
Abstract
It has been widely known that macrophages play critical roles during infection and immune responses. While as tumor-infiltrating myeloid cells, macrophages accumulated in the tumor microenvironment (TME) could either suppress tumor proliferation through producing pro-inflammatory cytokines or promote tumor growth via activating tumor proliferation, metastasis, and angiogenesis, tumor-associated macrophages (TAMs), the most widely infiltrating macrophages in the TME contributing to immunosuppressive microenvironment establishment, could promote tumor invasion and weaken conventional therapeutic effects. Thus, targeting TAMs is becoming a promising strategy for cancer therapy. Here we will discuss the role of TAMs and their crosstalk with tumor in TME, including immune suppression, tumor progression, tumor metastasis, and angiogenesis. In addition, the role of TAMs in cancer therapy, including chemotherapy, radiotherapy, and immunotherapy, will be presented. Next, TAMs as a promising therapeutic target for cancer treatment will be illustrated, including targeting recruitment and localization of TAMs, targeting TAMs inhibition, and targeting TAMs reprogramming. Finally, the potential of TAMs as drug delivery systems will also be summarized. The chapter highlighted TAMs as promising therapeutic targets of cancer, which might be useful for drug development and evaluation of targeting TAMs for cancer therapy.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Macrophages are white blood cells of the innate immune system that can reside in all tissues of the body and contribute to the development, tissue homeostasis, diseases, and tumor microenvironment (TME). They are given different names based upon their locations, for example, microglia cell in brain, Langerhans cells in skin, Osteoclasts in bone, alveolar macrophages in lung, and Kupffer cells in liver (Epelman et al. 2014; Ginhoux and Guilliams 2016). Tissue-resident macrophages are derived from at least three distinct sources: the yolk sac, the fetal liver, and the bone marrow (Guerriero 2018). Macrophages-mediated innate immunity plays a crucial role in maintaining tissue homeostasis by engulfing and digesting invading pathogens, bacteria, damaged tissue, and malignant cells (Haniffa et al. 2015; Yona and Gordon 2015; Guerriero 2019). They also contribute to specific adaptive immune responses of T lymphocytes activation by antigen presentation. Besides, macrophages can modulate immune system through the secretion of various cytokines, chemokines, and the activation of complement system (Guerriero 2019). Macrophages can be characterized in vitro into two types: classically activated M1 macrophages and alternatively activated M2 macrophages. While M1 type macrophages are induced by TH1 cytokines such as INF-γ, M2 type macrophages are induced by the TH2 cytokines such as IL-4/IL-13. Macrophages represent up to 50% of leukocytes in the TME. Clinical data revealed that the poor prognosis is associated with the abundance of tumor-associated macrophages (TAMs) in 80% of human cancer. It was originally thought the macrophages were recruited to malignant sites to kill the cancer cells. However, accumulating evidence demonstrated that tumor cells recruit macrophages to promote tumor malignancy and progression, like macrophages are recruited to a wound site to assist in healing. In this book chapter, we will discuss the role of TAMs and their interplay and crosstalk with T effector cells and tumor cells in TME, including immune suppression, tumor progression, tumor metastasis, and angiogenesis. In addition, the role of TAMs in cancer therapy, including chemotherapy, radiotherapy, and immunotherapy, will be presented. Moreover, TAMs as a therapeutic target for cancer treatment will be illustrated, including the recruitment of TAMs, the signaling pathways involved in TAMs activation, and the production of cytokines and chemokines that are involved in tumor angiogenesis. Then current strategies targeting TAMs for cancer therapy including targeting TAMs inhibition, recruitment, localization, and reprogram will be discussed. Lastly, the potential of TAMs as drug delivery systems will also be summarized.
2 Macrophages and Tumor-Associated Macrophages (TAMs)
Macrophages, the major phagocytic cells in immune system, are derived and matured from monocytes that leave from the circulation system and settle in spleen, lymph nodes, alveoli, and tonsils. Macrophages are also existing in the brain as microglia, in the skin as Langerhans cells, in bone as osteoclasts, and in the liver as Kupffer cells. These tissue-resident macrophages originate from at least three embryonic sources: erythro-myeloid progenitors in the yolk sac and in the fetal liver and macrophage/dendritic cell progenitor cells in the bone marrow that give rise to monocytes (Davies et al. 2013). Tissue-resident macrophages are primarily derived from both the yolk sac and the fetal liver. They reach their tissues during embryonic development, progressing to adulthood, and sustain through proliferation independent of hematopoietic stem cells. Borrow marrow-derived macrophages have a short life span and are only generated in the tissue under inflammatory conditions (Guerriero 2018). In cases of infection, macrophages, through their receptors on the surface, are able to undergo phagocytosis, the process of ingesting of foreign antigens by lysosomes, such as small particles, whole cells, and bacteria. In addition, macrophages in the peripheral lymphoid tissues serve as the major scavengers of abnormal cells and cellular debris. Importantly, macrophages present a vital role in processing antigen to T lymphocytes to activate specific adaptive immune responses. Activated macrophages can release many inflammatory mediators, such as cytokines, chemokines, enzymes, reactive oxidative species, coagulation factors, and growth factors (Ginhoux and Jung 2014; Olingy et al. 2019; Varol et al. 2015). All macrophages express colony-stimulating factor-1 receptor (CSF-1R), which binds to CSF-1 or alternatively to the interleukin IL-34 and regulates macrophage differentiation, proliferation, and survival. The presence and functional states of macrophages are mainly regulated through by CSF-1, granulocyte macrophage CSF (CSF2/GM-CSF), and chemokines.
The activation status of macrophages can be classified into two subpopulations: M1 and M2 macrophages. M1 (also known as classically activated) macrophages, induced by TH1-type cytokines such as INF-γ and GM-CSF and through Toll-like receptor 4 (TLR4) engagement of lipopolysaccharides (LPS) from gram negative bacteria, give rise to pro-inflammatory, antiviral, antibacterial, antitumoral phenotypes with powerful killing effects on invading pathogens and at the same time destructive effects on normal tissues (Fleetwood et al. 2007; Arnold et al. 2014). Activated macrophages are potent effector cells that can kill tumor cells and microorganisms, trigger massive proinflammatory cytokines production, and activate cytotoxic T lymphocytes. M2 (also known as alternatively activated) macrophages are stimulated by the TH2 cytokines IL-4/IL-13, IL-10, CSF-1 and play important roles in anti-inflammatory humoral responses and pro-repair, pro-tumoral, and antiparasitic phenotypes (Davies et al. 2013; Murray et al. 2014). Subgroups of M2 macrophages were divided into M2a (IL-4), M2b (INFγ + complexed immunoglobulin (Ig)), and M2c (dexamethasone) (Szulzewsky et al. 2015). The nomenclature of M1 or M2 macrophages describes two extremes states of macrophage activation and functions that have direct in vitro relevance during the infection of bacteria or parasites. In vivo, macrophages are not clearly divided into M1 and M2 classification. Indeed, a study demonstrated that over 60 percent of the upregulated genes in TAMs from brain tumors had no overlap with that from M1 or M2 (M2a, M2b, or M2c) ex vivo macrophages phenotypes, suggesting macrophages activated in TME may not be reflected by ex vivo stimulation of monocytes (Hambardzumyan et al. 2016). Recent studies revealed that there are populations of CD169+ and TCR+ macrophages in vivo, which cannot simply be described as M1 or M2 term and seem to play roles in maintaining homeostasis, immune regulation, and tolerance (Chavez-Galan et al. 2015; Crocker and Gordon 1986; Martinez-Pomares et al. 1996; Martinez-Pomares and Gordon 2012). TCRb gene was discovered rearrangement in the early stage of bone marrow macrophages differentiation. TCR+ macrophages express chemokine (C-C motif) ligand 2 (CCL2) with strong phagocytic functions, which are different from traditional macrophages (Kaminski et al. 2013). It was suggested to move away from the ambiguity of the M1-M2 characterization of TAMs and to define TAMs based on functional, transcriptional, or epigenetic status, which will demonstrate more clear characterization of TAMs (Guerriero 2018).
TAMs are usually referred to as macrophages recruited from circulating monocytes to tumors and influenced by the presence of cancer to promote tumor malignancy, survival, proliferation, angiogenesis, metastatic dissemination, and chemoresistance (Solinas et al. 2009; Qian and Pollard 2010; Kitamura et al. 2017). Rather than a homogenous population, TAMs actually can originate from different sources and exhibit either pro-tumoral or sometimes antitumoral roles. Each population has a unique transcriptional landscape based on the type, the stage, and the immune composition of the tumors they infiltrate. M2 macrophages are defined as TAMs in a narrow sense because active TAMs have various properties similar to M2 macrophages (Murray et al. 2014; Chavez-Galan et al. 2015). The TME consists of variouse cell types, such as tumor cells, granulocytes, macrophages (~50%), mast cells, fibroblast and epithelial cells. Secreted cytokines and chemokines, such as CCL2, CCL11, CCL16, and CCL21, are major determinants for the infiltration of macrophages. Increasing evidence demonstrated that tumor cells recruit macrophages to support tumor growth. Clinical data indicated that poor prognosis in over 80% of human tumor is associated with increased TAMs, which not only lack the phagocytic functions of tumor cells, also promote tumor cells dissemination. High density of TAMs in tumor has been associated with increased vascular density, chemotherapy resistance, and worse outcome in various cancer types, such as colorectal, breast, ovarian, nonsmall cell lung cancer, melanoma, Hodgkin’s lymphoma, and multiple myeloma (Guerriero 2018).
3 TAMs and Cancers
3.1 Crosstalk of TAMs and Cancer Cells in TME
Various molecular mechanisms have been documented to play roles in mediating cancer cells that escape form the attacks of macrophages. Programmed cell death protein (PD-1) belongs to CD28 superfamily and plays a significant role in immunosuppression on T effector cells and TAMs. PD-L1 on the surface of tumor cells binding to PD-1 on effector T-cells and macrophages helps tumor cells escape from the attacks of T effector cells as well as TAMs by inhibiting cytokine expression, activation, and proliferation of effector T-cells, and macrophage phagocytosis (Boussiotis et al. 2014; Yu et al. 2015; Gordon et al. 2017; Katsuya et al. 2016). The cluster of differentiation 47 (CD47) molecule, the “do-not-eat-me” signal, is recently characterized as a self-molecule that protects host cells from destruction by macrophages (Jaiswal et al. 2009; Zhao et al. 2016). CD47 expressed on the membrane of tumor cells binding to signal regulatory protein alpha1 (SIRP1α) on macrophages will inhibit phagocytosis by blocking accumulation of myosin IIA at the phagocytic synapse (Okazawa et al. 2005; Barclay and Van den Berg 2014). Antibody-mediated inhibition of human CD47 enhances macrophage-mediated phagocytosis (Tseng et al. 2013). Targeting CD47/SIRP1α signaling pathway is currently being tested in clinical trials (Pathria et al. 2019; Chao et al. 2012). In breast and ovarian cancer, CD24 was found as a dominant innate immune checkpoint, which could enhance tumor escape by providing “do-not-eat-me” signal through the interaction with inhibitory receptor sialic-acid-binding Ig-like lectin 10 (Siglec-10) expressed on TAMs. Suppressing the crosstalk between CD24 and Siglec-10 with respective antibodies or ablating the genes of CD24 or Siglec-10 can obviously promote phagocytic function of TAMs to tumors with CD24 expression (Barkal et al. 2019). Recent study revealed another recognition mechanism that can deter phagocytotic effects of macrophages on tumor cells, which is the interaction between leukocyte immunoglobulin like receptor subfamily B member 1 (LILRB1) on macrophages and major histocompatibility complex (MHC) class I component β2-microglobulin on tumor cells. Either blocking β2-microglobulin or anti-LILRB1 antibody can significantly enhance macrophage phagocytosis and inhibit tumor growth (Barkal et al. 2018). In TME, exosomes were recently discovered as important carriers for proteins, nucleic acid, which can affect survival, growth, and metastasis of cancer cells (Milane et al. 2015; Syn et al. 2016). A recent study found that TAMs characterized as M2-polarized phenotype can secrete exosomes and promote the metastasis of gastric cancer. Apolipoprotein E (ApoE) was identified richly in those exosomes, which can activate phosphatidylinositol 3-kinases (PI3k)-Akt signaling pathway to drive epithelial mesenchymal transition (EMT) and cytoskeleton rearrangement of cancer cells (Zheng et al. 2018). Another study found that TAMs-derived exosomes promote the resistance of PDAC to gemcitabine by transferring miR-365 into cancer cells (Binenbaum et al. 2018). Exploring the roles of macrophage-derived exosomes and their components in cancer development and metastasis will open a new door for the discovery of cancer treatment. Recent study revealed that complement-mediated inflammation could promote tumorigenic progression (Medler et al. 2018). In a mouse squamous cell carcinomas (SCC) model, TAMs-triggered plasminogen produces C5a that is independent of C3 activation. Activation of C5aR by C5a in macrophages and mast cells leads to immunosuppressive macrophages that can inhibit CD8+ T-cell activation (Medler et al. 2018). Combined therapy of C5aR antagonist PMX-53 and paclitaxel significantly inhibits tumor growth as compared to PMX-53 or paclitaxel alone. Moreover, inhibition of C5aR signaling with PMA-53 significantly enhances antitumor efficacy of checkpoint inhibitor anti-PD-L1 antibody, indicating anticomplement therapy might be beneficial for cancer immune therapy (Affara et al. 2014; Zha et al. 2017).
3.2 TAMs and Tumor Progression
It was originally considered that macrophages were recruited to tumor sites to kill the cancer cells. However, accumulating evidence has revealed that TAMs can cause tumor progression and development through secretions of various chemokines and cytokines, such as interleukin-6 (IL-6), interleukin-8 (IL-8), and interleukin-10 (IL-10) (Zhou et al. 2020). IL-6, secreted from tumor-associated endothelial cells and TAMs, has been involved in carcinogenesis and progression of tumors by regulating local inflammation, cell cycle, tumor angiogenesis, and stem cell self-renewal. IL-6 mediates the regulation of signal transducer and activator of transcription 3 (STAT3) phosphorylation, which leads to EMT, the overexpression of snail and Bcl-2, and antiapoptotic effect in cancers (Gao et al. 2018; Yadav et al. 2011). IL-8, highly secreted by TAMs, is involved in angiogenesis, tumor metastasis, and suppression of immunity (Williams et al. 2016). Monitoring IL-8 level can predict the outcome of immunotherapy of checkpoint blockade (Sanmamed et al. 2017). Exogenous IL-8 causes the activation of JAK2/STAT3/snail signaling pathway by enhancing the expression of p-JAK2 and p-STAT3, further promoting EMT and carcinogenesis (Deng et al. 2013). IL-8 and chemokines, strongly expressed in inflammatory breast cancer (IBC), promote macrophages recruitment and transforming to M2 macrophages, which can secrete high level of IL-8 and chemokines, leading to a feed-forward cytokine/chemokine loop that drives the EMT of IBC (Valeta-Magara et al. 2019). In addition, TAMs also secrete IL-10, transforming growth factor-β (TGF-β), and inflammatory mediators, such as prostaglandin E2 (PGE2) and matrix metalloprotease-7 (MMP-7), which can inhibit antigen-presenting process for T-cell activation. During chronic inflammation, toll-like receptor 4 (TLR4) stimulates M2 to secrete cytokine IL-10 and activation of TLR4 by LPS significantly enhanced the EMT of pancreatic cancer cells (Deng et al. 2013). IL-10 also increases the expression of inhibitor of CIP2A via the PI3K signaling pathway and promotes tumor aggressiveness in lung adenocarcinoma (Sung et al. 2013). IL-10 plasma level showed significant correlation with tumor progression, indicating IL-10 plays a critical role in tumor progression (Sato et al. 2011).
3.3 TAMs and Tumor Metastasis
The underlying mechanisms and processes of cancer metastasis are very complex. The cancer cells start from local invasion, intravasation, and ultimate extravasation at peripheral sites. The disseminated cancer cells need to escape the attack from immune system, survive in the blood or lymphatic circulation, settle down at a distant site, and proliferate in the new hostile environment (Ruffell and Coussens 2015; Cassetta and Pollard 2018). Macrophages have been implicated in all stages of the metastatic processes. High infiltration of macrophages, overexpression of CSF-1, and their chemoattractant CCL2 in human solid tumors are linked to poor clinical prognosis (Cassetta and Pollard 2020). Pollard and his colleagues have demonstrated that in a mouse mammary gland, tumor model genetic depletion of CSF-1 (similar to the depletion of macrophages) can slow the tumor progression and significantly inhibit metastasis, suggesting TAMs play crucial roles in cancer metastasis (Joyce and Pollard 2009). The phenomenon was recapitulated in other preclinical mouse model studies of the development of other cancer types, such as lung cancer, pancreatic cancer, and glioblastoma (Zabuawala et al. 2010; Welm et al. 2007; Rolny et al. 2011; Gocheva et al. 2010; Qian et al. 2011). Metastatic cancer cells recruit bone marrow-derived classical monocytes through CCL2-CCR2 signal mechanism (Qian et al. 2011). These extravasated monocytes differentiate into metastasis-associated macrophages (MAMs) through involvement of chemokine CCL3-CCR1 autocrine signaling (Mummalaneni et al. 2014; Kitamura et al. 2015). MAMs were characterized to express the markers CD11b, VEGF receptor 1, CXCR3, and CCR2, which are different from resident macrophages and MAM precursor cells. MAMs mediate cancer cells metastasis through promoting extravasation and tumor growth, as well as inhibiting cytotoxic T-cells function. Macrophages open up a gate for the cancer cells to escape when arriving at the circulating vessels. In addition, macrophages produce other molecules that help cancer cell invasion (Sangaletti et al. 2008). Cathepsin proteases can remodel the matrix and release sequestered growth factors and TGFβ generated from MAMs that drive epithelial to EMT of cancer cells (Laoui et al. 2011; Quail and Joyce 2013; Bonde et al. 2012). Macrophages in the lung interact with metastatic cancer cells and support their survival through vascular cell adhesion protein 1 (VCAM1)-dependent and Akt-dependent mechanism. Moreover, MAMs crosstalk to metastatic cancer cells to retain MAMs in the metastatic foci, which can further support cancer growth (Kitamura et al. 2015). TAMs inhibit the function of cytotoxic T-cells, which might be one of the mechanisms that help cancer cells escape from attack of T-cells and promote cancer metastasis (Kitamura et al. 2017). TAMs can directly inhibit T-cell cytotoxicity through the depletion of L-arginine via release of arginase 1, which is essential for the re-expression of the T-cell receptor (TCR) after antigen engagement on T-cells. In addition, TAMs generate cytokines such as IL-10 and TGFβ that lead to immunosuppressive microenvironment by inhibiting CD4+ and CD8+ T-cells and inducing Treg cells expansion in the TME. TAM-mediated release of chemokines, like CCL2, CCL3, CCL4, CCL5, and CCL20, contributes to the recruitment of Treg cells in the TME (Noy and Pollard 2014). Moreover, TAM-induced immune suppression is caused by the expression of inhibitory receptors, such as nonclassical major histocompatibility complex class I (MHC-I) molecules such as HLA-E and HLA-G, which can inhibit activation of NK cells and T-cells through the interaction with CD94 and leukocyte immunoglobulin-like receptor subfamily B member 1 (LIR1), respectively (Morandi and Pistoia 2014). The expression of T-cell immune checkpoint ligands by TAMs, such as PDL1, PDL2, B7–1, and B7–2, can directly inhibit T-cell functions (Santarpia and Karachaliou 2015; Buchbinder and Desai 2016). It is of great interest to target TAMs for anticancer metastasis therapy since both preclinical and clinical data have established the proof of principle.
