47, which interacts with the inhibitory receptor SIRPα on macrophages to transmit the “don’t eat me” signal (99). Studies have shown that CD47 mRNA expression levels in multiple types of tumors are inversely correlated with patient survival (100). CD47 blockade promotes the elimination of tumor cells through several mechanisms. The most well-investigated mechanism is that the inhibition of the CD47-SIRPα axis contributes to the restoration of macrophages’ phagocytic function. Besides, CD47 blockade enhances tumor engulfment and antigen presentation by antigen-presenting cells, which consequently facilitates the T cell priming and killing effects (101). In addition, anti-CD47 antibodies are reported to induce apoptosis of tumor cells directly and stimulate antibody-dependent cellular cytotoxicity (ADCC) to eliminate tumor cells (102, 103). By far, several antibodies or small molecules targeting CD47-SIRPα axis have entered clinical trials (104, 105). Magrolimab, an anti-CD47 monoclonal antibody, is undergoing a phase 3 ENHANCE trial for the treatment of myelodysplastic syndrome and has received FDA Breakthrough Therapy Designation (106). RRx-001, a small molecule that can downregulate CD47 and SIRP-α, is currently studied in phase 2 and 3 trials for the treatment of several tumor malignancies (107).
Although CD47 antagonists are promising in activating the anti-tumor activities of macrophages, the occurrence of adverse events such as anemia and thrombocytopenia following the systemic administration of the antibodies brings up a concern (108). To prevent post-surgical tumor recurrence and eliminate distant tumors, Chen et al. developed an anti-CD47-loaded in situ-formed gel (109). The antibody was loaded into calcium carbonate nanoparticles. The gel was formed in the tumor resection cavity following the simultaneous spraying and mixing of nanoparticles-loaded fibrinogen solution and thrombin solution. Calcium carbonate also functions as a proton scavenger that regulates the acidity in the TME. The in situ-formed gel elicited efficient anti-tumor activity of macrophages and T cells while reducing the systemic toxic effects.
In addition, blockade of the CD47-SIRPα axis can also be combined with other treatment modalities. Given that the chemodrug paclitaxel (PTX) can promote the infiltration of TAMs and foster the enrichment of CD47 on tumor cells, Wang et al. developed a PTX filament (PF) supramolecular hydrogel loaded with anti-CD47 antibody (aCD47) for the postoperative treatment of GBM (Fig. 8) (110). PTX molecules were conjugated to iRGD peptides through biodegradable linkers to form the amphiphilic PTX prodrug, which is capable of self-assembling into PF. The mixture of aCD47 and PF was infused into the resection cavity after the surgical removal of GBM. The solution-to-hydrogel phase transition could be induced by PBS or happen spontaneously under physiological conditions. The hydrogel acts as a reservoir for localized and sustained delivery of both aCD47 and PTX, enhancing the immune response against cancer cells and diminishing tumor recurrence. Current clinical treatments for GBM have shown limited efficacy due to both the blood–brain barrier that restricts the entry of T cells into the brain cells and the immunosuppressive factors released by cancer cells. As 30 to 50% of brain tumor mass is composed of TAMs, activating phagocytosis functions of TAM using these hydrogels is a promising approach to improve therapeutic efficacy.

Schematic illustration of PF hydrogel for localized delivery of PTX and aCD47 for the postoperative treatment of GBM. Reproduced with permission. (110) Copyright 2023, National Academy of Science
Moreover, the CD47-SIRPα blockade could be combined with the TAM repolarization strategy for the synergistic treatment efficacy. Kulkarni et al. developed a supramolecular assembly containing CSF-1R inhibitors and anti-SIRPα antibodies (111). BLZ945, the CSF-1R inhibitor in the assembly, depletes TAMs and promotes the M1 polarization. The supramolecule demonstrated robust anti-tumor and anti-metastatic efficacy in melanoma and breast cancer models. Feng et al. engineered bacteria-derived outer membrane vesicles (OMVs) with CD47 nanobody (CD47nb) (112). OMVs induced M1 polarization and sensitized TAMs for CD47-SIRPα blockade, thus evoking their phagocytosis ability.
Conclusion and Discussion
TAMs play a critical role in tumor progression, metastasis, and immunosuppression, forming a hindrance to effective therapeutic approaches against tumors. Insight into the factors that activate the anti-tumor function of TAMs has revealed new therapeutic avenues. Strategies to block inhibitory signaling or repolarization of macrophages can activate phagocytosis, transforming TAMs from a tumor promoter into a tumor suppressor. Depletion of TAM has been proven successful in the intervention of tumors in experimental settings, especially when combined with other therapy modalities. Engaging macrophages as drug carriers offer distinguishing advantages, including prolonged circulation, enhanced tumor-targeted delivery, and reduced immunogenicity. Specifically, due to the capability of macrophages to cross the BBB, macrophage-based delivery systems may open up opportunities for the treatment of brain tumors. Besides, CAR-M that can perform cytocidal tasks will expand the toolkit for the treatment of solid tumors.
