
Abstract
Antibody-drug conjugates (ADCs) are cancer drugs composed of a humanized antibody linked to a cytotoxic payload, allowing preferential release of payload in cancer cells expressing the antibody-targeted antigen. Here, a systems pharmacology model is used to simulate ADC transport from blood to tumor tissue and ADC uptake by tumor cells. The model includes effects of spatial gradients in drug concentration in a three-dimensional network of tumor blood vessels with realistic geometry and accounts for diffusion of ADC in the tumor extracellular space, binding to antigen, internalization, intracellular processing, and payload efflux from cells. Cells that process an internalized ADC-antigen complex may release payload that can be taken up by other “bystander” cells. Such bystander effects are included in the model. The model is used to simulate conditions in previous experiments, showing good agreement with experimental results. Simulations are used to analyze the relationship of bystander effects to payload properties and single-dose administrations. The model indicates that exposure of payload to cells distant from vessels is sensitive to the free payload diffusivity in the extracellular space. When antigen expression is heterogeneous, the model indicates that the amount of payload accumulating in non-antigen-expressing cells increases linearly with dose but depends only weakly on the percentage of antigen-expressing cells. The model provides an integrated mechanistic framework for understanding the effects of spatial gradients on drug distribution using ADCs and for designing ADCs to achieve more effective payload distribution in solid tumors, thereby increasing the therapeutic index of the ADC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
ADCs are a class of large molecular weight (~ 150 kDa) cancer drugs that have extremely potent small molecule drugs, referred to as the payload, conjugated to a humanized antibody via a chemical linker (1). The number of payload molecules per antibody is referred to as the drug-to-antibody ratio (DAR). It has been established that higher DAR values lead to both increased rates of DAR-dependent deconjugation of payload from the antibody in plasma and reductions in the half-life of the ADC in plasma (2,3,4). Therefore, most ADCs in current development typically have a DAR value of four or less (5). ADCs are transported to cancer cells by passing through the walls of microvessels, diffusing in the tumor tissue, and binding specifically to preferentially expressed target antigens on cancer cells. The antibody-antigen complex is internalized by the cell, where cellular processing of the antibody in lysosomes causes the toxic payload to be released in the cytosol to exert its cytotoxic effect (Fig. 1a).

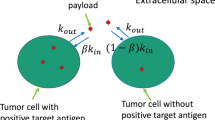
ADC transport and reactions including bystander effects. Smooth boundary denotes Ag− cell; wavy boundary denotes Ag+ cell; orange (blue) color denotes cell containing free (no) payload. a Steps in ADC transport and reaction. (1) ADC (green circles) is carried into the tumor via convection in plasma. (2) ADC passes across the vessel wall into the tumor interstitial space. (3) ADC diffuses through the interstitial space. (4) ADC binds to target antigen expressed on the cell membrane. (5) ADC-antigen complex is internalized by the cell and undergoes lysosomal degradation to release payload. (6) Payload passively effluxes from the cell (orange arrow) and diffuses to other cells, creating a possible bystander effect. (7) Payload is released by the cell, enters blood vessels, and is washed out in plasma. b–e Payload distribution in the absence and presence of bystander effects. b Heterogeneous antigen expression. Cells containing ADC do not release payload, so payload does not reach Ag– cells. c Heterogeneous antigen expression, with heterogeneous bystander effect. Cells containing ADC release payload, which diffuses and is taken up by Ag– cells, and also by more distant Ag+ cells. d Homogeneous antigen expression, no bystander effect. ADC transport into cells is restricted by the binding site barrier, and cells containing ADC do not release payload to reach distant cells. e Homogeneous antigen expression with spatial bystander effect. Cells containing ADC release payload, which diffuses and is taken up by more distant cells
Some ADCs can induce a bystander effect, defined here as non-targeted passive cellular uptake of payload released by other cells that had previously internalized and processed an ADC-antigen complex (6,7,8,9,10). The ability of an ADC to induce a bystander effect is dependent on the physicochemical properties of the linker, which must be designed so that the processed payload catabolite is neutrally charged, allowing for efflux from the cell and diffusion into neighboring cells (1). Typically, cleavable linkers leave the payload catabolite in a neutral form while non-cleavable linkers result in a charged catabolite that is unlikely to diffuse into neighboring cells (7,11).