3.4 TAMs and Angiogenesis
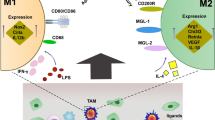
Angiogenesis is a key event in tumor growth and progression. Cancerous tissues are characterized by hypoxia and unequal vascularization, which actually can modify macrophage distribution and function. TAMs accumulate preferentially in the inadequately vascularization areas of tumors, which have low oxygen (Mantovani et al. 2002). Under hypoxic condition, macrophage migration is suppressed and TAMs are immobilized in nonvascular, necrotic, hypoxic regions of tumor, from which macrophages cooperate with cancer cells to promote angiogenesis (Grimshaw and Balkwill 2001; Leek et al. 1996, 1999; Lewis et al. 2000). In breast cancer, TAMs overexpress hypoxia-inducible factor-2α (HIF-2α) and HIF-1, which are transcription factors to induce the secretion of pro-angiogenic factors, such as endothelial growth factor VEGF, bFGF, and chemokines CXCL8 and CXCL12 to promote angiogenesis (Talks et al. 2000; Crowther et al. 2001; Lin and Pollard 2007; Hughes et al. 2015). Other factors such as TGFβ, WNT7B, TNF, and thymidine phosphorylase also contribute to angiogenic process by recruitment and activation of endothelial cells or other cells like fibroblast or pericytes that support the generation of vascular networks in microenvironment (Cassetta and Pollard 2018; Yeo et al. 2014; Hirano et al. 2001). A subpopulation of TAMs with the expression of the angiopoietin 1 receptor (TIE2) was identified to play essential roles in tumor angiogenesis. Depletion of these TAMs suppresses tumor growth and metastasis (De Palma et al. 2005; Mazzieri et al. 2011) (Fig. 1).

The role of tumor-associated macrophages (TAMs) in promoting tumor progression, tumor angiogenesis, tumor metastasis, and T-cell immunosuppression. Multiple mechanisms of molecular interactions (CSF-1/CSF-1R, VEGF/VEGFR1, CCL12/CXCR4, CCL2/CCR2, CCL3/CCR1, PD-1/PD-L1, LILRB1/MHC-I, Siglec-10/CD24, SIRPα/CD47, C5a/C5aR, and CD94/LIR1) and secreted growth factors (VEGF, EGF, and bFGF), cytokines (CSF-1, IL-6, IL-8, IL-10, TNFα, TGFβ), chemokines (CXCL8, CXCL12, and CCL22), and other mediators (TIE2, PGE2, MMP-7, exosome, thymidine phosphorylase) are involved in TAMs-mediated different stages of tumor progression and T-cell immunosuppression
4 TAMs in Cancer Therapy
TAMs are the dominant cell populations recruited into the TME and are shaped by cancer cells to present M2 properties. The considerable functional plasticity enables TAMs to rapidly adapt to microenvironmental perturbations from various cancer treatment modalities (ranging from radiotherapy, chemotherapy, targeted therapy to immunotherapy) in TME, and clinical consequences of such interactions between TAMs and cancer therapies vary across different cancer types and different therapeutic categories (Vitale et al. 2019; De Palma and Lewis 2013; Larionova et al. 2019; Neophytou et al. 2020). In some cases, therapeutic agents may achieve their pharmacological effects via the function of TAMs. For example, accumulating evidence indicated that metformin suppresses tumorigenesis, cancer cell proliferation, and tumor metastasis by modulating functional states of TAMs. Metformin induced the expression of M1-related cytokines IL-12 and TNF-α and reduced M2-related cytokines IL-8, IL-10, and TGF-β, to revert the polarization of TAMs to M2 phenotype by cancer cells and thus may recover tumor immune surveillance (Ding et al. 2015; Chiang et al. 2017; Ma et al. 2020; de Oliveira et al. 2019). However, in other cases, TAMs may hamper efficacy of drug treatment by forming an immunosuppressive TME.
4.1 TAMs in Radiotherapy
Radiotherapy is the most commonly used modality for clinical treatment of solid tumors. In addition to its direct cell-killing effects, radiotherapy profoundly modified TME, and especially, TAMs are usually reshaped by radiations to exhibit bidirectional consequences (pro-inflammatory or pro-tumorigenic phenotypes) depending on the radiation doses and cancer types (Mantovani and Allavena 2015). On the one hand, immunogenic cell death induced by radiation could activate innate and adaptive immune systems to trigger antitumor immune responses where TAMs are closely involved in (Golden et al. 2014). For example, local low-dose gamma irradiation programs TAMs to iNOS+ M1 state, which subsequently promotes the recruitment of tumor-specific T-cells to TME and then evokes T-cell immune response (Klug et al. 2013; Prakash et al. 2016). On the other hand, accumulating evidence has demonstrated that TAMs mediated radio-resistance in multiple cancers (Wu et al. 2017). Radiotherapy may polarize TAMs to their M2 state to allow immune escape and compromised antitumor effects. For instance, an in vivo study revealed that radiation at 3 Gy in prostate cancer upregulated CSF1 expression on cancer cells, which in turn recruited TAMs (express CSF-1R) to TME and then promoted tumor regrowth (Xu et al. 2013). In glioblastoma, irradiation increased the number of M2 TAMs by upregulating CSF-1R expression, which finally lowers the tumor sensitivity to radiotherapy (Stafford et al. 2016). Bone marrow-derived macrophages (BMDMφ) promoted tumor growth after ionizing radiation at 20 Gy. The underlying mechanism involved in the upregulation of VEGF expression resulted from autocrine or paracrine of TNFα in BMDMφ cells, and removing TNFα could re-sensitize tumors to ionizing radiation (Meng et al. 2010). Recent data demonstrated that fibroblast growth factor 2 (FGF2) is critical for the transition of TAMs toward M2 phenotype after radiotherapy, and blocking FGF2 increased percentage of M1-like TAMs, extended tumor growth delay, and prolonged long-term survival in mouse model (Im et al. 2020). In addition, the FGF2 receptor (FGF2R) has been reported to mediate radio-resistance in glioblastoma (Gouaze-Andersson et al. 2016). Those evidence indicated that reverting the M2 polarization either by direct depletion of TAMs or by targeting abovementioned key players (CSF-1/CSF-1R, TNFα, FGF2/FGF2R) represents a promising strategy for combinatory radiotherapy to improve clinical efficacy.
4.2 TAMs in Chemotherapy
The interactions between TAMs and chemotherapy have been widely cataloged to be involved in variations of clinical efficacy of a wide range of cancer therapeutics among patients (Larionova et al. 2019). Cancer cells can reshape cellular metabolism and thus shift the phenotype of TAMs to protect themselves from drug exposure. For example, TAMs in M2 state have been well documented to be involved in chemoresistance in the treatment of pancreatic ductal adenocarcinoma (PDA) and CCR2 and CSF-1R played key roles in driving chemoresistance (Mitchem et al. 2013). Metabolomics profiling of TAMs and PDA cells revealed the underlying mechanism that TAMs promote pyrimidine biosynthesis from glucose and release deoxycytidine molecules to interact with PDA cells and finally drive the gemcitabine resistance, and blocking deoxycytidine release by simply limiting glucose supply could sharply sensitize PDA cells to gemcitabine exposure, providing novel mechanism of gemcitabine resistance from an intra-tumoral metabolic crosstalk perspective (Halbrook et al. 2019). Further retrospective analysis in this study indicated that lower TAMs in microenvironment is a favorable factor for better drug response and improved disease-specific survival outcome of PDA patients. As deoxycytidine release was also observed in a panel of cancer cell lines and patient-derived cancer cells , release of pyrimidine molecules by TAMs may drive chemoresistance in other cancers.
Moreover, TAMs were revealed in vitro and in vivo to mediate chemotherapy resistance by enhancing survival factor activities or activating antiapoptotic programs in cancer cells (Correia and Bissell 2012; Castells et al. 2012; DeNardo et al. 2011). For instance, TAMs suppressed cytotoxic effects of Taxol, a widely used antimitotic agent, on multiple cancer cell lines and on in vivo model models (Olson et al. 2017). TAMs in M2 state are able to release nitric oxide (NO) to protect cancer cell from apoptosis induced by cisplatin, leading to resistance in several cancer cell lines (Perrotta et al. 2018). Coculture studies in vitro using macrophages derived from bone marrow (BM) with mammary carcinoma cell lines demonstrated that macrophages confer to chemotherapy resistance to paclitaxel, doxorubicin, and etoposide and to gemcitabine in PDA cells (Mitchem et al. 2013; Shree et al. 2011). The activation of signal transducer and activator of transcription 3 (STAT3) in TAMs has been shown to promote PDAC cancer cells proliferation and survival, resistance to carboplatin in vivo, and tumor cell growth when synergized with IL-6 and other macrophage-derived factors such as milk fat globule-epidermal growth factor VIII (Jinushi et al. 2011; Taniguchi and Karin 2014; Yu et al. 2014). IL-6 can be generated by co-culture of BM-derived macrophages with neoplastic cells as well as from TAMs in vivo (DeNardo et al. 2009; Movahedi et al. 2010; Song et al. 2009). IL-1β can also induce IL-6 production in monocytes and osteoblasts (Mori et al. 2011; Tosato and Jones 1990). In vivo evidence of IL-6 being chemoresistance was shown in a murine lymphoma model (Gilbert and Hemann 2010). Nevertheless, the source and relevance of IL-6 during chemotherapy for solid tumor remains partly described. In a subcutaneous MCF-7 breast cancer xenografts animal model, anti-CSF-1 neutralizing antibody can increase of chemosensitivity (Paulus et al. 2006). Soluble chemoprotective factors generated from TAMs depend on protease activity of cathepsin B and S. In vivo inhibition of cathepsin protease activity enhances the response of mammary carcinomas to paclitaxel (Shree et al. 2011). Macrophages are main source of tumor necrosis factor (TNF), which may be one of crucial factors mediating chemoresistance either directly through NF-κB activation of indirectly through induced IL-6 expression (Mori et al. 2011; Li and Sethi 2010). TNFα has been reported to mediate chemoresistance to MAPK inhibitors in melanoma through NF-κB-dependent expression of microphthalmia transcription factor (Smith et al. 2014). Taken together, it has a strong rationale to target TAMs that contribute chemoresistance for anticancer therapy.
4.3 TAMs in Cancer Immunotherapy
T-cell-based immunotherapies , such as adoptive T-cell therapy and immune checkpoint blockages (ICBs), are changing the traditional paradigm of cancer treatment in clinical practice (Galon and Bruni 2019). However, only a small fraction of cancer patients can benefit from those therapies. As the dominant immune cell population in TME, TAMs actively interact with other immune cells to interfere antitumor immunity derived from T-cell based immunotherapies, accounting for an important mechanism of immunotherapy failure in primary resistant patients (Li et al. 2019a). T-cell-based immunotherapies exert antitumor activity by boosting T-cell immunity in TME, while T-cells are closely influenced by TAMs. TAMs could not only inhibit T-cell function by suppressing naïve T-cell proliferation, but also inhibit cytotoxic T-cell responses via releasing various anti-inflammatory cytokines (e.g., IL-10, TGF-β, PGE2) or leveraging inhibitory receptors (checkpoint molecules on TAMs such as B7-H4 and VISTA), which finally established an immunosuppressive TME and therefore led to compromised clinical outcomes (De Palma and Lewis 2013; Ruffell et al. 2014).
4.4 TAMs Aid Precision Cancer Treatment
As discussed above, TAMs profoundly influence antitumor activities of cancer therapies. This provides a series of insights to develop predictive or prognostic biomarkers for customized therapeutic regimes and to dig into detailed mechanisms for the discovery of novel TAM-related therapeutic targets, which will finally facilitate precision cancer medicine.
High-throughput omics profiling studies proposed the possibility that using the functional landscape of TAMs as predictive biomarker to guide precise patient stratification for improved therapeutic outcomes. In the TRIBE and FIRE3 clinical trial cohorts, gene mutation sequencing analysis demonstrated that mutation features of TAMs regulating genes could robustly predict progression-free survival of patients with metastatic colorectal cancer treated with bevacizumab plus FOLFIRI (Sunakawa et al. 2015). A recent analysis of multiomics profiles of bulk tumors from 348 patients reported that the infiltration level of M1 macrophages significantly correlated with drug response of ICBs in metastatic urothelial cancer (Zeng et al. 2020), indicating that genomics profile that encoded TAMs states may be an effective biomarker for patient selection in ICB therapy. In a phase IV clinical trial study of NSCLC, DNA methylation microarray profiling revealed that nonresponders to ICBs were characterized as those patients who exhibit high enrichment of TAMs, neutrophils, and cancer-associated fibroblasts in TME, highlighting profiles of TAMs as complementary biomarkers in addition to PD-L1 staining score and tumor mutation burden (Duruisseaux et al. 2018). Transcriptomic profiling of monocytes isolated from breast cancer patients and healthy controls identified TAM signatures, which are highly associated with aggressive subtypes and poor survival, and elucidated that SIGLEC1 and CCL8 were key regulators to govern the interactions between TAMs and cancer cells in this scenario, suggesting novel biomarkers and potential therapeutic targets for breast cancer treatment (Cassetta et al. 2019).
Moreover, TAM transcriptomic profiling at single-cell level depicts more precise TAMs landscape and reveals more clues for personalized cancer therapy. Recent years, single cell sequencing technology is reforming our understanding of tumor heterogeneity. Transcriptomic landscape of tumor at single cell resolution is opening new revenue for personalized cancer treatment by providing individualized immune interaction profiles as novel evidence for tailored therapeutics design (Weissleder and Pittet 2020; Qian et al. 2020). Single-cell RNA-seq profiling of patients with lung cancer demonstrated that TAMs can be functionally categorized into up to 10 subtypes in lung cancer and especially, certain subtypes express both M1 and M2 markers (Weissleder and Pittet 2020), recapitulating that TAMs employ a spectrum of functional states rather than the conventional notion that TAMs simply consist of M1 and M2 subtypes to cope with the complexity of TME.
Interactions of TAMs and cancer therapeutics result in multifaceted effects on clinical outcomes of cancer therapies via various elucidated and unexplored molecular mechanisms. This provides promising opportunities to vanish immunosuppressive TME, sensitize resistant cancer cells to cancer treatment modalities, stratify patients to match optimal treatment strategies, and finally benefit cancer patients in clinical practice, by harnessing those interactions either via depleting tumor-promoting TAMs or re-directing them to their tumor-inhibiting phenotype.
5 Target TAMs for Cancer Therapy
TAMs play essential roles during tumor proliferation, angiogenesis, invasion, metastasis, and immunosuppression and interfere with most standard cancer therapy (Pathria et al. 2019). Elevated number of circulating monocytes from blood and massive macrophage infiltration into tumor tissues, especially accumulation of anti-inflammatory TAMs, is tightly associated with worse clinical outcome and resistance to therapy in various cancer types (La Fleur et al. 2020). Thus, manipulating TAMs in conjunction with chemotherapy, radiotherapy, or immunotherapy might improve standard cancer treatment response (Cheng et al. 2020). Moreover, the potential role of macrophages during cancer development has driven novel antitumor therapy development through targeting macrophages (Mantovani et al. 2017; Sawa-Wejksza and Kandefer-Szerszen 2018). Currently, several pharmacological strategies have been proposed either in preclinical study or in clinical trials through inhibiting TAMs recruitment, depleting TAMs, and reprogramming M2 TAMs to M1 TAMs in laboratory tumor models or clinical trials (Fig. 2) (Anfray et al. 2019). Besides, due to their intrinsic homing property, M1 macrophages or their derived components could also be used as drug delivery systems to actively carry payloads to the tumor sites (Guerra et al. 2017). Here the progress regarding TAMs as drug delivery systems, which mainly focusing on M1 macrophages, their derived exosomes, and membrane-coated nanoparticles will be summarized (Miller et al. 2015).

Targeting TAMs for cancer therapy. Currently, three strategies, including 1) targeting TAMs recruitment and localization, 2) targeting TAMs inhibition, and 3) targeting TAMs reprogram, have been proposed in different stage of studies. For targeting TAMs recruitment and localization, four signaling axis, that is, CSF-1/CSF-1R signaling axis, CCL-2/CCR-2 axis, CXCL-12/CXCR-4 axis, and VEGF/VEGFR signaling axis, were highlighted. For targeting TAMs inhibition, two chemical agents Bisphosphonates and Trabectedin and other targets for TAMs-specific surface markers were discussed. Targeting TLR, SIRPα/CD47 axis, CD40, and other strategies were involved in TAMs reprogramming
5.1 Target TAMs Recruitment and Localization for Cancer Therapy
Numerous evidences reveal that recruitment and localization of macrophage in tumors is driven by continuous accumulating monocytes from circulation due to some tumor-derived factors (Qian and Pollard 2010). These factors including colony-stimulating factor-1 (CSF-1), C-C motif chemokine ligand 2 (CCL-2), C-X-C motif chemokine ligand 12 (CXCL-12), and vascular endothelial growth factor (VEGF) have mediated interaction between monocytes and tumors cells; thus, suppressing the monocyte-chemotactic chemokines, cytokines, and their receptors could block the recruitment and localization of TAMs (Argyle and Kitamura 2018; Owen and Mohamadzadeh 2013; Zhang et al. 2020a).