Great endeavors have been made to target macrophages for tumor therapy. A CSF-1R inhibitor, PLX3397 (pexidartinib), has been approved by FDA for the treatment of tenosynovial giant cell tumors, and other kinds of CSF-1 inhibitors have also marched to advanced clinical trials (113). In addition, the initial data from the in-human study demonstrated a good safety profile of CAR-M CT-0508 in the phase 1 clinical trial (74). Moreover, progress in elucidating the precise mechanisms by which macrophages accelerate tumor progression will lay a solid foundation for the development of new anti-tumor therapies. Macrophages terminate inflammation through efferocytosis, which involves the process of the production of anti-inflammatory cytokines and the repression of pro-inflammatory mediators. A strong connection between tumors and efferocytosis has been revealed, and inhibition of efferocytosis has emerged as a promising strategy for tumor treatment (114). Myeloid-epithelial-reproductive tyrosine kinase (MerTK) of TAMs promotes efferocytosis and represents a target for efferocytosis. Inhibitors and antibodies targeting MerTK have been explored to block the activation of MerTK (115, 116). There is still plenty of room for the development of macrophage-centered tumor therapies.
Despite the success of targeting macrophages for anti-tumor therapy, many challenges remain that limit the translation of these treatments. The recruitment and polarization of macrophages in the TME are complicated and dynamic. M1-like macrophages in the tumor area may turn back into immunosuppressive M2 macrophages after treatment withdrawal, weakening their tumor-killing abilities and limiting the persistence of the therapy. Regulating and maintaining the M1 phenotypes of adoptively transferred macrophages in vivo is crucial in advancing adoptive cell therapies as conserved M1 phenotypes slow tumor growth and reduce metastasis. Therefore, it is necessary to develop therapeutics that maintain M1 polarization and monitor the phenotype of macrophages for a long time. Besides, macrophage heterogeneity is often explained utilizing an M1/M2 concept; however, complex macrophage activation is not elucidated with such a notion. Utilizing M2 markers such as CD163, CD204, and CD206, it is found that macrophages contain a wide range of polarization features or activation states (117, 118). Additional studies are crucial in further classifying macrophages beyond the M1/M2 identification approach as macrophage function is highly dependent upon ontogenesis, the environment, and the types of pathogens or injuries present (119). In addition, although M1 macrophages are regarded as tumoricidal in general, some studies also report its pro-tumor potential. Activation of pro-inflammatory pathways in TAMs can elevate the expression of inhibitory receptors and ligands such as PD-L1 to impact immune response (120). Phagocytosis of tumor cells by macrophages concomitantly leads to the upregulation of IDO and PD-L1 (121). Although the precise mechanisms remain to be discovered, it can be inferred that combination therapy (such as anti-PD-1/PD-L1+macrophage-based therapy) may generate synergistic effects to improve efficacy. Moreover, despite the encouraging data on combination therapy in preclinical and clinical models, the lack of understanding of the detailed mechanism of macrophage functions raises concerns regarding the safety of macrophage-targeted therapy, especially for the depletion of macrophages. As macrophages play a critical role in immune defense and tissue homeostasis, it is important to assess the systemic impact and side effects in view of the long-term safety of these therapies.
References
Underhill DM, Gordon S, Imhof BA, Nunez G, Bousso P. Elie Metchnikoff (1845-1916): celebrating 100 years of cellular immunology and beyond. Nat Rev Immunol. 2016;16(10):651–6. https://doi.org/10.1038/nri.2016.89.
Das A, Sinha M, Datta S, Abas M, Chaffee S, Sen CK, et al. Monocyte and macrophage plasticity in tissue repair and regeneration. Am J Pathol. 2015;185(10):2596–606. https://doi.org/10.1016/j.ajpath.2015.06.001.
Gordon S, Pluddemann A, Martinez EF. Macrophage heterogeneity in tissues: phenotypic diversity and functions. Immunol Rev. 2014;262(1):36–55. https://doi.org/10.1111/imr.12223.
Stein M, Keshav S, Harris N, Gordon S. Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. J Exp Med. 1992;176(1):287–92. https://doi.org/10.1084/jem.176.1.287.
Mills CD. M1 and M2 macrophages: oracles of health and disease. Crit Rev Immunol. 2012;32(6):463–88. https://doi.org/10.1615/critrevimmunol.v32.i6.10.
Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol. 2000;164(12):6166–73. https://doi.org/10.4049/jimmunol.164.12.6166.
Solinas G, Germano G, Mantovani A, Allavena P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J Leukocyte Biol. 2009;86(5):1065–73. https://doi.org/10.1189/jlb.0609385.
Zhao C, Pang X, Yang Z, Wang S, Deng H, Chen X. Nanomaterials targeting tumor associated macrophages for cancer immunotherapy. J Control Release. 2022;341:272–84. https://doi.org/10.1016/j.jconrel.2021.11.028.
Komohara Y, Fujiwara Y, Ohnishi K, Takeya M. Tumor-associated macrophages: potential therapeutic targets for anti-cancer therapy. Adv Drug Deliv Rev. 2016;99(Pt B):180–5. https://doi.org/10.1016/j.addr.2015.11.009.