The bystander effect provides a mechanism for exposure to payload of cells that are distant from vessels and for cells that do not express the antigen, i.e., antigen-negative cells (Ag–). We introduce the terms spatial bystander effect (SBE) and heterogeneous bystander effect (HBE), respectively, for these cases. The SBE is relevant because rapid binding of the ADC to the target antigen after extravasation from the vessels has been implicated in poor tumor penetration (12), resulting in accumulation of antibody around the periphery of the vessels, a phenomenon referred to as the binding site barrier (13) (Fig. 1d). Because of this, cells more distant from the vessels receive lower concentrations of ADC. However, low molecular weight payload released by antigen-positive (Ag+) cells close to the vessels may diffuse through the tissue to cells that are more distant from blood vessels (Fig. 1e). The potential of the SBE to contribute to the intratumoral distribution of payload has not to our knowledge been analyzed in detail, although its potential has been discussed (14).
Since antigen expression is often heterogeneous in solid tumors (15,16), the HBE has potential clinical utility by indirectly targeting Ag– cells in a tumor region. Several pre-clinical studies have been conducted to evaluate the HBE, using ADCs with cleavable linkers. Okeley et al. (8) demonstrated significant bystander killing of CD30+ and CD30– cell lines in vitro. Kovtun et al. (7) demonstrated cell killing of CanAg-positive and CanAg-negative cell lines both in vitro and in vivo. Li et al. (9) demonstrated bystander killing in vivo in admixed tumor models using various payloads and Golfier et al. (10) demonstrated bystander killing in vivo within tumor xenografts comprised of different percentages of Ag+ cells ranging from 20 to 100%.
For most ADCs targeting overexpressed antigens, a binding site barrier is likely to be present, and the SBE is expected to occur whenever free payload can be released and taken up by cells. If, in addition, antigen expression is heterogeneous, both types of bystander effect (SBE and HBE) can occur simultaneously, provided that antigen expression is high enough to induce a binding site barrier.
The release of free payload into the interstitial space by Ag+ cells creates a concentration gradient across blood vessel walls. If the walls are permeable to the payload, as is likely for neutrally charged payloads, then some of the payload released by cells can diffuse through the vessel wall, whereupon it is washed out in the flowing blood. Washout is particularly likely to occur for payload released by cells close to the vessel, because the diffusion distance is short and it is less likely to be taken up by other tumor cells before reaching the vessels. Thus, the conditions that facilitate a SBE are also likely to promote payload washout.
Various theoretical models have been developed to predict ADC payload exposure and cell kill in solid tumors. Studies by Shah and Singh (17,18,19) used mechanistic compartmental models to describe the transport and kinetics of a specific ADC including bystander effects. These models assume that the tissue compartment is well mixed with regard to ADC concentration and do not take into account the effects of vessel geometry and the possibility of steep gradients in ADC concentration. Models that do not account for gradients in concentration may lead to inaccurate predictions of cellular exposure. If a concentration threshold of payload is needed for sufficient exposure that would lead to cell death, well-mixed compartmental models will predict either all or none of the cells sufficiently exposed (within a compartment), while a model accounting for gradients will predict a percentage of cells sufficiently exposed. Studies by Cilliers et al. (20), Vasalou et al. (21), and Khera et al. (22) account for spatial effects by using a Krogh cylinder to represent the tissue surrounding a representative blood vessel. Both models assume one-dimensional outward radial diffusion of ADC and include ADC binding to antigen and intracellular trafficking kinetics of payload. Vasalou et al. (21) further examined the effects of bystander killing on tumor volume and its dependence on the ratio of cellular payload uptake to efflux. While these models represent the essential transport processes and kinetics of ADCs in solid tumors, the use of a Krogh cylinder model to represent solute transport from blood to tissue can result in unrealistic distributions of solute concentration (23,24). These models have, nonetheless, improved understanding of ADC transport and kinetics and supported decision making in ADC drug development.
Here, a systems pharmacology model based on a previously developed reaction diffusion model for solute transport (25) is developed to simulate the transport and reaction processes of ADCs in a three-dimensional tumor vascular network with realistic geometry, including bystander effects and payload washout. Model validation was established by simulating pre-clinical experiments from several previous studies (2,20,26) using parameter values from the literature and showing good agreement between model predictions and experimental results. The SBE is analyzed by predicting the dependence of cellular exposure over different distances to the nearest vessel for different payload diffusivities and different doses of ADC where the total payload quantity is fixed. The HBE is analyzed by predicting the dependence of cellular exposure to payload in Ag– cells on the percentage of Ag– cells in the tumor region and on dose.