5.1.1 Targeting CSF-1/CSF-1R Signaling
CSF-1/CSF-1 receptor (CSF-1R) signaling is one critical pathway related to TAMs recruitment, proliferation, differentiation, and survival and is also essential for stimulating chemotactic activity of monocytes and macrophages including the transition from M1 TAMs into M2 TAMs (Laoui et al. 2014; Dwyer et al. 2017). Studies have shown that CSF-1 enhances progression of mammary tumors to malignancy, high expression of CSF-1 in tumor tissues, or high expression of CSF-1R in TAMs which are associated with worse prognosis of tumor (Lin et al. 2001; Zhu et al. 2008; Koh et al. 2014). The absence of CSF-1R caused almost all macrophages to be dramatically depleted in mice (Erblich et al. 2011). Besides, G-CSF regulated macrophage phenotype, suppression of G-CSF enhanced circulating monocytes, and TAMs mutation and remarkably limited lung metastasis of triple-negative breast cancer, but if G-CSF was kept in high level, anti-CSF-1R therapy could strength anti-inflammatory TAMs and promote lung metastasis (Hollmen et al. 2016). Inhibition of M-CSF by both antibody and chemical inhibitor significantly suppressed tumor angiogenesis and lymphangiogenesis in mouse osteosarcoma model (Kubota et al. 2009). Antibody against CSF1-R depleted the resident subset of monocytes and TAMs without inhibiting inflammation, and macrophage blockade using a CSF-1R inhibitor also resulted in reduced infiltration of protumorigenic (M2) macrophages (MacDonald et al. 2010). Moreover, the mAb inhibiting the CSF-1R (RG7155) was proved both in vitro and in vivo to decrease F4/80+ TAMs accompanied by an increase in the CD8+/CD4+ T-cell ratio (Ries et al. 2014). Administration of RG7155 to patients led to striking reductions of CSF-1R + CD163+ macrophages in tumor tissues. Some clinical trials of RG7155 are underway in solid tumor treatment as monotherapy or combined with other therapies. Besides, targeting TAMs by CSF-1R blockade in breast cancer mice model could stimulate intratumoral type I interferon signaling, thus enhance the anticancer efficacy of platinum-based chemotherapeutics (Salvagno et al. 2019). Local targeting of lung TAMs with pulmonary delivery of a CSF-1R inhibitor PLX-3397 could treat lung metastases of breast tumor by decreasing M2 macrophages and increasing M1 macrophages in the tumor microenvironment (Alhudaithi et al. 2020). Researchers also reported a potent, highly selective, and orally bioavailable CSF-1R inhibitor, IACS-9439, which could lead to a dose-dependent reduction in macrophages and lead to tumor growth inhibition in MC38 and PANC02 syngeneic tumor models (Czako et al. 2020). In some other studies, CSF-1R inhibitors could reduce TAMs number in tumor tissue, inhibit tumor growth, and obviously decrease tumor angiogenesis and metastasis. JNJ-28312141, an FMS-related receptor tyrosine kinase-3 (FLT3) inhibitor, suppressed human nonsmall cell lung carcinoma growth and reduced tumor vasculature in a dose-dependent manner; these effects were highly correlated with marked reductions in F4/80+ TAMs. Another CSF-1R inhibitor, BLZ945, showed potentials as a potent antitumor drug in breast cancer, glioblastoma, and cervical carcinoma in preclinical studies. It was confirmed existing potent ability converting tumorigenic M2 macrophages into the antitumor M1 macrophages. Numerous studies revealed that targeting of TAMs through inhibiting CSF-1R could suppress tumor growth and metastasis, and currently massive clinical trials are launched to evaluate bio-safety and bio-activity of antibodies/small molecules targeting CSF-1/CSF-1R signaling alone or combined with other therapies in different types of tumors (Table 1). While researchers also found that anti-CSF-1R and anti-CSF-1 antibodies or CSF-1R small-molecule inhibitors treatment increased breast cancer metastasis, thus the CSF-1/CSF-1R signaling serving as the therapeutic targets for breast cancer still needs further investigation (Swierczak et al. 2014).
5.1.2 Targeting CCL-2/CCR-2 Axis
CCL-2, also widely known as monocyte chemoattractant protein-1(MCP-1) , is a C-C motif chemokine overexpressed in solid tumors and tightly associated with increased tumor adhesion, migration, invasion, progression, and angiogenesis (Hao et al. 2020). Besides, CCL-2 could also recruit monocytes into an immunosuppressive tumor microenvironment: Tumor cells recruited inflammatory monocytes to tumors sites through CCL-2/CCR-2 axis, and the recruited monocytes were polarized to TAMs to promote tumor survival and facilitate tumor metastasis (Qian et al. 2011; McClellan et al. 2012). As its main receptor, the upregulation of CCR-2 was also reported correlated with cancer metastasis and relapse (Nagarsheth et al. 2017) and recruitment of CCR-2 positive TAMs to sites of liver metastasis conferred a poor prognosis in human colorectal cancer (Grossman et al. 2018). Recent work suggested that during esophageal carcinogenesis, CCL-2/CCR-2 axis-mediated TAMs recruitment could induce immune evasion through PD-1 signaling (Yang et al. 2020). Blocking of the CCL-2/CCR-2 axis decreased macrophage infiltration and reduced tumor growth (Lim et al. 2016). Besides, disruption of CCL-2/CCR-2 axis could obviously reduce TAMs number in tumors, thereby suppressing tumor growth and dissemination in some certain cancer types (Fujimoto et al. 2009).
Anti-CCL-2 antibody or CCL-2 inhibitors were proved to block glioma, colon, prostate, and melanoma cancers proliferation in animal models (Loberg et al. 2007). Li et al. reported that blockade of CCL-2/CCR-2 axis by using CCR-2 antagonist or CCR-2 knockout could inhibit liver tumor malignant growth and metastasis, reduce postsurgical recurrence, and enhance survival through suppressing inflammatory monocytes recruitment, infiltration, and TAMs M2 polarization (Li et al. 2017). In a preclinical study, a CCL-2 neutralizing antibody led to a remarkable tumor growth suppression in hepatocellular cancer (Teng et al. 2017). Apart from small molecule inhibitors and antibodies, RNA aptamer CCL-2 inhibitor could also reduce M1 TAMs liver infiltration and pathogenic angiogenesis (Bartneck et al. 2019). In clinical trial, Carlumab (CNTO888), an anti-CCL-2 human monoclonal antibody, is being used either alone or in combination with standard chemotherapies for solid tumors patients’ treatment (Sandhu et al. 2013). The other approach to decrease the action of CCL-2 is to block CCR-2. Pharmacological CCR-2 inhibitors and humanized antibody that recognizes CCR-2 were examined. BMS-813160, a potent and selective CCR-2/CCR-5 antagonist, alone or combined with chemotherapy or Nivolumab in patients with advanced solid tumors such as colorectal cancer and pancreatic cancer, is launched in clinical trials (NCT03184870). A CCR-2 inhibitor RS 504393 could inhibit xenograft prostate tumor growth in SCID mice fed with high fat diet (Hao et al. 2020). An orally active, high-affinity CCR-2 antagonist CCX872 could also enhance anti-PD-1 effect to slow progression of gliomas and improve survival and is currently in the clinical trial for patients with advanced and metastatic pancreatic cancer (Flores-Toro et al. 2020). PF-04136309, small-molecule inhibitors of CCR-2 showed efficacy in preclinical study: In an orthotopic model of murine pancreatic cancer, CCR-2 blockade by PF-04136309 depleted inflammatory monocytes and macrophages from the primary tumor and premetastatic liver resulting in tumor growth inhibition, antitumor immunity enhancement, and metastasis reduction (Sanford et al. 2013); Currently, PF-04136309 has moved forward to clinical trials, and the inhibitor combined with chemotherapeutic regimen FOLRIRINOX presented favorable response in pancreatic cancer patients during phase 1b clinical trial (Nywening et al. 2016), while another clinical trial in PF-04136309 combined with nab-paclitaxel and gemcitabine for metastatic pancreatic patients was terminated in May 2017 (Jahchan et al. 2019). Plozalizumab (MLN1202), a humanized mAb to CCR-2, was conducted in phase 2 clinical trial for bone metastasis of unspecified tumors, while Plozalizumab combined with nivolumab (immune-checkpoint inhibitor) for patients with advanced melanoma in phase 1 clinical trial was terminated in May 2018 because of serious adverse events (Vela et al. 2015; Yumimoto et al. 2019). Thus, it should be noted that durable CCL2/CCR2 axis inhibition might lead to the compensation of other chemokine-dependent pathways, which facilitates the recruitment of TAMs in the tumor microenvironment.
5.1.3 Targeting CXCL-12/CXCR-4 Axis
CXCL-12, also known as stromal cell-derived factor-1 (SDF-1) , is widely expressed in many different types of tissues. CXCL-12 is the ligand for the chemokine receptor CXCR-4 (Liekens et al. 2010). Recent work also demonstrated that CXCL-12 could bind to another seven-transmembrane span receptor CXCR-7 with high affinity (Sun et al. 2010). Both CXCR-4 and CXCR-7 played critical roles on tumor growth and metastasis, and a lot researches have suggested that the CXCR-4 and its ligand CXCL-12 are potential targets for cancer therapy (Zhou et al. 2019). CXCL-12/CXCR-4 axis could directly or indirectly activate massive signaling pathways including PI3 kinase, stress-activated protein kinase, c-Jun N-terminal kinase, and mitogen-activated protein kinase (MAPK) to promote tumor growth, adhesion, migration, and survival (Dewan et al. 2006). Because of expressing CXCR-4, macrophages could migrate along CXCL-12 gradient. It was shown that CXCL-12 production by breast cancer cells results in increased macrophage number in breast tumor (Bonapace et al. 2014). Similar responses were also showed in monocytes: CXCL-12 released by multiple myeloma cells could trigger monocyte migration and anti-CXCR-4 antibodies could obviously suppress monocyte recruitment. BMS-936564/MDX-1338, a human mAb recognizing human CXCR-4, could significantly induce apoptosis in vitro and inhibit tumor growth in several established experimental tumor models (Kuhne et al. 2013). Plerixafor (AMD3100), CXCR-4 inhibitor, could suppress the postsepsis-induced melanoma progression, TAMs accumulation, and TAMs in situ proliferation in mice model. Plerixafor could also selectively reduce the number of M2 TAMs and suppress tumor re-vascularization and re-growth (Zhou et al. 2018). More specifically, cancer-associated fibroblast-derived CXCL12 attracted CXCR-4 expressing M2-like macrophages to infiltrate into tumors, which promoted cancer stem cells-like transition, proliferation, and migration of cancer cells in oral squamous cell carcinoma (Li et al. 2019b). Progranulin presents carcinogenic roles in breast cancer. Recent studies revealed that Progranulin KO TAMs inhibited invasion, migration, and EMT of breast cancer cells through their exosomes. miR-5100 in Progranulin KO TAMs-derived exosomes was upregulated, which might contribute to CXCL-12 regulation, thereby suppressing CXCL-12/CXCR-4 axis and finally inhibiting the invasion, migration, and EMT of breast cancer cells (Yue et al. 2021). Similar results were also shown in liver metastasis of colorectal cancer: Several miRNAs upregulated in colorectal cancer cells by suppressing CXCL-12/CXCR-4 axis activation could be transferred to TAMs via exosomes and mediate crosstalk between CXCR-4 overexpressing cancer cells and TAMs (Wang et al. 2020). Apart from antibodies, small-molecules, and microRNAs, small modified peptides designed as CXCR-4 antagonists were also widely tested: A novel CXCR-4 antagonist BKT140 showed potent therapeutic efficacy against human nonsmall cell lung cancer in vitro and in vivo. BKT140 could also augment therapeutic effects of chemotherapy and radiotherapy (Fahham et al. 2012). Similarly, a peptidic CXCR-4 inhibitor TF14016 also suppressed metastases of small cell lung cancer cells in mice (Otani et al. 2012). Similarly, as CXCR-4 antagonists, T140 analogs were also known as antimetastatic agents for breast cancer therapy (Tamamura et al. 2003). Targeting CXCL-12/CXCR-4 axis could also be in combination with standard cancer therapies: CXCL-12 upregulation in hepatocellular carcinoma models triggered hypoxia, immunosuppressive cells recruitment, T-regulatory cells, and M2 TAMs accumulation after sorafenib treatment. CXCR-4 inhibitor AMD3100 and Sorafenib enhanced PD-1 blockade-induced hepatocellular carcinoma suppression through inhibiting the polarization toward an immunosuppressive microenvironment (Chen et al. 2015).
5.1.4 Targeting VEGF/VEGFR Signaling
Tumor angiogenesis is a critical process for necessary nutrients and oxygen supply to support quickly tumor tissues growing (Nishida et al. 2006). Among them, the family of vascular endothelial growth factors (VEGFs) plays critical roles as regulators of angiogenesis. VEGF/VEGFR signaling pathway is upregulated in numerous cancer types and contributes to out-of-controlled angiogenesis and metastatic spreading (Ceci et al. 2020). VEGFs promote tumor angiogenesis via regulating endothelial cells proliferation and survival (Lee et al. 2015) and might promote tumor cell proliferation through activating VEGFR1 signaling (Bhattacharya et al. 2016). Tumor cells could also recruit and reprogram macrophages derived from circulating monocytes; these recruited and reprogrammed macrophages could also work as a main source of angiogenic factors (Cassetta et al. 2019). For example, angiopoietin-2 produced by tumor endothelium could enhance TIE2 receptor expressing monocytes recruiting in tumor cells. TAMs could also release angiogenic factors such as VEGFA as the response of hypoxia in tumor avascular areas; the released VEGFA could further stimulate macrophages and endothelial cells conversely (Mazzone and Bergers 2019). TAMs were able to release cytokines that indirectly contribute to tumor angiogenesis by the induction of a pro-angiogenic program in tumor cells. Tumor cells and recruited TAMs cooperated in the TME to amplify the production of pro-angiogenic factors resulting in an angiogenic switch. Apart from pro-angiogenesis, TAMs also tightly involved in lymph angiogenesis under inflammatory conditions or carcinogenetic conditions (Yang et al. 2018).
VEGFs can recruit macrophages to the tumor and promote TAMs development (Lapeyre-Prost et al. 2017; Linde et al. 2012). Thus, targeting VEGF/VEGFR has potential suppressing macrophage recruit and localize to the tumor (Yang et al. 2018). The potent proangiogenic activities of TAMs in the TME and the existing of angiogenesis-relevant macrophage subpopulation Tie2 expressing monocytes (TEMs) demonstrate novel antitumor strategies through targeting angiogenesis (Johansson-Percival et al. 2018; Chen et al. 2016). Dampening TME recruitment might be an effective method to block tumor angiogenesis. Inhibiting Ang2-Tie2 crosstalk by using Ang2-CovX-Bodies prevented TEMs infiltration (Huang et al. 2011). Anti-ANG2 monoclonal antibody (mAb) could also disrupt connection between TEMs and endothelial cells, leading to suppressed angiogenesis in pancreatic insulinomas and mammary carcinomas (Mazzieri et al. 2011). As ANG2 overexpression could induce anti-VEGF therapy resistance, antibodies targeting both ANG2 and VEGF could abrogate angiogenesis and promote antitumor efficacy (Kloepper et al. 2016; Scheuer et al. 2016). YKL-40, pro-angiogenic factor, could mediate angiogenesis both in vitro and in mice tumor models. Neutralizing mAbs targeting YKL-40 could suppress angiogenesis and progression of tumor (Faibish et al. 2011).
Besides, macrophages could mediate anti-VEGF therapy resistance through changing VEGFR expression level (Dalton et al. 2017), and TAMs might play critical roles during these processes (Itatani et al. 2018): TAMs have been shown to help escape from antiangiogenic therapy of glioblastoma both in preclinical models and in clinic and could work as a potential biomarker and therapeutic target (Lu-Emerson et al. 2013). Similarly, macrophages could be activated and recruited to the TME and further contribute to anti-VEGF resistance in regular ovarian cancer mice model, but there is no resistance anti-VEGF antibody when macrophages were depleted in a macrophage-deficient mouse model, while this resistance could still be triggered by macrophages injection (Dalton et al. 2017). Targeting TAMs might restore or enhance antiangiogenic therapeutic effects. As one of bisphosphonate drugs, Zoledronic was widely used for treating osteoporosis and bone metastases in clinic, while Zoledronic also showed ability to deplete macrophages. Researchers have investigated the role of macrophages depletion in anti-VEGF antibody therapy resistance in mice bearing ovarian tumor models: Macrophages inhibited by Zoledronic could overcome anti-VEGF antibody therapy resistance, enhance tumor growth suppression, and obviously extend survival when compared with vehicle and only anti-VEGF therapy groups (Dalton et al. 2017). Recent study also presented that expression of CCL-2 could promote antiangiogenic therapy resistance; mNOX-E36, a CCL-2 inhibitor inhibiting TAMs recruitment and angiogenesis, could enhance the efficacy of bevacizumab in glioma models (Cho et al. 2019). Similar strategies were also used by targeting CXCL-12/CXCR-4 axis: NOX-A12, a novel CXCL-12 inhibitor, could reverse TAMs recruitment and potentiate the antitumor effects of anti-VEGF therapy (Deng et al. 2017). Combination of CXCR-4 inhibitors and anti-VEGF therapy was also reported to slow GBM xenografts progression both in preclinic and in clinical trial: CXCR-4 antagonist PRX177561 increased the antitumor effects of bevacizumab in preclinical models of human glioblastoma (Gravina et al. 2017); another CXCR-4 antagonist POL5551 combined with VEGF inhibition could also improve survival in an intracranial mouse model of glioblastoma (Barone et al. 2014). AMD3100 against CXCR-4 was applied with a combination of bevacizumab in patients with recurrent high-grade glioma in clinical trial. Thus, suppressing VEGF/VEGFR signaling combined with other inhibiting TAMs strategies will bring more benefits for cancer therapies.
5.2 Target TAMs Inhibition for Cancer Therapy
There are two approaches to decrease the population of TAMs for cancer therapy: One is to reduce the monocytes number in the circulating system and the other is to suppress the population of macrophages already accumulated in the tumor tissues (Sawa-Wejksza and Kandefer-Szerszen 2018).
Bisphosphonates, including alendronate, ibandronate, risedronate, and zoledronic acid, are primary agents in the current pharmacological arsenal to prevent the loss of bone density and treat osteoporosis and related diseases (Drake et al. 2008). The use of bisphosphonates has been related to inhibit proliferation, migration, and invasion of macrophages, which share the same lineage with osteoclasts, causing apoptosis (Rogers and Holen 2011). As chemotherapeutics, no matter traditional free bisphosphonates or nanoparticles-captured bisphosphonates, both showed cytotoxicity to monocytes/macrophages and have been used to decrease their population in tumors (Farrell et al. 2018). In preclinical models, bisphosphonates could effectively inhibit tumor growth through suppressing TAMs infiltration in breast tumors (Holen and Coleman 2010). Some research also presented that Trabectedin , a registered antineoplastic agent that suppresses cancer cell growth, could also trigger specific death of circulating monocytes/macrophages through TNF-related apoptosis-inducing ligand (TRAIL)-dependent apoptosis, as circulating monocytes/macrophages expressed functional TRAIL receptors 1 and 2, which are susceptible to the cytotoxicity of trabectedin (Germano et al. 2013). Trabectedin-treated pancreatic ductal adenocarcinoma showed reduced TAMs, decreased angiogenesis, activated antitumoral CTLs, with promising clinical outcomes (Borgoni et al. 2018). Similarly, in cutaneous melanoma mice model, trabectedin showed significant activity of reducing TAMs and tumor blood vessel density and inhibited lung metastasis of melanoma (Carminati et al. 2019). Besides, a combination of checkpoint blockade and angiogenesis inhibitors could be an effective strategy to promote the curative effect of Trabectedin (Seliger 2019). Trabectedin also revealed a strategy of immunomodulation in chronic lymphocytic leukemia: Trabectedin depleted myeloid-derived suppressor cells and TAMs and increased memory T-cells in xenograft and immunocompetent chronic lymphocytic leukemia mouse models and could also suppress PD-1/PD-L1 axis through targeting PD-L1-positive chronic lymphocytic leukemia cells, monocytes/macrophages, and T-cells (Banerjee et al. 2019). Similar results were also shown in skeletal prostate cancer models: Trabectedin reduced prostate cancer bone resident tumor size, which was in associated with trabectedin-reduced M2 macrophages (Jones et al. 2019). Depletion of TAMs by trabectedin could also switch the epigenetic profile of pancreatic cancer infiltrating T-cells and restore their antitumor phenotype (Borgoni et al. 2018).