Zhou XF, Liu XR, Huang L. Macrophage-mediated tumor cell phagocytosis: opportunity for nanomedicine intervention. Adv Funct Mater. 2021;31(5):2006220. https://doi.org/10.1002/adfm.202006220.
Saeed M, Chen F, Ye J, Shi Y, Lammers T, De Geest BG, et al. From design to clinic: engineered nanobiomaterials for immune normalization therapy of cancer. Adv Mater. 2021;33(30):e2008094. https://doi.org/10.1002/adma.202008094.
Christofides A, Strauss L, Yeo A, Cao C, Charest A, Boussiotis VA. The complex role of tumor-infiltrating macrophages. Nat Immunol. 2022;23(8):1148–56. https://doi.org/10.1038/s41590-022-01267-2.
Li CX, Xu XF, Wei SH, Jiang P, Xue LX, Wang JJ. Tumor-associated macrophages: potential therapeutic strategies and future prospects in cancer. J Immunother Cancer. 2021;9(1):e001341. https://doi.org/10.1136/jitc-2020-001341.
Yang QY, Guo NN, Zhou Y, Chen JJ, Wei QC, Han M. The role of tumor-associated macrophages (TAMs) in tumor progression and relevant advance in targeted therapy. Acta Pharmacol Sin B. 2020;10(11):2156–70. https://doi.org/10.1016/j.apsb.2020.04.004.
Huang R, Wang S, Wang N, Zheng Y, Zhou J, Yang B, et al. CCL5 derived from tumor-associated macrophages promotes prostate cancer stem cells and metastasis via activating beta-catenin/STAT3 signaling. Cell Death Dis. 2020;11(4):234. https://doi.org/10.1038/s41419-020-2435-y.
Jinushi M, Chiba S, Yoshiyama H, Masutomi K, Kinoshita I, Dosaka-Akita H, et al. Tumor-associated macrophages regulate tumorigenicity and anticancer drug responses of cancer stem/initiating cells. Proc Natl Acad Sci U S A. 2011;108(30):12425–30. https://doi.org/10.1073/pnas.1106645108.
Chen J, McKay RM, Parada LF. Malignant glioma: lessons from genomics, mouse models, and stem cells. Cell. 2012;149(1):36–47. https://doi.org/10.1016/j.cell.2012.03.009.
Garner JM, Fan MY, Yang CH, Du ZY, Sims M, Davidoff AM, et al. Constitutive activation of signal transducer and activator of transcription 3 (STAT3) and nuclear factor kappa B signaling in glioblastoma cancer stem cells regulates the notch pathway. J Biolog Chem. 2013;288(36):26167–76. https://doi.org/10.1074/jbc.M113.477950.
Komohara Y, Horlad H, Ohnishi K, Fujiwara Y, Bai B, Nakagawa T, et al. Importance of direct macrophage - tumor cell interaction on progression of human glioma. Cancer Sci. 2012;103(12):2165–72. https://doi.org/10.1111/cas.12015.
Elinav E, Nowarski R, Thaiss CA, Hu B, Jin CC, Flavell RA. Inflammation-induced cancer: crosstalk between tumours, immune cells and microorganisms. Nat Rev Cancer. 2013;13(11):759–71. https://doi.org/10.1038/nrc3611.
Yang Y, Guo J, Huang L. Tackling TAMs for cancer immunotherapy: it's nano time. Trends Pharmacol Sci. 2020;41(10):701–14. https://doi.org/10.1016/j.tips.2020.08.003.
Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nature Rev Immunol. 2009;9(3):162–74. https://doi.org/10.1038/nri2506.
Geiger R, Rieckmann JC, Wolf T, Basso C, Feng YH, Fuhrer T, et al. L-Arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell. 2016;167(3):829–42. https://doi.org/10.1016/j.cell.2016.09.031.
Horlad H, Ma C, Yano H, Pan C, Ohnishi K, Fujiwara Y, et al. An IL-27/Stat3 axis induces expression of programmed cell death 1 ligands (PD-L1/2) on infiltrating macrophages in lymphoma. Cancer Sci. 2016;107(11):1696–704. https://doi.org/10.1111/cas.13065.
Kuang DM, Zhao Q, Peng C, Xu J, Zhang JP, Wu C, et al. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J Exp Med. 2009;206(6):1327–37. https://doi.org/10.1084/jem.20082173.
Columba-Cabezas S, Serafini B, Ambrosini E, Sanchez M, Penna G, Adorini L, et al. Induction of macrophage-derived chemokine/CCL22 expression in experimental autoimmune encephalomyelitis and cultured microglia: implications for disease regulation. J Neuroimmunol. 2002;130(1-2):10–21. https://doi.org/10.1016/S0165-5728(02)00170-4.
Wang D, Yang L, Yue DL, Cao L, Li LF, Wang D, et al. Macrophage-derived CCL22 promotes an immunosuppressive tumor microenvironment via IL-8 in malignant pleural effusion. Cancer Lett. 2019;452:244–53. https://doi.org/10.1016/j.canlet.2019.03.040.
Mantovani A, Allavena P, Marchesi F, Garlanda C. Macrophages as tools and targets in cancer therapy. Nature Rev Drug Discov. 2022;21(11):799–820. https://doi.org/10.1038/s41573-022-00520-5.
Kim IS, Gao Y, Welte T, Wang H, Liu J, Janghorban M, et al. Immuno-subtyping of breast cancer reveals distinct myeloid cell profiles and immunotherapy resistance mechanisms. Nat Cell Biol. 2019;21(9):1113–26. https://doi.org/10.1038/s41556-019-0373-7.
Koh MY, Sayegh N, Agarwal N. Seeing the forest for the trees-single-cell atlases link CD8(+) T cells and macrophages to disease progression and treatment response in kidney cancer. Cancer Cell. 2021;39(5):594–6. https://doi.org/10.1016/j.ccell.2021.03.008.
Yu J, Green MD, Li S, Sun Y, Journey SN, Choi JE, et al. Liver metastasis restrains immunotherapy efficacy via macrophage-mediated T cell elimination. Nat Med. 2021;27(1):152–64. https://doi.org/10.1038/s41591-020-1131-x.
Chow A, Schad S, Green MD, Hellmann MD, Allaj V, Ceglia N, et al. Tim-4(+) cavity-resident macrophages impair anti-tumor CD8(+) T cell immunity. Cancer Cell. 2021;39(7):973–88 e9. https://doi.org/10.1016/j.ccell.2021.05.006.
Chen Q, Li YF, Gao WJ, Chen L, Xu WL, Zhu XL. Exosome-mediated crosstalk between tumor and tumor-associated macrophages. Front Mol Biosci. 2021;8:764222. https://doi.org/10.3389/fmolb.2021.764222.
Jaiswal S, Chao MP, Majeti R, Weissman IL. Macrophages as mediators of tumor immunosurveillance. Trends Immunol. 2010;31(6):212–9. https://doi.org/10.1016/j.it.2010.04.001.
Zhu S, Yi M, Wu Y, Dong B, Wu K. Roles of tumor-associated macrophages in tumor progression: implications on therapeutic strategies. Exp Hematol Oncol. 2021;10(1):60. https://doi.org/10.1186/s40164-021-00252-z.
Chen J, Yao Y, Gong C, Yu F, Su S, Chen J, et al. CCL18 from tumor-associated macrophages promotes breast cancer metastasis via PITPNM3. Cancer Cell. 2011;19(4):541–55. https://doi.org/10.1016/j.ccr.2011.02.006.
Kitamura T, Qian BZ, Pollard JW. Immune cell promotion of metastasis. Nat Rev Immunol. 2015;15(2):73–86. https://doi.org/10.1038/nri3789.
Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141(1):39–51. https://doi.org/10.1016/j.cell.2010.03.014.
Franklin RA, Liao W, Sarkar A, Kim MV, Bivona MR, Liu K, et al. The cellular and molecular origin of tumor-associated macrophages. Science. 2014;344(6186):921–5. https://doi.org/10.1126/science.1252510.
Jeong H, Kim S, Hong BJ, Lee CJ, Kim YE, Bok S, et al. Tumor-associated macrophages enhance tumor hypoxia and aerobic glycolysis. Cancer Res. 2019;79(4):795–806. https://doi.org/10.1158/0008-5472.CAN-18-2545.
Murdoch C, Muthana M, Coffelt SB, Lewis CE. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer. 2008;8(8):618–31. https://doi.org/10.1038/nrc2444.
Mazzieri R, Pucci F, Moi D, Zonari E, Ranghetti A, Berti A, et al. Targeting the ANG2/TIE2 axis inhibits tumor growth and metastasis by impairing angiogenesis and disabling rebounds of proangiogenic myeloid cells. Cancer Cell. 2011;19(4):512–26. https://doi.org/10.1016/j.ccr.2011.02.005.
Tan YF, Wang M, Zhang Y, Ge SY, Zhong F, Xia GW, et al. Tumor-associated macrophages: a potential target for cancer therapy. Front Oncol. 2021;11:693517. https://doi.org/10.3389/fonc.2021.693517.
Uribe-Querol E, Rosales C. Phagocytosis: our current understanding of a universal biological process. Front Immunol. 2020;11:1066. https://doi.org/10.3389/fimmu.2020.01066.
Arandjelovic S, Ravichandran KS. Phagocytosis of apoptotic cells in homeostasis. Nat Immunol. 2015;16(9):907–17. https://doi.org/10.1038/ni.3253.
Li H, Somiya M, Kuroda S. Enhancing antibody-dependent cellular phagocytosis by re-education of tumor-associated macrophages with resiquimod-encapsulated liposomes. Biomaterials. 2021;268:120601. https://doi.org/10.1016/j.biomaterials.2020.120601.
Viola A, Munari F, Sanchez-Rodriguez R, Scolaro T, Castegna A. The metabolic signature of macrophage responses. Front Immunol. 2019;10:1462. https://doi.org/10.3389/fimmu.2019.01462.
Xia Y, Rao L, Yao H, Wang Z, Ning P, Chen X. Engineering macrophages for cancer immunotherapy and drug delivery. Adv Mater. 2020;32(40):e2002054. https://doi.org/10.1002/adma.202002054.