There also have other strategies to depleting TAMs through targeting their specific surface markers. Sialic acid receptors in the Siglec family are highly expressed on the surface of TAMs and most have immunosuppressive effects. Targeted delivery of zoledronic acid through the sialic acid-Siglec axis could kill and reversal M2-like TAMs and inhibit S180 tumor growth (Tang et al. 2020). Similarly, Folate receptor beta (FR beta) was found expressed on macrophages in human glioma and rat C6 glioma. A recombinant immunotoxin consisting of anti-FR beta mAbs and Pseudomonas exotoxin A could significantly reduce TAMs numbers and suppress tumor growth (Nagai et al. 2009). Apart from sialic acid receptors and FR beta, activated macrophages also expressed the pattern recognition receptor scavenger receptor A (SR-A), a small peptide SR-A ligand could compete with physiological SR-A ligand in vitro, and deficiency of SR-A suppressed progression and metastasis of ovarian and pancreatic cancer in vivo (Neyen et al. 2013). M2 macrophage-targeting peptide (M2pep), which could bind to murine M2 macrophages and M2-like TAMs, was fused with pro-apoptotic peptide KLA. This fusion peptide could also reduce TAM population in vivo but need high concentrations (Ngambenjawong et al. 2016).
Although targeting CSF-1/CSF-1R axis or CXCL-12/CXCR-4 axis could suppress TAMs through blocking the recruitment and localization of TAM, researchers also find that in some other studies, the use of these axis inhibitors decreased TAMs number in tumor tissue, inhibited tumor proliferation, and significantly decreased angiogenesis and metastasis (Anfray et al. 2019). Besides, the use of the anti-IL-6 antibody had a strong anticancer effect by decreasing CCL-2, VEGF, and CXCL-12. There was a significant decline in CCL-2, CXCL-12, and VEGF as well as the number of TAMs in tumor tissue in patients treated with anti-IL-6 antibody for 6 months (Chen and Chen 2015).
5.3 Reprogramming TAMs for Cancer Therapy
As the most abundant tumor-infiltrating immune cells in TME, TAMs play a crucial role in tumor development, invasion, and metastasis (Pathria et al. 2019). Emerging evidences demonstrate that the plasticity and diversity of TAMs allow them to be classified along the M1-M2 polarization axis (Genard et al. 2017). M2 macrophages, also known as alternatively activated macrophages, can be induced by the TH2 cytokines such as IL-4, IL-10, and IL-13 and promote tumor angiogenesis, immunosuppression, and drug resistance (Gordon and Taylor 2005). On the contrary, the TH1 cytokines such as IL12, IL-18, or activated TLRs stimulate macrophages to M1 macrophages, also known as classically activated macrophages (Biswas and Mantovani 2010). M1 macrophages present the ability to eliminate tumor cells by releasing reactive oxygen/nitrogen intermediates, together with pro-inflammatory cytokines such as TNF, IFN-γ. However, in TME of solid tumors, most TAMs are devoid of cytotoxic activity and closely related to the M2 phenotype with poor clinical outcome of patients. Thus, reprogramming the TAMs through chemotherapy, radiotherapy, and immunotherapy could be a promising strategy to eradicate tumors.
5.3.1 TLR Agonists to Reprogram TAMs
Currently, several options including TLRs agonists and compounds or mAb targeting inhibitory proteins of M1 phenotype are used to select M1 phenotype or reprogram TAMs from M2 to M1 phenotype (Mantovani et al. 2017). As pattern-recognition receptors in innate immunity, TLRs stimulate and activate M1-phenotype polarization via their engagement with the ligands. Thus, various agonists targeting TLRs, such as TLR3, TLR7, and TLR9, have been developed to evaluate their capacity to reprogram TAMs into antitumor effectors (Anfray et al. 2019).
Stimulation of TLR3 by poly I:C upregulated costimulatory molecules CD40, CD80, CD86, and M1-specific markers MHC-II on M2 macrophage. Simultaneously, the expression of pro-inflammatory cytokines and M2-specific markers Tim-3, CD206 was reduced (Shime et al. 2012). In MC38 tumor model, poly I:C reverted the M2 phenotype to M1 macrophages and eradicated the tumor in an IFN-α/β-dependent signaling pathway. Recently, Liu et al. developed novel polypeptide micelles to target TAM. Poly I:C-loaded polypeptide micelles showed property for targeting TAM, efficiently reeducated TAMs into M1 macrophages, substantially activated T lymphocytes and NK cells in melanoma models, indicating repolarizing TAMs into M1 phenotype in situ for effective immunotherapy of cancer (Liu et al. 2018). Ferumoxytol, a clinically approved nanoparticle, in combination with TLR3 agonist poly I:C induced pro-inflammatory macrophage polarization for regression of primary and metastatic murine melanoma (Zhao et al. 2018a; Zanganeh et al. 2016). In 2018, a phase II trial has been performed to evaluate the safety and therapeutic effect of combination of Poly I:C and immune checkpoint blockade in unresectable hepatocellular carcinoma, while its analog, poly-ICLC, has been tested as a tumor vaccine to boost antitumor immunity (Anfray et al. 2019).
TLR7 agonist was demonstrated to induce the nuclear translocation of NF-κB in macrophages with the production of pro-inflammatory cytokines (De Meyer et al. 2012). Topical administration of TLR7 agonist imiquimod with radiotherapy has been approved to synergistically inhibit tumor growth and cyclophosphamide further increased the therapeutic effect and induced immunologic memory in cutaneous breast cancer metastases. Currently, imiquimod is the only TLR agonist approved by Food and Drug Administration for topical injection in intraepidermal carcinoma and basal cell carcinoma.
In the past years, resiquimod has attracted more attention for its role to reeducate TAMs into M1 macrophages (Thauvin et al. 2019). Resiquimod , also known as R848, is a dual agonist of the toll-like receptors TLR7/8 (Chi et al. 2017). Similar to imiquimod, R488 showed more powerful as a TLR agonist, with more ability to elicit antitumor immune immunity. Despite several studies have showed promising results based on the topical injection in melanoma patients, there are no ongoing clinical trials due to that systemic administration of R848 is burdened with toxicities: anemia, inflammation, and flu-like symptoms (Perkins et al. 2012; Hasham et al. 2017). To overcome this issue, various methods have been introduced. MEDI9197, a formulation of R848 was designed with a lipid tail to increase lipid solubility and be retained at the injection sites, thus limiting systemic toxicity. As an alternative, R848 was covalently linked with vitamin E and modified by hyaluronic acid into a predrug nanopreparation (CDNP-R848), providing a sustained release of R848 by subcutaneous injection (Rodell et al. 2018). The predrug nanopreparation successfully delivered R848 to TAMs in vivo and altered the functional orientation of the TAMs toward M1 macrophage, leading to inhibited tumor growth and protecting the mice against tumor re-challenge. When CDNP-R848 was combined with anti-PD-1 blockade, improved immunotherapy response rates were observed, including an anti-PD-1 therapy-resistant tumor model. In addition, Rodel et al. identified R848 as a potent driver of M1 macrophage polarization and developed R848-loaded β-cyclodextrin nanoparticles to efficiently deliver R848 to TAM in MC38 colorectal tumors (Rodell et al. 2018). These strategies were able to trigger the IL-12 production of TAMs in TME, demonstrating that engineered R848-nanoparticle combinations could efficiently modulate TAMs for cancer immunotherapy.
As to the TLR9, preclinical data of the agonists singly or in combination with other agents showed efficacy, which led to clinical trials in patients with advanced malignancy (Karapetyan et al. 2020). In the preclinical models, intratumoral, subcutaneous, and intravenous routes of injection have been performed with potent antitumor responses in treated and metastatic sites with the repolarization of TAMs (Hofmann et al. 2008). In the clinic, despite TLR9 agonist monotherapy or combined with chemotherapy and targeted therapy showed no obvious efficacy in advanced solid tumors, especially with an intravenous injection, the greatest excitement has been reserved for TLR9 agonists in combination with immune checkpoint inhibitors. Currently, a clinical trial has been registered to evaluate the antitumor efficacy of TLR9 agonist CpG in combination with nivolumab in metastatic pancreatic cancer patients (NCT04612530).
Despite these in vitro data are promising, there is limited evidence for TLRs agonists as adjuvants in the field of cancer vaccination. In this regard, nanoparticles are considered as a potential strategy to load TLRs agonists to the target tumor sites. For example, nanoparticles loaded with poly I:C or R848 presented the power to elicit potent innate immune responses in lymph nodes, resulting in therapeutic effect without systemic release of inflammatory cytokines (Bocanegra Gondan et al. 2018; Da Silva et al. 2019).
5.3.2 Reprogramming TAMs by mAb and Fusion Protein
In the TME , reprogramming M2 macrophages into M1 proinflammatory phenotype is a potential antitumor immunotherapy which is involved in increased activity of macrophage phagocytosis. The macrophage phagocytosis is balanced by activating and inhibitory signals. CD47 is an immune checkpoint that confers a “do-not-eat-me” signal to host immune surveillance via binding to its ligand, SIRPα on the surface of phagocytic cells, such as macrophages and dendritic cells (Zhang et al. 2017). SIRPα binds to CD47 and transmits intracellular signals through its cytoplasmic domain. The cytoplasmic tail of SIRPα contains four immunoreceptor tyrosine-based inhibition motifs (ITIMs) and becomes phosphorylated after binding of CD47, leading to prevention of myosin-IIA accumulation at the phagocytic synapse and inhibition of phagocytosis (Tsai and Discher 2008; Barclay and Brown 2006; van Beek et al. 2005). The “do-not-eat-me” signals play key roles for tissue homeostasis. However, CD47 overexpression has been adopted by various cancer cells to escape the innate immune surveillance. In this sense, pharmacological blockade of CD47-SIRPα axis by monoclonal antibody or fusion protein could reprogram macrophage phagocytosis in multiple preclinical models (Galli et al. 2015; Weiskopf et al. 2016; Zhang et al. 2018a). For instance, reprogramming of TAMs into M1 macrophages has been achieved by the combination of anti-SIRPα antibody with CSF-1R inhibitor (Kulkarni et al. 2018). TTI-621, a CD47-blocking SIRPαFc fusion protein, triggers tumor cell phagocytosis by M1 macrophages in vitro and exhibits antitumor activity in vivo. Combinational therapies have the potential to increase the effect (Petrova et al. 2017). Data from a phase 1b trial showed that the macrophage checkpoint inhibitor Hu5F9-G4 combined with anti-CD20 antibody rituximab demonstrated promising activity in advanced lymphoma patients (Advani et al. 2018). Based on these promising evidences, several clinical trials are performed to evaluate the safety and activity of anti-CD47/SIRPα monotherapy, also in combination with other agents (Table 2).
As an alternative target to favor cytotoxic functions of TAMs, CD40 is a costimulatory protein found on antigen-presenting cells including macrophages and dendritic cells. CD40L on TH cells binding to CD40 activates antigen presenting cells and triggers various downstream effects, including upregulation of the MHC molecule expression, secretion of proinflammatory cytokines, and finally cytotoxic T-cell activation (Zippelius et al. 2015; Zhang et al. 2018b). Re-educating macrophages using CD40 agonistic antibodies could recover tumor immune surveillance and reprogram TAMs toward M1-polarization in various tumor models (Vonderheide 2020). However, the antitumor activities have been moderate. Since anti-CD40 therapy induced PD-L1 upregulation in TAM, combining CD40 agonists with anti-PD-1 therapy resulted in significantly lower tumor burden than either monotherapy alone (Diggs et al. 2020). Concurrent CSF-1R blockade and CD40 agonists lead to significant changes in the composition of immune infiltrates, causing an increased differentiation of proinflammatory M1 macrophages and also priming potent cytotoxic T-cell response in the draining lymph nodes of “cold” tumor models (Wiehagen et al. 2017).
5.3.3 Other Strategies to Reprogram TAMs
With the progress of oligonucleotide delivery, gene therapies, including microRNA (miRNA), small interfering RNA (siRNA), and messenger RNA (mRNA), are promising strategies to re-educate macrophages for cancer treatment. MiRNAs are tiny, single-stranded noncoding RNA molecule containing 19–24 nucleotides. Substantial research data depict that miRNAs are capable of silencing the gene expression either by degrading mRNA or repressing the gene transcription (Chatterjee et al. 2020). By performing miRNA profiling studies on macrophages, Graff and colleagues identified the miRNAs were associated with macrophage responses to inflammatory stimuli and demonstrated that miR-125a-3p and miR-26a-2 promote M1-like phenotype (Graff et al. 2012). Cai et al. presented the first evidence that miRNA-155 was a key molecule re-educating TAMs into pro-inflammatory M1 macrophages. In a mouse sarcoma model, delivering miRNA-155 by lipid-coated calcium phosphonate nanoparticles to TAMs successfully increased IL-12 expression with controlled tumor growth and extended survival (Cai et al. 2012). SiRNA has been employed to block the expression of immunosuppressive genes in TAMs. Shi et al. developed a robust nanoparticle platform for efficient siRNA delivery and gene silencing in macrophages (Liang et al. 2018). Silencing CCL-18 in macrophages remarkably suppressed breast cancer cells migration via regulation of the TAMs behavior.
In addition, researchers also exploited new strategies and carriers to increase the target gene expression in TAMs, such as charge-altering releasable transporters. This method has been used to deliver a combination of CD80, CD86, and OX40L mRNAs in several tumor models and induced systemic antitumor responses in vivo (Haabeth et al. 2019). As far as we know, no RNA delivery systems have been approved to reprogram TAMs, and more potent and promising strategies are still needed to initiate RNA delivery technology for TAMs reprogramming.
5.4 TAM as Drug Delivery Systems
As a novel strategy to deliver payloads, the biomimetic delivery system recently showed its superiority on cancer targeting drug delivery (Yoo et al. 2011). Neutrophils, red blood cells, and macrophages are three common drug carriers (Fang et al. 2018). Through binding to IL-6 and TNF-α, surface adhesion molecules expressed on neutrophils lead to the intrinsic target to inflammation. Red blood cells can increase the long-circulation time of cargo nanoparticles in blood circulation without tumor-targeting ability. Macrophages not only circulate in the blood circulation like neutrophils and red blood cells, but also specifically target tumors through integrin on macrophages binding to VCAM-1 of tumor cells. Herein, macrophage-based drug deliveries are more versatile (Xia et al. 2020).
Given the intrinsic homing property, M1 macrophages can be employed as a biomimetic delivery system to actively carry payloads to the tumor sites (Guerra et al. 2017). Up to now, researchers have mainly designed strategies to utilize M1 macrophage and M1 macrophage-derived exosomes as tumor-targeted drug carriers. Furthermore, membrane fragments have also been extracted to coat nanoparticles to increase the targeting ability. Here, these three strategies will be detailed analyzed: M1 macrophages, M1 macrophages-derived exosomes, and M1 macrophage membrane-coated nanoparticles (Fig. 3).

TAMs as drug delivery systems. Three strategies were highlighted for TAMs as drug delivery systems: 1) M1 macrophages as drug carriers through direct drug loading, indirect drug loading, or genetical/chemical modification; 2) M1 macrophages-derived exosomes as drug carriers; and 3) M1 macrophage membrane-coated nanoparticles as drug carriers
5.4.1 M1 Macrophages as Drug Carriers
M1 macrophages including RAW264.7 cells and bone-marrow-derived macrophages (BMDMs) are extensively used for tumor-targeted delivery (Xia et al. 2020). Up to now, there are three M1 macrophage-based drug delivery systems. The first system is using macrophage as a direct antitumor drug carrier. Drug-loaded M1 macrophages were developed by simply incubation. For example, Fu and colleagues constructed a doxorubicin-loaded RAW264.7 cells to treat 4 T1 tumors. Data showed that doxorubicin did not significantly affect and alter the tumor-tropic capacity of M1 macrophages to tumor cells in vitro and in vivo. Importantly, the doxorubicin-loaded M1 macrophages potently inhibited the tumor metastasis and prolonged the life span, with reduced toxicity (Fu et al. 2015). The second one is using macrophage as an indirect antitumor drug carrier. Instead of directly loading drug, macrophages were employed to load nanoparticles containing drugs. This strategy could reduce the cytotoxicity of drugs on macrophages and further increase the drug load. Tao et al. used BMDMs to load nano/paclitaxel (paclitaxel-loaded nano-formulations) and found that nano/paclitaxel-loaded M1 macrophages showed better efficacy in malignant glioma (Tao et al. 2013). The third one is to modify macrophage as a drug carrier. To further potentiate the property of these carriers, macrophages can be further modified. M1 macrophages can be genetically or chemically modified to enhance immunological activity. Zhang et al. developed induced pluripotent stem cells-derived engineered chimeric antigen receptor-expressing macrophage and conferred the macrophage antigen-dependent functions such as secretion of cytokines, polarization toward the pro-inflammatory state (Zhang et al. 2020b).
5.4.2 M1 Macrophages-Derived Exosomes as Drug Carriers
Exosomes are membrane-bound extracellular vesicles that originated from the endosomal compartment of cells (Bunggulawa et al. 2018). Compared to M1 macrophage, M1 macrophage-derived exosomes present similar membrane properties and can be utilized to load drugs for cancer treatment. Kim and colleagues used M1 macrophage-derived exosomes to load paclitaxel via a sonication method. This delivery system increased the cytotoxicity more than 50 times in multidrug-resistant cancer cells (Kim et al. 2016). M1 macrophage-derived exosomes also could be a cancer vaccine adjuvant owing to the specific proinflammatory function (Wang et al. 2019). M1 macrophage-derived exosomes not only potentiated tumor-targeted drug delivery, but also could be engineered with liposomes including different components (Rayamajhi et al. 2019). In short, M1 macrophages and M1 macrophage-derived exosomes retained the targeting ability and proinflammatory function. However, the yield of M1 macrophage-derived exosomes is still low, and proper engineering of M1 macrophages and constructing exosome-mimetic vesicles may be the potential strategies to overcome this issue.
5.4.3 Nanoparticles Coated with M1 Macrophage Membrane as Drug Carriers
Considering the excellent tumor-targeting abilities and tumor infiltration of M1 macrophages, cell membranes of M1 macrophages can be extracted to coat nanoparticles and increase the targeting ability of nanoparticles in tumors. M1 macrophage membrane-coated nanoparticles could be constructed by coextruding the isolated macrophage membranes with nanoparticles and exhibited more persistent circulation and higher tumor accumulation (Cao et al. 2016; Zhao et al. 2018b). Cao et al. reported drug-loaded liposomes with isolated macrophage membranes to increase the drug delivery to metastasis sites. The macrophage membranes potentiated the cellular uptake of liposomes in cancer cells and presented potent suppression on metastatic breast cancer (Cao et al. 2016). This is due to the high expression of integrin α4 and β1 on macrophages, which could bind to VCAM-1 on breast cancer cells. Herein, M1 macrophage-derived membrane has the targeting property for tumors. Xuan and colleagues engineered macrophage cell membrane-coated mesoporous silica nano-capsules (MSNCs) by a biomimetic drug-delivery system. The macrophage membrane reduced the uptake percentage of MSNCs by immune cells and tissues, effectively prolong the half-life in blood circulation. Compared to the totally cleared noncoated MSNCs, the macrophage membrane-coated MSNCs showed 32% retention in blood circulation and the targeting accumulation of macrophage membrane-coated MSNCs led to complete tumor eradication (Xuan et al. 2015). In summary, membranes from M1 macrophage could also be used to modify various nanoparticles for drug delivery.