Qian Y, Qiao S, Dai Y, Xu G, Dai B, Lu L, et al. Molecular-targeted immunotherapeutic strategy for melanoma via dual-targeting nanoparticles delivering small interfering RNA to tumor-associated macrophages. ACS Nano. 2017;11(9):9536–49. https://doi.org/10.1021/acsnano.7b05465.
Ries CH, Cannarile MA, Hoves S, Benz J, Wartha K, Runza V, et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell. 2014;25(6):846–59. https://doi.org/10.1016/j.ccr.2014.05.016.
Shen S, Li HJ, Chen KG, Wang YC, Yang XZ, Lian ZX, et al. Spatial targeting of tumor-associated macrophages and tumor cells with a pH-sensitive cluster nanocarrier for cancer chemoimmunotherapy. Nano Lett. 2017;17(6):3822–9. https://doi.org/10.1021/acs.nanolett.7b01193.
Li Z, Ding Y, Liu J, Wang J, Mo F, Wang Y, et al. Depletion of tumor associated macrophages enhances local and systemic platelet-mediated anti-PD-1 delivery for post-surgery tumor recurrence treatment. Nat Commun. 2022;13(1):1845. https://doi.org/10.1038/s41467-022-29388-0.
Zhan X, Jia L, Niu Y, Qi H, Chen X, Zhang Q, et al. Targeted depletion of tumour-associated macrophages by an alendronate-glucomannan conjugate for cancer immunotherapy. Biomat. 2014;35(38):10046–57. https://doi.org/10.1016/j.biomaterials.2014.09.007.
Miselis NR, Wu ZJ, Van Rooijen N, Kane AB. Targeting tumor-associated macrophages in an orthotopic murine model of diffuse malignant mesothelioma. Mol Cancer Ther. 2008;7(4):788–99. https://doi.org/10.1158/1535-7163.MCT-07-0579.
Liu XS, Xie XD, Jiang JH, Lin M, Zheng E, Qiu WV, et al. Use of nanoformulation to target macrophages for disease treatment. Adv Funct Mater. 2021;31(38):2104487. https://doi.org/10.1002/adfm.202104487.
Lepland A, Asciutto EK, Malfanti A, Simon-Gracia L, Sidorenko V, Vicent MJ, et al. Targeting pro-tumoral macrophages in early primary and metastatic breast tumors with the CD206-binding mUNO peptide. Mol Pharm. 2020;17(7):2518–31. https://doi.org/10.1021/acs.molpharmaceut.0c00226.
Zang X, Zhang X, Hu H, Qiao M, Zhao X, Deng Y, et al. Targeted delivery of zoledronate to tumor-associated macrophages for cancer immunotherapy. Mol Pharm. 2019;16(5):2249–58. https://doi.org/10.1021/acs.molpharmaceut.9b00261.
Bonavita E, Galdiero MR, Jaillon S, Mantovani A. Phagocytes as corrupted policemen in cancer-related inflammation. Adv Cancer Res. 2015;128:141–71. https://doi.org/10.1016/bs.acr.2015.04.013.
Halama N, Zoernig I, Berthel A, Kahlert C, Klupp F, Suarez-Carmona M, et al. Tumoral immune cell exploitation in colorectal cancer metastases can be targeted effectively by anti-CCR5 therapy in cancer patients. Cancer Cell. 2016;29(4):587–601. https://doi.org/10.1016/j.ccell.2016.03.005.
Mantovani A, Allavena P, Sozzani S, Vecchi A, Locati M, Sica A. Chemokines in the recruitment and shaping of the leukocyte infiltrate of tumors. Semin Cancer Biol. 2004;14(3):155–60. https://doi.org/10.1016/j.semcancer.2003.10.001.
Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. 2017;14(7):399–416. https://doi.org/10.1038/nrclinonc.2016.217.
Qian BZ, Li JF, Zhang H, Kitamura T, Zhang JH, Campion LR, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011;475(7355):222–5. https://doi.org/10.1038/nature10138.
Radtke F, Fasnacht N, Macdonald HR. Notch signaling in the immune system. Immunity. 2010;32(1):14–27. https://doi.org/10.1016/j.immuni.2010.01.004.
Wang YC, He F, Feng F, Liu XW, Dong GY, Qin HY, et al. Notch signaling determines the M1 versus M2 polarization of macrophages in antitumor immune responses. Cancer Res. 2010;70(12):4840–9. https://doi.org/10.1158/0008-5472.CAN-10-0269.
Sterner RC, Sterner RM. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J. 2021;11(4):69. https://doi.org/10.1038/s41408-021-00459-7.
Jensen MC, Riddell SR. Designing chimeric antigen receptors to effectively and safely target tumors. Curr Opin Immunol. 2015;33:9–15. https://doi.org/10.1016/j.coi.2015.01.002.
Zhang C, Zhuang Q, Liu J, Liu X. Synthetic biology in chimeric antigen receptor T (CAR T) cell engineering. ACS Synth Biol. 2022;11(1):1–15. https://doi.org/10.1021/acssynbio.1c00256.
Wang SH, Yang YQ, Ma PW, Zha Y, Zhang J, Lei AH, et al. CAR-macrophage: an extensive immune enhancer to fight cancer. Ebiomedicine. 2022;76:103873. https://doi.org/10.1016/j.ebiom.2022.103873.
Sloas C, Gill S, Klichinsky M. Engineered CAR-macrophages as adoptive immunotherapies for solid tumors. Frontiers in Immunology. 2021;12:783305. https://doi.org/10.3389/fimmu.2021.783305.
Morrissey MA, Williamson AP, Steinbach AM, Roberts EW, Kern N, Headley MB, et al. Chimeric antigen receptors that trigger phagocytosis. Elife. 2018;7:e36688. https://doi.org/10.7554/eLife.36688.
Chen Y, Yu Z, Tan X, Jiang H, Xu Z, Fang Y, et al. CAR-macrophage: a new immunotherapy candidate against solid tumors. Biomed Pharmacother. 2021;139:111605. https://doi.org/10.1016/j.biopha.2021.111605.
Villanueva MT. Macrophages get a CAR. Nat Rev Immunol. 2020;20(5):273. https://doi.org/10.1038/s41577-020-0302-9.
Klichinsky M, Ruella M, Shestova O, Lu XM, Best A, Zeeman M, et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat Biotechnol. 2020;38(8):947–53. https://doi.org/10.1038/s41587-020-0462-y.
Reiss KA, Yuan Y, Ueno NT, Johnson ML, Gill S, Dees EC, et al. A phase 1, first-in-human (FIH) study of the anti-HER2 CAR macrophage CT-0508 in subjects with HER2 overexpressing solid tumors. J Clin Oncol. 2022;40(16):2533. https://doi.org/10.1200/JCO.2022.40.16_suppl.2533.
Chen C, Jing W, Chen Y, Wang G, Abdalla M, Gao L, et al. Intracavity generation of glioma stem cell-specific CAR macrophages primes locoregional immunity for postoperative glioblastoma therapy. Sci Transl Med. 2022;14(656):eabn1128. https://doi.org/10.1126/scitranslmed.abn1128.
Parihar A, Eubank TD, Doseff AI. Monocytes and macrophages regulate immunity through dynamic networks of survival and cell death. J Innate Immun. 2010;2(3):204–15. https://doi.org/10.1159/000296507.
Zheng Y, Han Y, Sun Q, Li Z. Harnessing anti-tumor and tumor-tropism functions of macrophages via nanotechnology for tumor immunotherapy. Exploration (Beijing). 2022;2(3):20210166. https://doi.org/10.1002/EXP.20210166.
Shields CWT, Evans MA, Wang LL, Baugh N, Iyer S, Wu D, et al. Cellular backpacks for macrophage immunotherapy. Sci Adv. 2020;6(18):eaaz6579. https://doi.org/10.1126/sciadv.aaz6579.
Gao C, Wang Q, Li J, Kwong CHT, Wei J, Xie B, et al. In vivo hitchhiking of immune cells by intracellular self-assembly of bacteria-mimetic nanomedicine for targeted therapy of melanoma. Sci Adv. 2022;8(19):eabn1805. https://doi.org/10.1126/sciadv.abn1805.
Cao HQ, Dan ZL, He XY, Zhang ZW, Yu HJ, Yin Q, et al. Liposomes coated with isolated macrophage membrane can target lung metastasis of breast cancer. Acs Nano. 2016;10(8):7738–48. https://doi.org/10.1021/acsnano.6b03148.
Shan S, Chen J, Sun Y, Wang Y, Xia B, Tan H, et al. Functionalized macrophage exosomes with panobinostat and PPM1D-siRNA for diffuse intrinsic pontine gliomas therapy. Adv Sci (Weinh). 2022;9(21):e2200353. https://doi.org/10.1002/advs.202200353.
Wu T, Liu Y, Cao Y, Liu Z. Engineering macrophage exosome disguised biodegradable nanoplatform for enhanced sonodynamic therapy of glioblastoma. Adv Mater. 2022;34(15):e2110364. https://doi.org/10.1002/adma.202110364.
Gunassekaran GR, Poongkavithai Vadevoo SM, Baek MC, Lee B. M1 macrophage exosomes engineered to foster M1 polarization and target the IL-4 receptor inhibit tumor growth by reprogramming tumor-associated macrophages into M1-like macrophages. Biomaterials. 2021;278:121137. https://doi.org/10.1016/j.biomaterials.2021.121137.
Hou X, Zhang X, Zhao W, Zeng C, Deng B, McComb DW, et al. Vitamin lipid nanoparticles enable adoptive macrophage transfer for the treatment of multidrug-resistant bacterial sepsis. Nat Nanotechnol. 2020;15(1):41–6. https://doi.org/10.1038/s41565-019-0600-1.
Li R, He Y, Zhu Y, Jiang L, Zhang S, Qin J, et al. Route to rheumatoid arthritis by macrophage-derived microvesicle-coated nanoparticles. Nano Lett. 2019;19(1):124–34. https://doi.org/10.1021/acs.nanolett.8b03439.