6 Conclusions
Pro-tumoral functions of TAMs make them attractive targets for cancer immunotherapy. As described in this book chapter, several preclinical and clinical studies revealed that depletion of TAMs significantly augments the response of immunotherapy and chemo−/radiotherapy and suppresses the cancer metastasis. However, as we know, TAMs represent a heterogeneous population with distinct functions that vary according to cancer types and the stages of cancer progression. One of the future works need to be done to define the subsets of TAMs involving tumorigenesis, which can be targeted for effective therapies, while saving the macrophages with antitumor functions (Guerriero 2018; Cassetta and Pollard 2018). One big challenge is to investigate metastasis-associated macrophage (MAM) phenotypes involved in cancer metastasis, which is major cause of death of cancer patients. To define those subsets of macrophages, new technologies, such as single cell sequencing, digital spatial profiling, multiplex immunofluorescence, and metabolomics, can be applied (Pathria et al. 2019). Despite multiple early stages of clinical trials are going on, scarce knowledge about TAMs in clinic human cancers is available since much of these data were generated from in vitro cellular models or mouse models. TAMs may possess more potential for cancer target therapy than previously thought. More studies should emphasis on the regulation of TAMs, such as reprogramming TAMs, targeting inhibitory molecules, which will help improve the efficacy of current cancer therapeutics, such as immunotherapy, chemotherapy, and radiotherapy, and ultimately open a new door for targeting TAMs immunotherapy (Cassetta and Pollard 2018).
References
Advani R, Flinn I, Popplewell L, Forero A, Bartlett NL, Ghosh N, et al. CD47 blockade by Hu5F9-G4 and rituximab in non-Hodgkin’s lymphoma. N Engl J Med. 2018;379(18):1711–21. https://doi.org/10.1056/NEJMoa1807315.
Affara NI, Ruffell B, Medler TR, Gunderson AJ, Johansson M, Bornstein S, et al. B cells regulate macrophage phenotype and response to chemotherapy in squamous carcinomas. Cancer Cell. 2014;25(6):809–21. https://doi.org/10.1016/j.ccr.2014.04.026.
Alhudaithi SS, Almuqbil RM, Zhang H, Bielski ER, Du W, Sunbul FS, et al. Local targeting of lung-tumor-associated macrophages with pulmonary delivery of a CSF-1R inhibitor for the treatment of breast cancer lung metastases. Mol Pharm. 2020;17(12):4691–703. https://doi.org/10.1021/acs.molpharmaceut.0c00983.
Anfray C, Ummarino A, Andon FT, Allavena P. Current Strategies to Target Tumor-Associated-Macrophages to Improve Anti-Tumor Immune Responses. Cells. 2019;9(1). doi: https://doi.org/10.3390/cells9010046.
Argyle D, Kitamura T. Targeting macrophage-recruiting chemokines as a novel therapeutic strategy to prevent the progression of solid tumors. Front Immunol. 2018;9:2629. https://doi.org/10.3389/fimmu.2018.02629.
Arnold CE, Whyte CS, Gordon P, Barker RN, Rees AJ, Wilson HM. A critical role for suppressor of cytokine signalling 3 in promoting M1 macrophage activation and function in vitro and in vivo. Immunology. 2014;141(1):96–110. https://doi.org/10.1111/imm.12173.
Banerjee P, Zhang R, Ivan C, Galletti G, Clise-Dwyer K, Barbaglio F, et al. Trabectedin reveals a strategy of immunomodulation in chronic lymphocytic leukemia. Cancer Immunol Res. 2019;7(12):2036–51. https://doi.org/10.1158/2326-6066.CIR-19-0152.
Barclay AN, Brown MH. The SIRP family of receptors and immune regulation. Nat Rev Immunol. 2006;6(6):457–64. https://doi.org/10.1038/nri1859.
Barclay AN, Van den Berg TK. The interaction between signal regulatory protein alpha (SIRPalpha) and CD47: structure, function, and therapeutic target. Annu Rev Immunol. 2014;32:25–50. https://doi.org/10.1146/annurev-immunol-032713-120142.
Barkal AA, Weiskopf K, Kao KS, Gordon SR, Rosental B, Yiu YY, et al. Engagement of MHC class I by the inhibitory receptor LILRB1 suppresses macrophages and is a target of cancer immunotherapy. Nat Immunol. 2018;19(1):76–84. https://doi.org/10.1038/s41590-017-0004-z.
Barkal AA, Brewer RE, Markovic M, Kowarsky M, Barkal SA, Zaro BW, et al. CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature. 2019;572(7769):392–6. https://doi.org/10.1038/s41586-019-1456-0.
Barone A, Sengupta R, Warrington NM, Smith E, Wen PY, Brekken RA, et al. Combined VEGF and CXCR4 antagonism targets the GBM stem cell population and synergistically improves survival in an intracranial mouse model of glioblastoma. Oncotarget. 2014;5(20):9811–22. doi: https://doi.org/10.18632/oncotarget.2443.
Bartneck M, Schrammen PL, Mockel D, Govaere O, Liepelt A, Krenkel O, et al. The CCR2(+) macrophage subset promotes pathogenic angiogenesis for tumor vascularization in fibrotic livers. Cell Mol Gastroenterol Hepatol. 2019;7(2):371–90. https://doi.org/10.1016/j.jcmgh.2018.10.007.
Bhattacharya R, Ye XC, Wang R, Ling X, McManus M, Fan F, et al. Intracrine VEGF signaling mediates the activity of Prosurvival pathways in human colorectal cancer cells. Cancer Res. 2016;76(10):3014–24. https://doi.org/10.1158/0008-5472.CAN-15-1605.
Binenbaum Y, Fridman E, Yaari Z, Milman N, Schroeder A, Ben David G, et al. Transfer of miRNA in macrophage-derived exosomes induces drug resistance in pancreatic adenocarcinoma. Cancer Res. 2018;78(18):5287–99. https://doi.org/10.1158/0008-5472.CAN-18-0124.
Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010;11(10):889–96. https://doi.org/10.1038/ni.1937.
Bocanegra Gondan AI, Ruiz-de-Angulo A, Zabaleta A, Gomez Blanco N, Cobaleda-Siles BM, Garcia-Granda MJ, et al. Effective cancer immunotherapy in mice by polyIC-imiquimod complexes and engineered magnetic nanoparticles. Biomaterials. 2018;170:95–115. https://doi.org/10.1016/j.biomaterials.2018.04.003.
Bonapace L, Coissieux MM, Wyckoff J, Mertz KD, Varga Z, Junt T, et al. Cessation of CCL2 inhibition accelerates breast cancer metastasis by promoting angiogenesis. Nature. 2014;515(7525):130–3. https://doi.org/10.1038/nature13862.
Bonde AK, Tischler V, Kumar S, Soltermann A, Schwendener RA. Intratumoral macrophages contribute to epithelial-mesenchymal transition in solid tumors. BMC Cancer. 2012;12:35. https://doi.org/10.1186/1471-2407-12-35.
Borgoni S, Iannello A, Cutrupi S, Allavena P, D’Incalci M, Novelli F, et al. Depletion of tumor-associated macrophages switches the epigenetic profile of pancreatic cancer infiltrating T cells and restores their anti-tumor phenotype. Onco Targets Ther. 2018;7(2):e1393596. https://doi.org/10.1080/2162402X.2017.1393596.
Boussiotis VA, Chatterjee P, Li L. Biochemical signaling of PD-1 on T cells and its functional implications. Cancer J. 2014;20(4):265–71. https://doi.org/10.1097/PPO.0000000000000059.
Buchbinder EI, Desai A. CTLA-4 and PD-1 pathways: similarities, differences, and implications of their inhibition. Am J Clin Oncol. 2016;39(1):98–106. https://doi.org/10.1097/COC.0000000000000239.
Bunggulawa EJ, Wang W, Yin T, Wang N, Durkan C, Wang Y, et al. Recent advancements in the use of exosomes as drug delivery systems. J Nanobiotechnology. 2018;16(1):81. https://doi.org/10.1186/s12951-018-0403-9.
Cai X, Yin Y, Li N, Zhu D, Zhang J, Zhang CY, et al. Re-polarization of tumor-associated macrophages to pro-inflammatory M1 macrophages by microRNA-155. J Mol Cell Biol. 2012;4(5):341–3. https://doi.org/10.1093/jmcb/mjs044.
Cao H, Dan Z, He X, Zhang Z, Yu H, Yin Q, et al. Liposomes coated with isolated macrophage membrane can target lung metastasis of breast cancer. ACS Nano. 2016;10(8):7738–48. https://doi.org/10.1021/acsnano.6b03148.
Carminati L, Pinessi D, Borsotti P, Minoli L, Giavazzi R, D’Incalci M, et al. Antimetastatic and antiangiogenic activity of trabectedin in cutaneous melanoma. Carcinogenesis. 2019;40(2):303–12. https://doi.org/10.1093/carcin/bgy177.
Cassetta L, Pollard JW. Targeting macrophages: therapeutic approaches in cancer. Nat Rev Drug Discov. 2018;17(12):887–904. https://doi.org/10.1038/nrd.2018.169.
Cassetta L, Pollard JW. Tumor-associated macrophages. Curr Biol. 2020;30(6):R246–R8. https://doi.org/10.1016/j.cub.2020.01.031.
Cassetta L, Fragkogianni S, Sims AH, Swierczak A, Forrester LM, Zhang H, et al. Human tumor-associated macrophage and monocyte transcriptional landscapes reveal cancer-specific reprogramming, biomarkers, and therapeutic targets. Cancer Cell. 2019;35(4):588–602. e10. https://doi.org/10.1016/j.ccell.2019.02.009.
Castells M, Thibault B, Delord JP, Couderc B. Implication of tumor microenvironment in chemoresistance: tumor-associated stromal cells protect tumor cells from cell death. Int J Mol Sci. 2012;13(8):9545–71. https://doi.org/10.3390/ijms13089545.
Ceci C, Atzori MG, Lacal PM, Graziani G. Role of VEGFs/VEGFR-1 Signaling and its Inhibition in Modulating Tumor Invasion: Experimental Evidence in Different Metastatic Cancer Models. Int J Mol Sci. 2020;21(4). doi: https://doi.org/10.3390/ijms21041388.
Chao MP, Weissman IL, Majeti R. The CD47-SIRPalpha pathway in cancer immune evasion and potential therapeutic implications. Curr Opin Immunol. 2012;24(2):225–32. https://doi.org/10.1016/j.coi.2012.01.010.
Chatterjee B, Saha P, Bose S, Shukla D, Chatterjee N, Kumar S, et al. MicroRNAs: As Critical Regulators of Tumor- Associated Macrophages. Int J Mol Sci. 2020;21(19). doi: https://doi.org/10.3390/ijms21197117.
Chavez-Galan L, Olleros ML, Vesin D, Garcia I. Much more than M1 and M2 macrophages, there are also CD169(+) and TCR(+) macrophages. Front Immunol. 2015;6:263. https://doi.org/10.3389/fimmu.2015.00263.
Chen R, Chen B. Siltuximab (CNTO 328): a promising option for human malignancies. Drug Des Devel Ther. 2015;9:3455–8. https://doi.org/10.2147/DDDT.S86438.
Chen Y, Ramjiawan RR, Reiberger T, Ng MR, Hato T, Huang Y, et al. CXCR4 inhibition in tumor microenvironment facilitates anti-programmed death receptor-1 immunotherapy in sorafenib-treated hepatocellular carcinoma in mice. Hepatology. 2015;61(5):1591–602. https://doi.org/10.1002/hep.27665.
Chen L, Li J, Wang F, Dai C, Wu F, Liu X, et al. Tie2 expression on macrophages is required for blood vessel reconstruction and tumor relapse after chemotherapy. Cancer Res. 2016;76(23):6828–38. https://doi.org/10.1158/0008-5472.CAN-16-1114.
Cheng N, Bai X, Shu Y, Ahmad O, Shen P. Targeting tumor-associated macrophages as an antitumor strategy. Biochem Pharmacol. 2020;183:114354. https://doi.org/10.1016/j.bcp.2020.114354.
Chi H, Li C, Zhao FS, Zhang L, Ng TB, Jin G, et al. Anti-tumor activity of toll-like receptor 7 agonists. Front Pharmacol. 2017;8:304. https://doi.org/10.3389/fphar.2017.00304.
Chiang CF, Chao TT, Su YF, Hsu CC, Chien CY, Chiu KC, et al. Metformin-treated cancer cells modulate macrophage polarization through AMPK-NF-kappaB signaling. Oncotarget. 2017;8(13):20706–18. doi: https://doi.org/10.18632/oncotarget.14982.
Cho HR, Kumari N, Thi Vu H, Kim H, Park CK, Choi SH. Increased antiangiogenic effect by blocking CCL2-dependent macrophages in a rodent glioblastoma model: correlation study with dynamic susceptibility contrast perfusion MRI. Sci Rep. 2019;9(1):11085. https://doi.org/10.1038/s41598-019-47438-4.
Correia AL, Bissell MJ. The tumor microenvironment is a dominant force in multidrug resistance. Drug Resist Updat. 2012;15(1–2):39–49. https://doi.org/10.1016/j.drup.2012.01.006.
Crocker PR, Gordon S. Properties and distribution of a lectin-like hemagglutinin differentially expressed by murine stromal tissue macrophages. J Exp Med. 1986;164(6):1862–75. https://doi.org/10.1084/jem.164.6.1862.
Crowther M, Brown NJ, Bishop ET, Lewis CE. Microenvironmental influence on macrophage regulation of angiogenesis in wounds and malignant tumors. J Leukoc Biol. 2001;70(4):478–90.
Czako B, Marszalek JR, Burke JP, Mandal P, Leonard PG, Cross JB, et al. Discovery of IACS-9439, a potent, exquisitely selective, and orally bioavailable inhibitor of CSF1R. J Med Chem. 2020;63(17):9888–911. https://doi.org/10.1021/acs.jmedchem.0c00936.
Da Silva CG, Camps MGM, Li T, Chan AB, Ossendorp F, Cruz LJ. Co-delivery of immunomodulators in biodegradable nanoparticles improves therapeutic efficacy of cancer vaccines. Biomaterials. 2019;220:119417. https://doi.org/10.1016/j.biomaterials.2019.119417.
Dalton HJ, Pradeep S, McGuire M, Hailemichael Y, Ma S, Lyons Y, et al. Macrophages facilitate resistance to anti-VEGF therapy by altered VEGFR expression. Clin Cancer Res. 2017;23(22):7034–46. https://doi.org/10.1158/1078-0432.CCR-17-0647.
Davies LC, Jenkins SJ, Allen JE, Taylor PR. Tissue-resident macrophages. Nat Immunol. 2013;14(10):986–95. https://doi.org/10.1038/ni.2705.
De Meyer I, Martinet W, Schrijvers DM, Timmermans JP, Bult H, De Meyer GR. Toll-like receptor 7 stimulation by imiquimod induces macrophage autophagy and inflammation in atherosclerotic plaques. Basic Res Cardiol. 2012;107(3):269. https://doi.org/10.1007/s00395-012-0269-1.
de Oliveira S, Houseright RA, Graves AL, Golenberg N, Korte BG, Miskolci V, et al. Metformin modulates innate immune-mediated inflammation and early progression of NAFLD-associated hepatocellular carcinoma in zebrafish. J Hepatol. 2019;70(4):710–21. https://doi.org/10.1016/j.jhep.2018.11.034.
De Palma M, Lewis CE. Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell. 2013;23(3):277–86. https://doi.org/10.1016/j.ccr.2013.02.013.
De Palma M, Venneri MA, Galli R, Sergi L, Politi LS, Sampaolesi M, et al. Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell. 2005;8(3):211–26. https://doi.org/10.1016/j.ccr.2005.08.002.
DeNardo DG, Barreto JB, Andreu P, Vasquez L, Tawfik D, Kolhatkar N, et al. CD4(+) T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell. 2009;16(2):91–102. https://doi.org/10.1016/j.ccr.2009.06.018.
DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011;1(1):54–67. https://doi.org/10.1158/2159-8274.CD-10-0028.
Deng J, Liang H, Zhang R, Sun D, Pan Y, Liu Y, et al. STAT3 is associated with lymph node metastasis in gastric cancer. Tumour Biol. 2013;34(5):2791–800. https://doi.org/10.1007/s13277-013-0837-5.
Deng L, Stafford JH, Liu SC, Chernikova SB, Merchant M, Recht L, et al. SDF-1 blockade enhances anti-VEGF therapy of glioblastoma and can be monitored by MRI. Neoplasia. 2017;19(1):1–7. https://doi.org/10.1016/j.neo.2016.11.010.
Dewan MZ, Ahmed S, Iwasaki Y, Ohba K, Toi M, Yamamoto N. Stromal cell-derived factor-1 and CXCR4 receptor interaction in tumor growth and metastasis of breast cancer. Biomed Pharmacother. 2006;60(6):273–6. https://doi.org/10.1016/j.biopha.2006.06.004.
Diggs LP, Ruf B, Ma C, Heinrich B, Cui L, Zhang Q, et al. CD40-mediated immune cell activation enhances response to anti-PD1 in murine intrahepatic cholangiocarcinoma. J Hepatol. 2020; https://doi.org/10.1016/j.jhep.2020.11.037.
Ding L, Liang G, Yao Z, Zhang J, Liu R, Chen H, et al. Metformin prevents cancer metastasis by inhibiting M2-like polarization of tumor associated macrophages. Oncotarget. 2015;6(34):36441–55. doi: https://doi.org/10.18632/oncotarget.5541.
Drake MT, Clarke BL, Khosla S. Bisphosphonates: mechanism of action and role in clinical practice. Mayo Clin Proc. 2008;83(9):1032–45. https://doi.org/10.4065/83.9.1032.
Duruisseaux M, Martinez-Cardus A, Calleja-Cervantes ME, Moran S. Castro de Moura M, Davalos V, et al. epigenetic prediction of response to anti-PD-1 treatment in non-small-cell lung cancer: a multicentre, retrospective analysis. Lancet Respir Med. 2018;6(10):771–81. https://doi.org/10.1016/S2213-2600(18)30284-4.
Dwyer AR, Greenland EL, Pixley FJ. Promotion of Tumor Invasion by Tumor-Associated Macrophages: The Role of CSF-1-Activated Phosphatidylinositol 3 Kinase and Src Family Kinase Motility Signaling. Cancers (Basel). 2017;9(6). doi: https://doi.org/10.3390/cancers9060068.
Epelman S, Lavine KJ, Randolph GJ. Origin and functions of tissue macrophages. Immunity. 2014;41(1):21–35. https://doi.org/10.1016/j.immuni.2014.06.013.
Erblich B, Zhu L, Etgen AM, Dobrenis K, Pollard JW. Absence of colony stimulation factor-1 receptor results in loss of microglia, disrupted brain development and olfactory deficits. PLoS One. 2011;6(10):e26317. https://doi.org/10.1371/journal.pone.0026317.
Fahham D, Weiss ID, Abraham M, Beider K, Hanna W, Shlomai Z, et al. In vitro and in vivo therapeutic efficacy of CXCR4 antagonist BKT140 against human non-small cell lung cancer. J Thorac Cardiovasc Surg. 2012;144(5):1167–75. e1. https://doi.org/10.1016/j.jtcvs.2012.07.031.