Gao C, Huang Q, Liu C, Kwong CHT, Yue L, Wan JB, et al. Treatment of atherosclerosis by macrophage-biomimetic nanoparticles via targeted pharmacotherapy and sequestration of proinflammatory cytokines. Nat Commun. 2020;11(1):2622. https://doi.org/10.1038/s41467-020-16439-7.
Jaguin M, Houlbert N, Fardel O, Lecureur V. Polarization profiles of human M-CSF-generated macrophages and comparison of M1-markers in classically activated macrophages from GM-CSF and M-CSF origin. Cell Immunol. 2013;281(1):51–61. https://doi.org/10.1016/j.cellimm.2013.01.010.
Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25(12):677–86. https://doi.org/10.1016/j.it.2004.09.015.
Duan Z, Luo Y. Targeting macrophages in cancer immunotherapy. Signal Transduct Target Ther. 2021;6(1):127. https://doi.org/10.1038/s41392-021-00506-6.
Boutilier AJ, Elsawa SF. Macrophage polarization states in the tumor microenvironment. Int J Mol Sci. 2021;22(13):6995. https://doi.org/10.3390/ijms22136995.
Mosser DM, Hamidzadeh K, Goncalves R. Macrophages and the maintenance of homeostasis. Cell Mol Immunol. 2021;18(3):579–87. https://doi.org/10.1038/s41423-020-00541-3.
Cassetta L, Pollard JW. Targeting macrophages: therapeutic approaches in cancer. Nat Rev Drug Discov. 2018;17(12):887–904. https://doi.org/10.1038/nrd.2018.169.
Genard G, Lucas S, Michiels C. Reprogramming of tumor-associated macrophages with anticancer therapies: radiotherapy versus chemo- and immunotherapies. Front Immunol. 2017;8:828. https://doi.org/10.3389/fimmu.2017.00828.
Klug F, Prakash H, Huber PE, Seibel T, Bender N, Halama N, et al. Low-dose irradiation programs macrophage differentiation to an iNOS(+)/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell. 2013;24(5):589–602. https://doi.org/10.1016/j.ccr.2013.09.014.
He XY, Liu BY, Wu JL, Ai SL, Zhuo RX, Cheng SX. A dual macrophage targeting nanovector for delivery of oligodeoxynucleotides to overcome cancer-associated immunosuppression. ACS Appl Mater Interfaces. 2017;9(49):42566–76. https://doi.org/10.1021/acsami.7b13594.
Stanczak MA, Rodrigues Mantuano N, Kirchhammer N, Sanin DE, Jacob F, Coelho R, et al. Targeting cancer glycosylation repolarizes tumor-associated macrophages allowing effective immune checkpoint blockade. Sci Transl Med. 2022;14(669):eabj1270. https://doi.org/10.1126/scitranslmed.abj1270.
Nie W, Wu G, Zhang J, Huang LL, Ding J, Jiang A, et al. Responsive exosome nano-bioconjugates for synergistic cancer therapy. Angew Chem Int Ed Engl. 2020;59(5):2018–22. https://doi.org/10.1002/anie.201912524.
Deng G, Sun Z, Li S, Peng X, Li W, Zhou L, et al. Cell-membrane immunotherapy based on natural killer cell membrane coated nanoparticles for the effective inhibition of primary and abscopal tumor growth. ACS Nano. 2018;12(12):12096–108. https://doi.org/10.1021/acsnano.8b05292.
Wang Y, Zhao C, Liu Y, Wang C, Jiang H, Hu Y, et al. Recent advances of tumor therapy based on the CD47-SIRPalpha axis. Mol Pharm. 2022;19(5):1273–93. https://doi.org/10.1021/acs.molpharmaceut.2c00073.
Willingham SB, Volkmer JP, Gentles AJ, Sahoo D, Dalerba P, Mitra SS, et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. P Natl Acad Sci USA. 2012;109(17):6662–7. https://doi.org/10.1073/pnas.1121623109.
Tseng D, Volkmer JP, Willingham SB, Contreras-Trujillo H, Fathman JW, Fernhoff NB, et al. Anti-CD47 antibody-mediated phagocytosis of cancer by macrophages primes an effective antitumor T-cell response. Proc Natl Acad Sci U S A. 2013;110(27):11103–8. https://doi.org/10.1073/pnas.1305569110.
Kurdi AT, Glavey SV, Bezman NA, Jhatakia A, Guerriero JL, Manier S, et al. Antibody-dependent cellular phagocytosis by macrophages is a novel mechanism of action of elotuzumab. Mol Cancer Ther. 2018;17(7):1454–63. https://doi.org/10.1158/1535-7163.Mct-17-0998.
Mantovani A, Caprioli V, Gritti P, Spreafico F. Human mature macrophages mediate antibody-dependent cellular cytotoxicity on tumour cells. Transplantation. 1977;24(4):291–3. https://doi.org/10.1097/00007890-197710000-00010.
Zhang WT, Huang QH, Xiao WW, Zhao Y, Pi J, Xu H, et al. Advances in anti-tumor treatments targeting the CD47/SIRP alpha axis. Front Immunol. 2020;11:18. https://doi.org/10.3389/fimmu.2020.00018.