Faibish M, Francescone R, Bentley B, Yan W, Shao R. A YKL-40-neutralizing antibody blocks tumor angiogenesis and progression: a potential therapeutic agent in cancers. Mol Cancer Ther. 2011;10(5):742–51. https://doi.org/10.1158/1535-7163.MCT-10-0868.
Fang RH, Kroll AV, Gao W, Zhang L. Cell membrane coating nanotechnology. Adv Mater. 2018;30(23):e1706759. https://doi.org/10.1002/adma.201706759.
Farrell KB, Karpeisky A, Thamm DH, Zinnen S. Bisphosphonate conjugation for bone specific drug targeting. Bone Rep. 2018;9:47–60. https://doi.org/10.1016/j.bonr.2018.06.007.
Fleetwood AJ, Lawrence T, Hamilton JA, Cook AD. Granulocyte-macrophage colony-stimulating factor (CSF) and macrophage CSF-dependent macrophage phenotypes display differences in cytokine profiles and transcription factor activities: implications for CSF blockade in inflammation. J Immunol. 2007;178(8):5245–52. https://doi.org/10.4049/jimmunol.178.8.5245.
Flores-Toro JA, Luo D, Gopinath A, Sarkisian MR, Campbell JJ, Charo IF, et al. CCR2 inhibition reduces tumor myeloid cells and unmasks a checkpoint inhibitor effect to slow progression of resistant murine gliomas. Proc Natl Acad Sci U S A. 2020;117(2):1129–38. https://doi.org/10.1073/pnas.1910856117.
Fu J, Wang D, Mei D, Zhang H, Wang Z, He B, et al. Macrophage mediated biomimetic delivery system for the treatment of lung metastasis of breast cancer. J Control Release. 2015;204:11–9. https://doi.org/10.1016/j.jconrel.2015.01.039.
Fujimoto H, Sangai T, Ishii G, Ikehara A, Nagashima T, Miyazaki M, et al. Stromal MCP-1 in mammary tumors induces tumor-associated macrophage infiltration and contributes to tumor progression. Int J Cancer. 2009;125(6):1276–84. https://doi.org/10.1002/ijc.24378.
Galli S, Zlobec I, Schurch C, Perren A, Ochsenbein AF, Banz Y. CD47 protein expression in acute myeloid leukemia: a tissue microarray-based analysis. Leuk Res. 2015;39(7):749–56. https://doi.org/10.1016/j.leukres.2015.04.007.
Galon J, Bruni D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat Rev Drug Discov. 2019;18(3):197–218. https://doi.org/10.1038/s41573-018-0007-y.
Gao S, Hu J, Wu X, Liang Z. PMA treated THP-1-derived-IL-6 promotes EMT of SW48 through STAT3/ERK-dependent activation of Wnt/beta-catenin signaling pathway. Biomed Pharmacother. 2018;108:618–24. https://doi.org/10.1016/j.biopha.2018.09.067.
Genard G, Lucas S, Michiels C. Reprogramming of tumor-associated macrophages with anticancer therapies: radiotherapy versus chemo- and immunotherapies. Front Immunol. 2017;8:828. https://doi.org/10.3389/fimmu.2017.00828.
Germano G, Frapolli R, Belgiovine C, Anselmo A, Pesce S, Liguori M, et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell. 2013;23(2):249–62. https://doi.org/10.1016/j.ccr.2013.01.008.
Gilbert LA, Hemann MT. DNA damage-mediated induction of a chemoresistant niche. Cell. 2010;143(3):355–66. https://doi.org/10.1016/j.cell.2010.09.043.
Ginhoux F, Guilliams M. Tissue-resident macrophage ontogeny and homeostasis. Immunity. 2016;44(3):439–49. https://doi.org/10.1016/j.immuni.2016.02.024.
Ginhoux F, Jung S. Monocytes and macrophages: developmental pathways and tissue homeostasis. Nat Rev Immunol. 2014;14(6):392–404. https://doi.org/10.1038/nri3671.
Gocheva V, Wang HW, Gadea BB, Shree T, Hunter KE, Garfall AL, et al. IL-4 induces cathepsin protease activity in tumor-associated macrophages to promote cancer growth and invasion. Genes Dev. 2010;24(3):241–55. https://doi.org/10.1101/gad.1874010.
Golden EB, Frances D, Pellicciotta I, Demaria S, Helen Barcellos-Hoff M, Formenti SC. Radiation fosters dose-dependent and chemotherapy-induced immunogenic cell death. Onco Targets Ther. 2014;3:e28518. https://doi.org/10.4161/onci.28518.
Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5(12):953–64. https://doi.org/10.1038/nri1733.
Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature. 2017;545(7655):495–9. https://doi.org/10.1038/nature22396.
Gouaze-Andersson V, Delmas C, Taurand M, Martinez-Gala J, Evrard S, Mazoyer S, et al. FGFR1 induces glioblastoma Radioresistance through the PLCgamma/Hif1alpha pathway. Cancer Res. 2016;76(10):3036–44. https://doi.org/10.1158/0008-5472.CAN-15-2058.
Graff JW, Dickson AM, Clay G, McCaffrey AP, Wilson ME. Identifying functional microRNAs in macrophages with polarized phenotypes. J Biol Chem. 2012;287(26):21816–25. https://doi.org/10.1074/jbc.M111.327031.
Gravina GL, Mancini A, Marampon F, Colapietro A, Delle Monache S, Sferra R, et al. The brain-penetrating CXCR4 antagonist, PRX177561, increases the antitumor effects of bevacizumab and sunitinib in preclinical models of human glioblastoma. J Hematol Oncol. 2017;10(1):5. https://doi.org/10.1186/s13045-016-0377-8.
Grimshaw MJ, Balkwill FR. Inhibition of monocyte and macrophage chemotaxis by hypoxia and inflammation--a potential mechanism. Eur J Immunol. 2001;31(2):480–9. https://doi.org/10.1002/1521-4141(200102)31:2<480::aid-immu480>3.0.co;2-l.
Grossman JG, Nywening TM, Belt BA, Panni RZ, Krasnick BA, DeNardo DG, et al. Recruitment of CCR2(+) tumor associated macrophage to sites of liver metastasis confers a poor prognosis in human colorectal cancer. Onco Targets Ther. 2018;7(9):e1470729. https://doi.org/10.1080/2162402X.2018.1470729.
Guerra AD, Yeung OWH, Qi X, Kao WJ, Man K. The anti-tumor effects of M1 macrophage-loaded poly (ethylene glycol) and gelatin-based hydrogels on hepatocellular carcinoma. Theranostics. 2017;7(15):3732–44. https://doi.org/10.7150/thno.20251.
Guerriero JL. Macrophages: the road less traveled. Changing Anticancer Therapy Trends Mol Med. 2018;24(5):472–89. https://doi.org/10.1016/j.molmed.2018.03.006.
Guerriero JL. Macrophages: their untold story in T cell activation and function. Int Rev Cell Mol Biol. 2019;342:73–93. https://doi.org/10.1016/bs.ircmb.2018.07.001.
Haabeth OAW, Blake TR, McKinlay CJ, Tveita AA, Sallets A, Waymouth RM, et al. Local delivery of Ox40l, Cd80, and Cd86 mRNA kindles global anticancer immunity. Cancer Res. 2019;79(7):1624–34. https://doi.org/10.1158/0008-5472.CAN-18-2867.
Halbrook CJ, Pontious C, Kovalenko I, Lapienyte L, Dreyer S, Lee HJ, et al. Macrophage-released pyrimidines inhibit gemcitabine therapy in pancreatic cancer. Cell Metab. 2019;29(6):1390–9. e6. https://doi.org/10.1016/j.cmet.2019.02.001.
Hambardzumyan D, Gutmann DH, Kettenmann H. The role of microglia and macrophages in glioma maintenance and progression. Nat Neurosci. 2016;19(1):20–7. https://doi.org/10.1038/nn.4185.
Haniffa M, Bigley V, Collin M. Human mononuclear phagocyte system reunited. Semin Cell Dev Biol. 2015;41:59–69. https://doi.org/10.1016/j.semcdb.2015.05.004.
Hao Q, Vadgama JV, Wang P. CCL2/CCR2 signaling in cancer pathogenesis. Cell Commun Signal. 2020;18(1):82. https://doi.org/10.1186/s12964-020-00589-8.
Hasham MG, Baxan N, Stuckey DJ, Branca J, Perkins B, Dent O, et al. Systemic autoimmunity induced by the TLR7/8 agonist Resiquimod causes myocarditis and dilated cardiomyopathy in a new mouse model of autoimmune heart disease. Dis Model Mech. 2017;10(3):259–70. https://doi.org/10.1242/dmm.027409.
Hirano H, Tanioka K, Yokoyama S, Akiyama S, Kuratsu J. Angiogenic effect of thymidine phosphorylase on macrophages in glioblastoma multiforme. J Neurosurg. 2001;95(1):89–95. https://doi.org/10.3171/jns.2001.95.1.0089.
Hofmann MA, Kors C, Audring H, Walden P, Sterry W, Trefzer U. Phase 1 evaluation of intralesionally injected TLR9-agonist PF-3512676 in patients with basal cell carcinoma or metastatic melanoma. J Immunother. 2008;31(5):520–7. https://doi.org/10.1097/CJI.0b013e318174a4df.
Holen I, Coleman RE. Anti-tumour activity of bisphosphonates in preclinical models of breast cancer. Breast Cancer Res. 2010;12(6):214. https://doi.org/10.1186/bcr2769.
Hollmen M, Karaman S, Schwager S, Lisibach A, Christiansen AJ, Maksimow M, et al. G-CSF regulates macrophage phenotype and associates with poor overall survival in human triple-negative breast cancer. Onco Targets Ther. 2016;5(3):e1115177. https://doi.org/10.1080/2162402X.2015.1115177.
Huang H, Lai JY, Do J, Liu D, Li L, Del Rosario J, et al. Specifically targeting angiopoietin-2 inhibits angiogenesis, Tie2-expressing monocyte infiltration, and tumor growth. Clin Cancer Res. 2011;17(5):1001–11. https://doi.org/10.1158/1078-0432.CCR-10-2317.
Hughes R, Qian BZ, Rowan C, Muthana M, Keklikoglou I, Olson OC, et al. Perivascular M2 macrophages stimulate tumor relapse after chemotherapy. Cancer Res. 2015;75(17):3479–91. https://doi.org/10.1158/0008-5472.CAN-14-3587.
Im JH, Buzzelli JN, Jones K, Franchini F, Gordon-Weeks A, Markelc B, et al. FGF2 alters macrophage polarization, tumour immunity and growth and can be targeted during radiotherapy. Nat Commun. 2020;11(1):4064. https://doi.org/10.1038/s41467-020-17914-x.
Itatani Y, Kawada K, Yamamoto T, Sakai Y. Resistance to Anti-Angiogenic Therapy in Cancer-Alterations to Anti-VEGF Pathway. Int J Mol Sci. 2018;19(4). doi: https://doi.org/10.3390/ijms19041232.
Jahchan NS, Mujal AM, Pollack JL, Binnewies M, Sriram V, Reyno L, et al. Tuning the tumor myeloid microenvironment to fight cancer. Front Immunol. 2019;10:1611. https://doi.org/10.3389/fimmu.2019.01611.
Jaiswal S, Jamieson CH, Pang WW, Park CY, Chao MP, Majeti R, et al. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell. 2009;138(2):271–85. https://doi.org/10.1016/j.cell.2009.05.046.
Jinushi M, Chiba S, Yoshiyama H, Masutomi K, Kinoshita I, Dosaka-Akita H, et al. Tumor-associated macrophages regulate tumorigenicity and anticancer drug responses of cancer stem/initiating cells. Proc Natl Acad Sci U S A. 2011;108(30):12425–30. https://doi.org/10.1073/pnas.1106645108.
Johansson-Percival A, He B, Ganss R. Immunomodulation of tumor vessels: it takes two to tango. Trends Immunol. 2018;39(10):801–14. https://doi.org/10.1016/j.it.2018.08.001.
Jones JD, Sinder BP, Paige D, Soki FN, Koh AJ, Thiele S, et al. Trabectedin reduces skeletal prostate cancer tumor size in association with effects on M2 macrophages and Efferocytosis. Neoplasia. 2019;21(2):172–84. https://doi.org/10.1016/j.neo.2018.11.003.
Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer. 2009;9(4):239–52. https://doi.org/10.1038/nrc2618.
Kaminski WE, Beham AW, Kzhyshkowska J, Gratchev A, Puellmann K. On the horizon: flexible immune recognition outside lymphocytes. Immunobiology. 2013;218(3):418–26. https://doi.org/10.1016/j.imbio.2012.05.024.
Karapetyan L, Luke JJ, Davar D. Toll-like receptor 9 agonists in cancer. Onco Targets Ther. 2020;13:10039–60. https://doi.org/10.2147/OTT.S247050.
Katsuya Y, Horinouchi H, Asao T, Kitahara S, Goto Y, Kanda S, et al. Expression of programmed death 1 (PD-1) and its ligand (PD-L1) in thymic epithelial tumors: impact on treatment efficacy and alteration in expression after chemotherapy. Lung Cancer. 2016;99:4–10. https://doi.org/10.1016/j.lungcan.2016.05.007.
Kim MS, Haney MJ, Zhao Y, Mahajan V, Deygen I, Klyachko NL, et al. Development of exosome-encapsulated paclitaxel to overcome MDR in cancer cells. Nanomedicine. 2016;12(3):655–64. https://doi.org/10.1016/j.nano.2015.10.012.
Kitamura T, Qian BZ, Soong D, Cassetta L, Noy R, Sugano G, et al. CCL2-induced chemokine cascade promotes breast cancer metastasis by enhancing retention of metastasis-associated macrophages. J Exp Med. 2015;212(7):1043–59. https://doi.org/10.1084/jem.20141836.
Kitamura T, Doughty-Shenton D, Cassetta L, Fragkogianni S, Brownlie D, Kato Y, et al. Monocytes differentiate to immune suppressive precursors of metastasis-associated macrophages in mouse models of metastatic breast cancer. Front Immunol. 2017;8:2004. https://doi.org/10.3389/fimmu.2017.02004.
Kloepper J, Riedemann L, Amoozgar Z, Seano G, Susek K, Yu V, et al. Ang-2/VEGF bispecific antibody reprograms macrophages and resident microglia to anti-tumor phenotype and prolongs glioblastoma survival. Proc Natl Acad Sci U S A. 2016;113(16):4476–81. https://doi.org/10.1073/pnas.1525360113.
Klug F, Prakash H, Huber PE, Seibel T, Bender N, Halama N, et al. Low-dose irradiation programs macrophage differentiation to an iNOS(+)/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell. 2013;24(5):589–602. https://doi.org/10.1016/j.ccr.2013.09.014.
Koh YW, Park C, Yoon DH, Suh C, Huh J. CSF-1R expression in tumor-associated macrophages is associated with worse prognosis in classical Hodgkin lymphoma. Am J Clin Pathol. 2014;141(4):573–83. https://doi.org/10.1309/AJCPR92TDDFARISU.
Kubota Y, Takubo K, Shimizu T, Ohno H, Kishi K, Shibuya M, et al. M-CSF inhibition selectively targets pathological angiogenesis and lymphangiogenesis. J Exp Med. 2009;206(5):1089–102. https://doi.org/10.1084/jem.20081605.
Kuhne MR, Mulvey T, Belanger B, Chen S, Pan C, Chong C, et al. BMS-936564/MDX-1338: a fully human anti-CXCR4 antibody induces apoptosis in vitro and shows antitumor activity in vivo in hematologic malignancies. Clin Cancer Res. 2013;19(2):357–66. https://doi.org/10.1158/1078-0432.CCR-12-2333.
Kulkarni A, Chandrasekar V, Natarajan SK, Ramesh A, Pandey P, Nirgud J, et al. A designer self-assembled supramolecule amplifies macrophage immune responses against aggressive cancer. Nat Biomed Eng. 2018;2(8):589–99. https://doi.org/10.1038/s41551-018-0254-6.
La Fleur L, Botling J, He F, Pelicano C, Zhou C, He C, et al. Targeting MARCO and IL-37R on immunosuppressive macrophages in lung cancer blocks regulatory T cells and supports cytotoxic lymphocyte function. Cancer Res. 2020; https://doi.org/10.1158/0008-5472.CAN-20-1885.
Laoui D, Movahedi K, Van Overmeire E, Van den Bossche J, Schouppe E, Mommer C, et al. Tumor-associated macrophages in breast cancer: distinct subsets, distinct functions. Int J Dev Biol. 2011;55(7–9):861–7. https://doi.org/10.1387/ijdb.113371dl.
Laoui D, Van Overmeire E, De Baetselier P, Van Ginderachter JA, Raes G. Functional relationship between tumor-associated macrophages and macrophage Colony-stimulating factor as contributors to cancer progression. Front Immunol. 2014;5:489. https://doi.org/10.3389/fimmu.2014.00489.
Lapeyre-Prost A, Terme M, Pernot S, Pointet AL, Voron T, Tartour E, et al. Immunomodulatory activity of VEGF in cancer. Int Rev Cell Mol Biol. 2017;330:295–342. https://doi.org/10.1016/bs.ircmb.2016.09.007.
Larionova I, Cherdyntseva N, Liu T, Patysheva M, Rakina M, Kzhyshkowska J. Interaction of tumor-associated macrophages and cancer chemotherapy. Onco Targets Ther. 2019;8(7):1596004. https://doi.org/10.1080/2162402X.2019.1596004.
Lee SH, Jeong D, Han YS, Baek MJ. Pivotal role of vascular endothelial growth factor pathway in tumor angiogenesis. Ann Surg Treat Res. 2015;89(1):1–8. https://doi.org/10.4174/astr.2015.89.1.1.
Leek RD, Lewis CE, Whitehouse R, Greenall M, Clarke J, Harris AL. Association of macrophage infiltration with angiogenesis and prognosis in invasive breast carcinoma. Cancer Res. 1996;56(20):4625–9.
Leek RD, Landers RJ, Harris AL, Lewis CE. Necrosis correlates with high vascular density and focal macrophage infiltration in invasive carcinoma of the breast. Br J Cancer. 1999;79(5–6):991–5. https://doi.org/10.1038/sj.bjc.6690158.
Lewis JS, Landers RJ, Underwood JC, Harris AL, Lewis CE. Expression of vascular endothelial growth factor by macrophages is up-regulated in poorly vascularized areas of breast carcinomas. J Pathol. 2000;192(2):150–8. https://doi.org/10.1002/1096-9896(2000)9999:9999<::AID-PATH687>3.0.CO;2-G.
Li F, Sethi G. Targeting transcription factor NF-kappaB to overcome chemoresistance and radioresistance in cancer therapy. Biochim Biophys Acta. 2010;1805(2):167–80. https://doi.org/10.1016/j.bbcan.2010.01.002.
Li X, Yao W, Yuan Y, Chen P, Li B, Li J, et al. Targeting of tumour-infiltrating macrophages via CCL2/CCR2 signalling as a therapeutic strategy against hepatocellular carcinoma. Gut. 2017;66(1):157–67. https://doi.org/10.1136/gutjnl-2015-310514.