Bouwstra R, van Meerten T, Bremer E. CD47-SIRP alpha blocking-based immunotherapy: current and prospective therapeutic strategies. Clin Transl Med. 2022;12(8):e943. https://doi.org/10.1002/ctm2.943.
Koenig KL, Borate U. New investigational combinations for higher-risk MDS. Hematol-Am Soc Hemat. 2022;1:368–74. https://doi.org/10.1182/hematology.2022000351.
Cabrales P, Carter C, Oronsky B, Reid T. Rrx-001 is a phase 3 small molecule dual inhibitor of CD47 and Sirp alpha with activity in multiple myeloma. Blood. 2018;132:5623. https://doi.org/10.1182/blood-2018-99-116947.
Huang Y, Ma Y, Gao P, Yao Z. Targeting CD47: the achievements and concerns of current studies on cancer immunotherapy. J Thorac Dis. 2017;9(2):E168–E74. https://doi.org/10.21037/jtd.2017.02.30.
Chen Q, Wang C, Zhang X, Chen G, Hu Q, Li H, et al. In situ sprayed bioresponsive immunotherapeutic gel for post-surgical cancer treatment. Nat Nanotechnol. 2019;14(1):89–97. https://doi.org/10.1038/s41565-018-0319-4.
Wang F, Huang Q, Su H, Sun M, Wang Z, Chen Z, et al. Self-assembling paclitaxel-mediated stimulation of tumor-associated macrophages for postoperative treatment of glioblastoma. Proc Natl Acad Sci U S A. 2023;120(18):e2204621120. https://doi.org/10.1073/pnas.2204621120.
Kulkarni A, Chandrasekar V, Natarajan SK, Ramesh A, Pandey P, Nirgud J, et al. A designer self-assembled supramolecule amplifies macrophage immune responses against aggressive cancer. Nat Biomed Eng. 2018;2(8):589–99. https://doi.org/10.1038/s41551-018-0254-6.
Feng QQ, Ma XT, Cheng KM, Liu GN, Li Y, Yue YL, et al. Engineered bacterial outer membrane vesicles as controllable two-way adaptors to activate macrophage phagocytosis for improved tumor immunotherapy. Adv Mater. 2022;34(40):e2206200. https://doi.org/10.1002/adma.202206200.
Wen J, Wang S, Guo R, Liu D. CSF1R inhibitors are emerging immunotherapeutic drugs for cancer treatment. Eur J Med Chem. 2023;245(Pt 1):114884. https://doi.org/10.1016/j.ejmech.2022.114884.
Mehrotra P, Ravichandran KS. Drugging the efferocytosis process: concepts and opportunities. Nat Rev Drug Discov. 2022;21(8):601–20. https://doi.org/10.1038/s41573-022-00470-y.
Bae SH, Kim JH, Park TH, Lee K, Lee BI, Jang H. BMS794833 inhibits macrophage efferocytosis by directly binding to MERTK and inhibiting its activity. Exp Mol Med. 2022;54(9):1450–60. https://doi.org/10.1038/s12276-022-00840-x.
Zhuang WR, Wang Y, Nie W, Lei Y, Liang C, He J, et al. Bacterial outer membrane vesicle based versatile nanosystem boosts the efferocytosis blockade triggered tumor-specific immunity. Nat Commun. 2023;14(1):1675. https://doi.org/10.1038/s41467-023-37369-0.
Komohara Y, Niino D, Ohnishi K, Ohshima K, Takeya M. Role of tumor-associated macrophages in hematological malignancies. Pathol Int. 2015;65(4):170–6. https://doi.org/10.1111/pin.12259.
Saito Y, Komohara Y, Niino D, Horlad H, Ohnishi K, Takeya H, et al. Role of CD204-positive tumor-associated macrophages in adult T-cell leukemia/lymphoMa. J Clin Exp Hematop. 2014;54(1):59–65. https://doi.org/10.3960/jslrt.54.59.
Komohara Y, Fujiwara Y, Ohnishi K, Shiraishi D, Takeya M. Contribution of macrophage polarization to metabolic diseases. J Atheroscler Thromb. 2016;23(1):10–7. https://doi.org/10.5551/jat.32359.
Cai H, Zhang Y, Wang J, Gu J. Defects in macrophage reprogramming in cancer therapy: the negative impact of PD-L1/PD-1. Front Immunol. 2021;12:690869. https://doi.org/10.3389/fimmu.2021.690869.
Su S, Zhao J, Xing Y, Zhang X, Liu J, Ouyang Q, et al. Immune checkpoint inhibition overcomes ADCP-induced immunosuppression by macrophages. Cell. 2018;175(2):442–57 e23. https://doi.org/10.1016/j.cell.2018.09.007.
Acknowledgements
Q. H. acknowledges the start-up package support from the University of Wisconsin-Madison.
Author information
Authors and Affiliations
Contributions
Writing: Y.W. and A.B.
Funding acquisition: Q.H.
Supervision: Q.H.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Additional information
Responsible Editor: Aliasger Salem
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Wang, Y., Barrett, A. & Hu, Q. Targeting Macrophages for Tumor Therapy. AAPS J 25, 80 (2023). https://doi.org/10.1208/s12248-023-00845-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1208/s12248-023-00845-y