Li X, Liu R, Su X, Pan Y, Han X, Shao C, et al. Harnessing tumor-associated macrophages as aids for cancer immunotherapy. Mol Cancer. 2019a;18(1):177. https://doi.org/10.1186/s12943-019-1102-3.
Li X, Bu W, Meng L, Liu X, Wang S, Jiang L, et al. CXCL12/CXCR4 pathway orchestrates CSC-like properties by CAF recruited tumor associated macrophage in OSCC. Exp Cell Res. 2019b;378(2):131–8. https://doi.org/10.1016/j.yexcr.2019.03.013.
Liang S, Zheng J, Wu W, Li Q, Saw PE, Chen J, et al. A robust nanoparticle platform for RNA interference in macrophages to suppress tumor cell migration. Front Pharmacol. 2018;9:1465. https://doi.org/10.3389/fphar.2018.01465.
Liekens S, Schols D, Hatse S. CXCL12-CXCR4 axis in angiogenesis, metastasis and stem cell mobilization. Curr Pharm Des. 2010;16(35):3903–20. https://doi.org/10.2174/138161210794455003.
Lim SY, Yuzhalin AE, Gordon-Weeks AN, Muschel RJ. Targeting the CCL2-CCR2 signaling axis in cancer metastasis. Oncotarget. 2016;7(19):28697–710. doi: https://doi.org/10.18632/oncotarget.7376.
Lin EY, Pollard JW. Tumor-associated macrophages press the angiogenic switch in breast cancer. Cancer Res. 2007;67(11):5064–6. https://doi.org/10.1158/0008-5472.CAN-07-0912.
Lin EY, Nguyen AV, Russell RG, Pollard JW. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J Exp Med. 2001;193(6):727–40. https://doi.org/10.1084/jem.193.6.727.
Linde N, Lederle W, Depner S, van Rooijen N, Gutschalk CM, Mueller MM. Vascular endothelial growth factor-induced skin carcinogenesis depends on recruitment and alternative activation of macrophages. J Pathol. 2012;227(1):17–28. https://doi.org/10.1002/path.3989.
Liu L, He H, Liang R, Yi H, Meng X, Chen Z, et al. ROS-inducing micelles sensitize tumor-associated macrophages to TLR3 stimulation for potent immunotherapy. Biomacromolecules. 2018;19(6):2146–55. https://doi.org/10.1021/acs.biomac.8b00239.
Loberg RD, Ying C, Craig M, Day LL, Sargent E, Neeley C, et al. Targeting CCL2 with systemic delivery of neutralizing antibodies induces prostate cancer tumor regression in vivo. Cancer Res. 2007;67(19):9417–24. https://doi.org/10.1158/0008-5472.CAN-07-1286.
Lu-Emerson C, Snuderl M, Kirkpatrick ND, Goveia J, Davidson C, Huang Y, et al. Increase in tumor-associated macrophages after antiangiogenic therapy is associated with poor survival among patients with recurrent glioblastoma. Neuro-Oncology. 2013;15(8):1079–87. https://doi.org/10.1093/neuonc/not082.
Ma R, Yi B, Riker AI, Xi Y. Metformin and cancer immunity. Acta Pharmacol Sin. 2020;41(11):1403–9. https://doi.org/10.1038/s41401-020-00508-0.
MacDonald KP, Palmer JS, Cronau S, Seppanen E, Olver S, Raffelt NC, et al. An antibody against the colony-stimulating factor 1 receptor depletes the resident subset of monocytes and tissue- and tumor-associated macrophages but does not inhibit inflammation. Blood. 2010;116(19):3955–63. https://doi.org/10.1182/blood-2010-02-266296.
Mantovani A, Allavena P. The interaction of anticancer therapies with tumor-associated macrophages. J Exp Med. 2015;212(4):435–45. https://doi.org/10.1084/jem.20150295.
Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23(11):549–55. https://doi.org/10.1016/s1471-4906(02)02302-5.
Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. 2017;14(7):399–416. https://doi.org/10.1038/nrclinonc.2016.217.
Martinez-Pomares L, Gordon S. CD169+ macrophages at the crossroads of antigen presentation. Trends Immunol. 2012;33(2):66–70. https://doi.org/10.1016/j.it.2011.11.001.
Martinez-Pomares L, Kosco-Vilbois M, Darley E, Tree P, Herren S, Bonnefoy JY, et al. Fc chimeric protein containing the cysteine-rich domain of the murine mannose receptor binds to macrophages from splenic marginal zone and lymph node subcapsular sinus and to germinal centers. J Exp Med. 1996;184(5):1927–37. https://doi.org/10.1084/jem.184.5.1927.
Mazzieri R, Pucci F, Moi D, Zonari E, Ranghetti A, Berti A, et al. Targeting the ANG2/TIE2 axis inhibits tumor growth and metastasis by impairing angiogenesis and disabling rebounds of proangiogenic myeloid cells. Cancer Cell. 2011;19(4):512–26. https://doi.org/10.1016/j.ccr.2011.02.005.
Mazzone M, Bergers G. Regulation of blood and lymphatic vessels by immune cells in tumors and metastasis. Annu Rev Physiol. 2019;81:535–60. https://doi.org/10.1146/annurev-physiol-020518-114721.
McClellan JL, Davis JM, Steiner JL, Enos RT, Jung SH, Carson JA, et al. Linking tumor-associated macrophages, inflammation, and intestinal tumorigenesis: role of MCP-1. Am J Physiol Gastrointest Liver Physiol. 2012;303(10):G1087–95. https://doi.org/10.1152/ajpgi.00252.2012.
Medler TR, Murugan D, Horton W, Kumar S, Cotechini T, Forsyth AM, et al. Complement C5a fosters squamous carcinogenesis and limits T cell response to chemotherapy. Cancer Cell. 2018;34(4):561–78. e6. https://doi.org/10.1016/j.ccell.2018.09.003.
Meng Y, Beckett MA, Liang H, Mauceri HJ, van Rooijen N, Cohen KS, et al. Blockade of tumor necrosis factor alpha signaling in tumor-associated macrophages as a radiosensitizing strategy. Cancer Res. 2010;70(4):1534–43. https://doi.org/10.1158/0008-5472.CAN-09-2995.
Milane L, Singh A, Mattheolabakis G, Suresh M, Amiji MM. Exosome mediated communication within the tumor microenvironment. J Control Release. 2015;219:278–94. https://doi.org/10.1016/j.jconrel.2015.06.029.
Miller MA, Zheng YR, Gadde S, Pfirschke C, Zope H, Engblom C, et al. Tumour-associated macrophages act as a slow-release reservoir of nano-therapeutic Pt(IV) pro-drug. Nat Commun. 2015;6:8692. https://doi.org/10.1038/ncomms9692.
Mitchem JB, Brennan DJ, Knolhoff BL, Belt BA, Zhu Y, Sanford DE, et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013;73(3):1128–41. https://doi.org/10.1158/0008-5472.CAN-12-2731.
Morandi F, Pistoia V. Interactions between HLA-G and HLA-E in physiological and pathological conditions. Front Immunol. 2014;5:394. https://doi.org/10.3389/fimmu.2014.00394.
Mori T, Miyamoto T, Yoshida H, Asakawa M, Kawasumi M, Kobayashi T, et al. IL-1beta and TNFalpha-initiated IL-6-STAT3 pathway is critical in mediating inflammatory cytokines and RANKL expression in inflammatory arthritis. Int Immunol. 2011;23(11):701–12. https://doi.org/10.1093/intimm/dxr077.
Movahedi K, Laoui D, Gysemans C, Baeten M, Stange G, Van den Bossche J, et al. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer Res. 2010;70(14):5728–39. https://doi.org/10.1158/0008-5472.CAN-09-4672.
Mummalaneni S, Qian J, Phan TH, Rhyu MR, Heck GL, DeSimone JA, et al. Effect of ENaC modulators on rat neural responses to NaCl. PLoS One. 2014;9(5):e98049. https://doi.org/10.1371/journal.pone.0098049.
Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41(1):14–20. https://doi.org/10.1016/j.immuni.2014.06.008.
Nagai T, Tanaka M, Tsuneyoshi Y, Xu B, Michie SA, Hasui K, et al. Targeting tumor-associated macrophages in an experimental glioma model with a recombinant immunotoxin to folate receptor beta. Cancer Immunol Immunother. 2009;58(10):1577–86. https://doi.org/10.1007/s00262-009-0667-x.
Nagarsheth N, Wicha MS, Zou W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat Rev Immunol. 2017;17(9):559–72. https://doi.org/10.1038/nri.2017.49.
Neophytou CM, Pierides C, Christodoulou MI, Costeas P, Kyriakou TC, Papageorgis P. The role of tumor-associated myeloid cells in modulating cancer therapy. Front Oncol. 2020;10:899. https://doi.org/10.3389/fonc.2020.00899.
Neyen C, Pluddemann A, Mukhopadhyay S, Maniati E, Bossard M, Gordon S, et al. Macrophage scavenger receptor a promotes tumor progression in murine models of ovarian and pancreatic cancer. J Immunol. 2013;190(7):3798–805. https://doi.org/10.4049/jimmunol.1203194.
Ngambenjawong C, Cieslewicz M, Schellinger JG, Pun SH. Synthesis and evaluation of multivalent M2pep peptides for targeting alternatively activated M2 macrophages. J Control Release. 2016;224:103–11. https://doi.org/10.1016/j.jconrel.2015.12.057.
Nishida N, Yano H, Nishida T, Kamura T, Kojiro M. Angiogenesis in cancer. Vasc Health Risk Manag. 2006;2(3):213–9. https://doi.org/10.2147/vhrm.2006.2.3.213.
Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014;41(1):49–61. https://doi.org/10.1016/j.immuni.2014.06.010.
Nywening TM, Wang-Gillam A, Sanford DE, Belt BA, Panni RZ, Cusworth BM, et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: a single-Centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. 2016;17(5):651–62. https://doi.org/10.1016/S1470-2045(16)00078-4.
Okazawa H, Motegi S, Ohyama N, Ohnishi H, Tomizawa T, Kaneko Y, et al. Negative regulation of phagocytosis in macrophages by the CD47-SHPS-1 system. J Immunol. 2005;174(4):2004–11. https://doi.org/10.4049/jimmunol.174.4.2004.
Olingy CE, Dinh HQ, Hedrick CC. Monocyte heterogeneity and functions in cancer. J Leukoc Biol. 2019;106(2):309–22. https://doi.org/10.1002/JLB.4RI0818-311R.
Olson OC, Kim H, Quail DF, Foley EA, Joyce JA. Tumor-associated macrophages suppress the cytotoxic activity of antimitotic agents. Cell Rep. 2017;19(1):101–13. https://doi.org/10.1016/j.celrep.2017.03.038.
Otani Y, Kijima T, Kohmo S, Oishi S, Minami T, Nagatomo I, et al. Suppression of metastases of small cell lung cancer cells in mice by a peptidic CXCR4 inhibitor TF14016. FEBS Lett. 2012;586(20):3639–44. https://doi.org/10.1016/j.febslet.2012.08.011.
Owen JL, Mohamadzadeh M. Macrophages and chemokines as mediators of angiogenesis. Front Physiol. 2013;4:159. https://doi.org/10.3389/fphys.2013.00159.
Pathria P, Louis TL, Varner JA. Targeting tumor-associated macrophages in cancer. Trends Immunol. 2019;40(4):310–27. https://doi.org/10.1016/j.it.2019.02.003.
Paulus P, Stanley ER, Schafer R, Abraham D, Aharinejad S. Colony-stimulating factor-1 antibody reverses chemoresistance in human MCF-7 breast cancer xenografts. Cancer Res. 2006;66(8):4349–56. https://doi.org/10.1158/0008-5472.CAN-05-3523.
Perkins H, Khodai T, Mechiche H, Colman P, Burden F, Laxton C, et al. Therapy with TLR7 agonists induces lymphopenia: correlating pharmacology to mechanism in a mouse model. J Clin Immunol. 2012;32(5):1082–92. https://doi.org/10.1007/s10875-012-9687-y.
Perrotta C, Cervia D, Di Renzo I, Moscheni C, Bassi MT, Campana L, et al. Nitric oxide generated by tumor-associated macrophages is responsible for cancer resistance to cisplatin and correlated with Syntaxin 4 and acid sphingomyelinase inhibition. Front Immunol. 2018;9:1186. https://doi.org/10.3389/fimmu.2018.01186.
Petrova PS, Viller NN, Wong M, Pang X, Lin GH, Dodge K, et al. TTI-621 (SIRPalphaFc): a CD47-blocking innate immune checkpoint inhibitor with broad antitumor activity and minimal erythrocyte binding. Clin Cancer Res. 2017;23(4):1068–79. https://doi.org/10.1158/1078-0432.CCR-16-1700.
Prakash H, Klug F, Nadella V, Mazumdar V, Schmitz-Winnenthal H, Umansky L. Low doses of gamma irradiation potentially modifies immunosuppressive tumor microenvironment by retuning tumor-associated macrophages: lesson from insulinoma. Carcinogenesis. 2016;37(3):301–13. https://doi.org/10.1093/carcin/bgw007.
Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141(1):39–51. https://doi.org/10.1016/j.cell.2010.03.014.
Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011;475(7355):222–5. https://doi.org/10.1038/nature10138.
Qian J, Olbrecht S, Boeckx B, Vos H, Laoui D, Etlioglu E, et al. A pan-cancer blueprint of the heterogeneous tumor microenvironment revealed by single-cell profiling. Cell Res. 2020;30(9):745–62. https://doi.org/10.1038/s41422-020-0355-0.
Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19(11):1423–37. https://doi.org/10.1038/nm.3394.
Rayamajhi S, Nguyen TDT, Marasini R, Aryal S. Macrophage-derived exosome-mimetic hybrid vesicles for tumor targeted drug delivery. Acta Biomater. 2019;94:482–94. https://doi.org/10.1016/j.actbio.2019.05.054.
Ries CH, Cannarile MA, Hoves S, Benz J, Wartha K, Runza V, et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell. 2014;25(6):846–59. https://doi.org/10.1016/j.ccr.2014.05.016.
Rodell CB, Arlauckas SP, Cuccarese MF, Garris CS, Li R, Ahmed MS, et al. TLR7/8-agonist-loaded nanoparticles promote the polarization of tumour-associated macrophages to enhance cancer immunotherapy. Nat Biomed Eng. 2018;2(8):578–88. https://doi.org/10.1038/s41551-018-0236-8.
Rogers TL, Holen I. Tumour macrophages as potential targets of bisphosphonates. J Transl Med. 2011;9:177. https://doi.org/10.1186/1479-5876-9-177.
Rolny C, Mazzone M, Tugues S, Laoui D, Johansson I, Coulon C, et al. HRG inhibits tumor growth and metastasis by inducing macrophage polarization and vessel normalization through downregulation of PlGF. Cancer Cell. 2011;19(1):31–44. https://doi.org/10.1016/j.ccr.2010.11.009.
Ruffell B, Coussens LM. Macrophages and therapeutic resistance in cancer. Cancer Cell. 2015;27(4):462–72. https://doi.org/10.1016/j.ccell.2015.02.015.
Ruffell B, Chang-Strachan D, Chan V, Rosenbusch A, Ho CM, Pryer N, et al. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell. 2014;26(5):623–37. https://doi.org/10.1016/j.ccell.2014.09.006.
Salvagno C, Ciampricotti M, Tuit S, Hau CS, van Weverwijk A, Coffelt SB, et al. Therapeutic targeting of macrophages enhances chemotherapy efficacy by unleashing type I interferon response. Nat Cell Biol. 2019;21(4):511–21. https://doi.org/10.1038/s41556-019-0298-1.
Sandhu SK, Papadopoulos K, Fong PC, Patnaik A, Messiou C, Olmos D, et al. A first-in-human, first-in-class, phase I study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 in patients with solid tumors. Cancer Chemother Pharmacol. 2013;71(4):1041–50. https://doi.org/10.1007/s00280-013-2099-8.
Sanford DE, Belt BA, Panni RZ, Mayer A, Deshpande AD, Carpenter D, et al. Inflammatory monocyte mobilization decreases patient survival in pancreatic cancer: a role for targeting the CCL2/CCR2 axis. Clin Cancer Res. 2013;19(13):3404–15. https://doi.org/10.1158/1078-0432.CCR-13-0525.
Sangaletti S, Di Carlo E, Gariboldi S, Miotti S, Cappetti B, Parenza M, et al. Macrophage-derived SPARC bridges tumor cell-extracellular matrix interactions toward metastasis. Cancer Res. 2008;68(21):9050–9. https://doi.org/10.1158/0008-5472.CAN-08-1327.
Sanmamed MF, Perez-Gracia JL, Schalper KA, Fusco JP, Gonzalez A, Rodriguez-Ruiz ME, et al. Changes in serum interleukin-8 (IL-8) levels reflect and predict response to anti-PD-1 treatment in melanoma and non-small-cell lung cancer patients. Ann Oncol. 2017;28(8):1988–95. https://doi.org/10.1093/annonc/mdx190.
Santarpia M, Karachaliou N. Tumor immune microenvironment characterization and response to anti-PD-1 therapy. Cancer Biol Med. 2015;12(2):74–8. https://doi.org/10.7497/j.issn.2095-3941.2015.0022.
Sato T, Terai M, Tamura Y, Alexeev V, Mastrangelo MJ, Selvan SR. Interleukin 10 in the tumor microenvironment: a target for anticancer immunotherapy. Immunol Res. 2011;51(2–3):170–82. https://doi.org/10.1007/s12026-011-8262-6.
Sawa-Wejksza K, Kandefer-Szerszen M. Tumor-associated macrophages as target for antitumor therapy. Arch Immunol Ther Exp. 2018;66(2):97–111. https://doi.org/10.1007/s00005-017-0480-8.
Scheuer W, Thomas M, Hanke P, Sam J, Osl F, Weininger D, et al. Anti-tumoral, anti-angiogenic and anti-metastatic efficacy of a tetravalent bispecific antibody (TAvi6) targeting VEGF-A and angiopoietin-2. MAbs. 2016;8(3):562–73. https://doi.org/10.1080/19420862.2016.1147640.
Seliger B. Combinatorial approaches with checkpoint inhibitors to enhance anti-tumor immunity. Front Immunol. 2019;10:999. https://doi.org/10.3389/fimmu.2019.00999.
Shime H, Matsumoto M, Oshiumi H, Tanaka S, Nakane A, Iwakura Y, et al. Toll-like receptor 3 signaling converts tumor-supporting myeloid cells to tumoricidal effectors. Proc Natl Acad Sci U S A. 2012;109(6):2066–71. https://doi.org/10.1073/pnas.1113099109.
Shree T, Olson OC, Elie BT, Kester JC, Garfall AL, Simpson K, et al. Macrophages and cathepsin proteases blunt chemotherapeutic response in breast cancer. Genes Dev. 2011;25(23):2465–79. https://doi.org/10.1101/gad.180331.111.
Smith MP, Sanchez-Laorden B, O’Brien K, Brunton H, Ferguson J, Young H, et al. The immune microenvironment confers resistance to MAPK pathway inhibitors through macrophage-derived TNFalpha. Cancer Discov. 2014;4(10):1214–29. https://doi.org/10.1158/2159-8290.CD-13-1007.
Solinas G, Germano G, Mantovani A, Allavena P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J Leukoc Biol. 2009;86(5):1065–73. https://doi.org/10.1189/jlb.0609385.
Song L, Asgharzadeh S, Salo J, Engell K, Wu HW, Sposto R, et al. Valpha24-invariant NKT cells mediate antitumor activity via killing of tumor-associated macrophages. J Clin Invest. 2009;119(6):1524–36. https://doi.org/10.1172/JCI37869.
Stafford JH, Hirai T, Deng L, Chernikova SB, Urata K, West BL, et al. Colony stimulating factor 1 receptor inhibition delays recurrence of glioblastoma after radiation by altering myeloid cell recruitment and polarization. Neuro-Oncology. 2016;18(6):797–806. https://doi.org/10.1093/neuonc/nov272.
Sun X, Cheng G, Hao M, Zheng J, Zhou X, Zhang J, et al. CXCL12 / CXCR4 / CXCR7 chemokine axis and cancer progression. Cancer Metastasis Rev. 2010;29(4):709–22. https://doi.org/10.1007/s10555-010-9256-x.
Sunakawa Y, Stintzing S, Cao S, Heinemann V, Cremolini C, Falcone A, et al. Variations in genes regulating tumor-associated macrophages (TAMs) to predict outcomes of bevacizumab-based treatment in patients with metastatic colorectal cancer: results from TRIBE and FIRE3 trials. Ann Oncol. 2015;26(12):2450–6. https://doi.org/10.1093/annonc/mdv474.
Sung WW, Wang YC, Lin PL, Cheng YW, Chen CY, Wu TC, et al. IL-10 promotes tumor aggressiveness via upregulation of CIP2A transcription in lung adenocarcinoma. Clin Cancer Res. 2013;19(15):4092–103. https://doi.org/10.1158/1078-0432.CCR-12-3439.
Swierczak A, Cook AD, Lenzo JC, Restall CM, Doherty JP, Anderson RL, et al. The promotion of breast cancer metastasis caused by inhibition of CSF-1R/CSF-1 signaling is blocked by targeting the G-CSF receptor. Cancer Immunol Res. 2014;2(8):765–76. https://doi.org/10.1158/2326-6066.CIR-13-0190.
Syn N, Wang L, Sethi G, Thiery JP, Goh BC. Exosome-mediated metastasis: from epithelial-mesenchymal transition to escape from Immunosurveillance. Trends Pharmacol Sci. 2016;37(7):606–17. https://doi.org/10.1016/j.tips.2016.04.006.
Szulzewsky F, Pelz A, Feng X, Synowitz M, Markovic D, Langmann T, et al. Glioma-associated microglia/macrophages display an expression profile different from M1 and M2 polarization and highly express Gpnmb and Spp1. PLoS One. 2015;10(2):e0116644. https://doi.org/10.1371/journal.pone.0116644.
Talks KL, Turley H, Gatter KC, Maxwell PH, Pugh CW, Ratcliffe PJ, et al. The expression and distribution of the hypoxia-inducible factors HIF-1alpha and HIF-2alpha in normal human tissues, cancers, and tumor-associated macrophages. Am J Pathol. 2000;157(2):411–21. https://doi.org/10.1016/s0002-9440(10)64554-3.
Tamamura H, Hori A, Kanzaki N, Hiramatsu K, Mizumoto M, Nakashima H, et al. T140 analogs as CXCR4 antagonists identified as anti-metastatic agents in the treatment of breast cancer. FEBS Lett. 2003;550(1–3):79–83. https://doi.org/10.1016/s0014-5793(03)00824-x.
Tang X, Sui D, Liu M, Zhang H, Liu M, Wang S, et al. Targeted delivery of zoledronic acid through the sialic acid - Siglec axis for killing and reversal of M2 phenotypic tumor-associated macrophages - a promising cancer immunotherapy. Int J Pharm. 2020;590:119929. https://doi.org/10.1016/j.ijpharm.2020.119929.
Taniguchi K, Karin M. IL-6 and related cytokines as the critical lynchpins between inflammation and cancer. Semin Immunol. 2014;26(1):54–74. https://doi.org/10.1016/j.smim.2014.01.001.
Tao Y, Ning M, Dou H. A novel therapeutic system for malignant glioma: nanoformulation, pharmacokinetic, and anticancer properties of cell-nano-drug delivery. Nanomedicine. 2013;9(2):222–32. https://doi.org/10.1016/j.nano.2012.10.006.
Teng KY, Han J, Zhang X, Hsu SH, He S, Wani NA, et al. Blocking the CCL2-CCR2 Axis using CCL2-neutralizing antibody is an effective therapy for hepatocellular cancer in a mouse model. Mol Cancer Ther. 2017;16(2):312–22. https://doi.org/10.1158/1535-7163.MCT-16-0124.
Thauvin C, Widmer J, Mottas I, Hocevar S, Allemann E, Bourquin C, et al. Development of resiquimod-loaded modified PLA-based nanoparticles for cancer immunotherapy: a kinetic study. Eur J Pharm Biopharm. 2019;139:253–61. https://doi.org/10.1016/j.ejpb.2019.04.007.
Tosato G, Jones KD. Interleukin-1 induces interleukin-6 production in peripheral blood monocytes. Blood. 1990;75(6):1305–10.
Tsai RK, Discher DE. Inhibition of "self" engulfment through deactivation of myosin-II at the phagocytic synapse between human cells. J Cell Biol. 2008;180(5):989–1003. https://doi.org/10.1083/jcb.200708043.
Tseng D, Volkmer JP, Willingham SB, Contreras-Trujillo H, Fathman JW, Fernhoff NB, et al. Anti-CD47 antibody-mediated phagocytosis of cancer by macrophages primes an effective antitumor T-cell response. Proc Natl Acad Sci U S A. 2013;110(27):11103–8. https://doi.org/10.1073/pnas.1305569110.
Valeta-Magara A, Gadi A, Volta V, Walters B, Arju R, Giashuddin S, et al. Inflammatory breast cancer promotes development of M2 tumor-associated macrophages and cancer mesenchymal cells through a complex chemokine network. Cancer Res. 2019;79(13):3360–71. https://doi.org/10.1158/0008-5472.CAN-17-2158.
van Beek EM, Cochrane F, Barclay AN, van den Berg TK. Signal regulatory proteins in the immune system. J Immunol. 2005;175(12):7781–7. https://doi.org/10.4049/jimmunol.175.12.7781.
Varol C, Mildner A, Jung S. Macrophages: development and tissue specialization. Annu Rev Immunol. 2015;33:643–75. https://doi.org/10.1146/annurev-immunol-032414-112220.
Vela M, Aris M, Llorente M, Garcia-Sanz JA, Kremer L. Chemokine receptor-specific antibodies in cancer immunotherapy: achievements and challenges. Front Immunol. 2015;6:12. https://doi.org/10.3389/fimmu.2015.00012.
Vitale I, Manic G, Coussens LM, Kroemer G, Galluzzi L. Macrophages and metabolism in the tumor microenvironment. Cell Metab. 2019;30(1):36–50. https://doi.org/10.1016/j.cmet.2019.06.001.
Vonderheide RH. CD40 agonist antibodies in cancer immunotherapy. Annu Rev Med. 2020;71:47–58. https://doi.org/10.1146/annurev-med-062518-045435.
Wang P, Wang H, Huang Q, Peng C, Yao L, Chen H, et al. Exosomes from M1-polarized macrophages enhance paclitaxel antitumor activity by activating macrophages-mediated inflammation. Theranostics. 2019;9(6):1714–27. https://doi.org/10.7150/thno.30716.
Wang D, Wang X, Si M, Yang J, Sun S, Wu H, et al. Exosome-encapsulated miRNAs contribute to CXCL12/CXCR4-induced liver metastasis of colorectal cancer by enhancing M2 polarization of macrophages. Cancer Lett. 2020;474:36–52. https://doi.org/10.1016/j.canlet.2020.01.005.
Weiskopf K, Jahchan NS, Schnorr PJ, Cristea S, Ring AM, Maute RL, et al. CD47-blocking immunotherapies stimulate macrophage-mediated destruction of small-cell lung cancer. J Clin Invest. 2016;126(7):2610–20. https://doi.org/10.1172/JCI81603.
Weissleder R, Pittet MJ. The expanding landscape of inflammatory cells affecting cancer therapy. Nat Biomed Eng. 2020;4(5):489–98. https://doi.org/10.1038/s41551-020-0524-y.
Welm AL, Sneddon JB, Taylor C, Nuyten DS, van de Vijver MJ, Hasegawa BH, et al. The macrophage-stimulating protein pathway promotes metastasis in a mouse model for breast cancer and predicts poor prognosis in humans. Proc Natl Acad Sci U S A. 2007;104(18):7570–5. https://doi.org/10.1073/pnas.0702095104.
Wiehagen KR, Girgis NM, Yamada DH, Smith AA, Chan SR, Grewal IS, et al. Combination of CD40 Agonism and CSF-1R blockade reconditions tumor-associated macrophages and drives potent antitumor immunity. Cancer Immunol Res. 2017;5(12):1109–21. https://doi.org/10.1158/2326-6066.CIR-17-0258.
Williams CB, Yeh ES, Soloff AC. Tumor-associated macrophages: unwitting accomplices in breast cancer malignancy. NPJ Breast Cancer. 2016;2 https://doi.org/10.1038/npjbcancer.2015.25.
Wu Q, Allouch A, Martins I, Modjtahedi N, Deutsch E, Perfettini JL. Macrophage biology plays a central role during ionizing radiation-elicited tumor response. Biom J. 2017;40(4):200–11. https://doi.org/10.1016/j.bj.2017.06.003.
Xia Y, Rao L, Yao H, Wang Z, Ning P, Chen X. Engineering macrophages for cancer immunotherapy and drug delivery. Adv Mater. 2020;32(40):e2002054. https://doi.org/10.1002/adma.202002054.
Xu J, Escamilla J, Mok S, David J, Priceman S, West B, et al. CSF1R signaling blockade stanches tumor-infiltrating myeloid cells and improves the efficacy of radiotherapy in prostate cancer. Cancer Res. 2013;73(9):2782–94. https://doi.org/10.1158/0008-5472.CAN-12-3981.
Xuan M, Shao J, Dai L, He Q, Li J. Macrophage cell membrane camouflaged mesoporous silica nanocapsules for in vivo cancer therapy. Adv Healthc Mater. 2015;4(11):1645–52. https://doi.org/10.1002/adhm.201500129.
Yadav A, Kumar B, Datta J, Teknos TN, Kumar P. IL-6 promotes head and neck tumor metastasis by inducing epithelial-mesenchymal transition via the JAK-STAT3-SNAIL signaling pathway. Mol Cancer Res. 2011;9(12):1658–67. https://doi.org/10.1158/1541-7786.MCR-11-0271.
Yang J, Yan J, Liu B. Targeting VEGF/VEGFR to modulate antitumor immunity. Front Immunol. 2018;9:978. https://doi.org/10.3389/fimmu.2018.00978.
Yang H, Zhang Q, Xu M, Wang L, Chen X, Feng Y, et al. CCL2-CCR2 axis recruits tumor associated macrophages to induce immune evasion through PD-1 signaling in esophageal carcinogenesis. Mol Cancer. 2020;19(1):41. https://doi.org/10.1186/s12943-020-01165-x.
Yeo EJ, Cassetta L, Qian BZ, Lewkowich I, Li JF, Stefater JA 3rd, et al. Myeloid WNT7b mediates the angiogenic switch and metastasis in breast cancer. Cancer Res. 2014;74(11):2962–73. https://doi.org/10.1158/0008-5472.CAN-13-2421.
Yona S, Gordon S. From the reticuloendothelial to mononuclear phagocyte system - the unaccounted years. Front Immunol. 2015;6:328. https://doi.org/10.3389/fimmu.2015.00328.
Yoo JW, Irvine DJ, Discher DE, Mitragotri S. Bio-inspired, bioengineered and biomimetic drug delivery carriers. Nat Rev Drug Discov. 2011;10(7):521–35. https://doi.org/10.1038/nrd3499.
Yu H, Lee H, Herrmann A, Buettner R, Jove R. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer. 2014;14(11):736–46. https://doi.org/10.1038/nrc3818.
Yu GT, Bu LL, Huang CF, Zhang WF, Chen WJ, Gutkind JS, et al. PD-1 blockade attenuates immunosuppressive myeloid cells due to inhibition of CD47/SIRPalpha axis in HPV negative head and neck squamous cell carcinoma. Oncotarget. 2015;6(39):42067–80. doi: https://doi.org/10.18632/oncotarget.5955.
Yue S, Ye X, Zhou T, Gan D, Qian H, Fang W, et al. PGRN(−/−) TAMs-derived exosomes inhibit breast cancer cell invasion and migration and its mechanism exploration. Life Sci. 2021;264:118687. https://doi.org/10.1016/j.lfs.2020.118687.
Yumimoto K, Sugiyama S, Mimori K, Nakayama KI. Potentials of C-C motif chemokine 2-C-C chemokine receptor type 2 blockers including propagermanium as anticancer agents. Cancer Sci. 2019;110(7):2090–9. https://doi.org/10.1111/cas.14075.
Zabuawala T, Taffany DA, Sharma SM, Merchant A, Adair B, Srinivasan R, et al. An ets2-driven transcriptional program in tumor-associated macrophages promotes tumor metastasis. Cancer Res. 2010;70(4):1323–33. https://doi.org/10.1158/0008-5472.CAN-09-1474.
Zanganeh S, Hutter G, Spitler R, Lenkov O, Mahmoudi M, Shaw A, et al. Iron oxide nanoparticles inhibit tumour growth by inducing pro-inflammatory macrophage polarization in tumour tissues. Nat Nanotechnol. 2016;11(11):986–94. https://doi.org/10.1038/nnano.2016.168.
Zeng D, Ye Z, Wu J, Zhou R, Fan X, Wang G, et al. Macrophage correlates with immunophenotype and predicts anti-PD-L1 response of urothelial cancer. Theranostics. 2020;10(15):7002–14. https://doi.org/10.7150/thno.46176.
Zha H, Han X, Zhu Y, Yang F, Li Y, Li Q, et al. Blocking C5aR signaling promotes the anti-tumor efficacy of PD-1/PD-L1 blockade. Onco Targets Ther. 2017;6(10):e1349587. https://doi.org/10.1080/2162402X.2017.1349587.
Zhang X, Fan J, Wang S, Li Y, Wang Y, Li S, et al. Targeting CD47 and autophagy elicited enhanced antitumor effects in non-small cell lung cancer. Cancer Immunol Res. 2017;5(5):363–75. https://doi.org/10.1158/2326-6066.CIR-16-0398.
Zhang X, Chen W, Fan J, Wang S, Xian Z, Luan J, et al. Disrupting CD47-SIRPalpha axis alone or combined with autophagy depletion for the therapy of glioblastoma. Carcinogenesis. 2018a;39(5):689–99. https://doi.org/10.1093/carcin/bgy041.
Zhang JQ, Zeng S, Vitiello GA, Seifert AM, Medina BD, Beckman MJ, et al. Macrophages and CD8(+) T cells mediate the antitumor efficacy of combined CD40 ligation and Imatinib therapy in gastrointestinal stromal tumors. Cancer Immunol Res. 2018b;6(4):434–47. https://doi.org/10.1158/2326-6066.CIR-17-0345.
Zhang SY, Song XY, Li Y, Ye LL, Zhou Q, Yang WB. Tumor-associated macrophages: a promising target for a cancer immunotherapeutic strategy. Pharmacol Res. 2020a;161:105111. https://doi.org/10.1016/j.phrs.2020.105111.
Zhang L, Tian L, Dai X, Yu H, Wang J, Lei A, et al. Pluripotent stem cell-derived CAR-macrophage cells with antigen-dependent anti-cancer cell functions. J Hematol Oncol. 2020b;13(1):153. https://doi.org/10.1186/s13045-020-00983-2.
Zhao H, Wang J, Kong X, Li E, Liu Y, Du X, et al. CD47 promotes tumor invasion and metastasis in non-small cell lung cancer. Sci Rep. 2016;6:29719. https://doi.org/10.1038/srep29719.
Zhao J, Zhang Z, Xue Y, Wang G, Cheng Y, Pan Y, et al. Anti-tumor macrophages activated by ferumoxytol combined or surface-functionalized with the TLR3 agonist poly (I : C) promote melanoma regression. Theranostics. 2018a;8(22):6307–21. https://doi.org/10.7150/thno.29746.
Zhao H, Li L, Zhang J, Zheng C, Ding K, Xiao H, et al. C-C chemokine ligand 2 (CCL2) recruits macrophage-membrane-camouflaged hollow bismuth selenide nanoparticles to facilitate photothermal sensitivity and inhibit lung metastasis of breast cancer. ACS Appl Mater Interfaces. 2018b;10(37):31124–35. https://doi.org/10.1021/acsami.8b11645.
Zheng P, Luo Q, Wang W, Li J, Wang T, Wang P, et al. Tumor-associated macrophages-derived exosomes promote the migration of gastric cancer cells by transfer of functional apolipoprotein E. Cell Death Dis. 2018;9(4):434. https://doi.org/10.1038/s41419-018-0465-5.
Zhou Y, Cao HB, Li WJ, Zhao L. The CXCL12 (SDF-1)/CXCR4 chemokine axis: oncogenic properties, molecular targeting, and synthetic and natural product CXCR4 inhibitors for cancer therapy. Chin J Nat Med. 2018;16(11):801–10. https://doi.org/10.1016/S1875-5364(18)30122-5.
Zhou W, Guo S, Liu M, Burow ME, Wang G. Targeting CXCL12/CXCR4 Axis in tumor immunotherapy. Curr Med Chem. 2019;26(17):3026–41. https://doi.org/10.2174/0929867324666170830111531.
Zhou J, Tang Z, Gao S, Li C, Feng Y, Zhou X. Tumor-associated macrophages: recent insights and therapies. Front Oncol. 2020;10:188. https://doi.org/10.3389/fonc.2020.00188.
Zhu XD, Zhang JB, Zhuang PY, Zhu HG, Zhang W, Xiong YQ, et al. High expression of macrophage colony-stimulating factor in peritumoral liver tissue is associated with poor survival after curative resection of hepatocellular carcinoma. J Clin Oncol. 2008;26(16):2707–16. https://doi.org/10.1200/JCO.2007.15.6521.
Zippelius A, Schreiner J, Herzig P, Muller P. Induced PD-L1 expression mediates acquired resistance to agonistic anti-CD40 treatment. Cancer Immunol Res. 2015;3(3):236–44. https://doi.org/10.1158/2326-6066.CIR-14-0226.
Acknowledgements
The work was supported by grants from the National Natural Science Foundation of China (81901232 (to Y.L.), 32000479 (to X.Z.), 82002915 (to S.W.)).
Conflict of Interests
The authors declared that there is no conflict of interests.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Li, Y., Zhang, X., Zeng, X., Wang, S., Wang, H. (2022). Tumor-Associated Macrophages: Therapeutic Targets of Cancer. In: Gupta, S., Pathak, Y.V. (eds) Macrophage Targeted Delivery Systems. Springer, Cham. https://doi.org/10.1007/978-3-030-84164-5_13
Download citation
DOI: https://doi.org/10.1007/978-3-030-84164-5_13
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-84163-8
Online ISBN: 978-3-030-84164-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)