Materials and Methods
Model Overview
The ADC and payload distribution in tumor tissue are analyzed by considering several diffusible and non-diffusible solute concentrations: ADC in the extracellular space (diffusible), target antigen (non-diffusible), ADC-antigen complex (non-diffusible), internalized ADC-antigen complex (non-diffusible), intracellular payload of Ag+ cells (non-diffusible), extracellular payload (diffusible), and intracellular payload of Ag– cells (non-diffusible). These solutes may be restricted to specific tissue domains (e.g., intracellular space) and are subject to differing transport and reaction mechanisms in the vessels and in the tissue space. Solute concentration is calculated as function of position and time within a region of tumor tissue supplied by a network of microvessels with a given three-dimensional geometry and given blood flow rate in each segment.
Vessel Network and Tumor Cell
Compartments. Solutions are computed for the microvessel network shown in Fig. 2a, which was previously mapped in a 550 μm × 520 μm × 230 μm region of a mammary carcinoma implanted in a rat dorsal skin flap. The tumor was imaged using confocal microscopy, and the vasculature was digitally reconstructed (27). The distribution of distances from the nearest vessel to a representative array of tissue points within the domain is shown in Fig. 2b, together with the corresponding distribution for a Krogh cylinder model with the same ratio of vessel length to tissue volume. The network model gives a much broader distribution of distances than the Krogh model, including a significant fraction (12%) of tissue points more than 80 μm from the nearest vessel. By contrast, the maximum distance from the nearest vessel in the Krogh model is 53 μm.

Microvessel network. a Network used for computational model. Flow rates in nl/min and flow directions are indicated for each vessel segment. Vessel segment color indicates the magnitude of flow rate. b Distribution of tissue point distances from the nearest vessel for the microvessel network. For comparison, the distribution of distances for the Krogh cylinder geometry with the same mean vessel diameter and the same vessel length per tissue volume is also shown. The Krogh cylinder has a tissue radius of 52.96 μm and a capillary radius of 5.59 μm
Flows were estimated based on flow measurements in the same tissue for vessels of corresponding diameters, with the net flow entering and leaving each vessel branch point constrained to be zero. Computationally, the vessels are represented as a set of distributed sources or sinks for solutes and are treated as part of the tissue space. The intracellular and extracellular regions of the tissue space are treated as two coexisting homogeneous media, each with its own volume fraction. A continuum representation of the cellular tissue structure is used since the length scale of a typical cell (≈ 10 μm) is small compared with the length scale of the vessel network (≈ 500 μm). In the case of heterogeneous antigen-expressing cells, it is assumed that Ag+ and Ag– cells are interspersed, consistent with observation (20,28). It is assumed that the volume fraction of cells remains constant throughout the simulation. Changes in cell numbers resulting from cell kill or cell growth are not included in the model.
Governing Equations
The governing equations for the ADC diffusion in the tumor tissue and for reaction kinetics including bystander effects are as follows:
The unknown variables CADC, CAg, CB, CI, \( {C}_{ip}^{+} \), \( {C}_{ip}^{-} \), and Cep are the concentrations of extracellular ADC, antigen, ADC bound to antigen, internalized ADC bound to antigen, intracellular payload within Ag+ cells, intracellular payload within Ag– cells, and extracellular payload, respectively. The fixed total antigen concentration is CAgtot = CAg + CB. The function DAR(t) represents the time course of DAR and accounts for deconjugation of payload from the antibody in plasma and was used in the model validation case studies. The constant φe is the extracellular volume fraction, and φ+ and φ– are the volume fractions of antigen positive and negative cells, respectively, where φ+ + φ– = 1 – φe. To ensure consistency with the numerical method, all concentrations are defined with respect to the total volume of the tissue region. Defining concentration in this way requires the inclusion of these volume fractions. Solutions are then converted to concentrations with respect to the volume of interest. The HBE is explicitly represented in the last two equations, which describe the efflux, extracellular diffusion, and uptake of extracellular payload. The SBE is implicitly represented by accounting for diffusive transport and binding of ADC to target antigen. Payload washout is included in the diffusive term in the last equation. Other parameters are described in Table II. The system of equations is solved using the unsteady Green’s function method (25), which is summarized in the Supplementary Information.
Parameter Values
The model parameters derived from measured values in the literature are shown in Table I. Parameter values used for model validation are shown in Table II. When possible, kinetic parameter values were chosen to match the experimental setup in the study of interest. For example, if several association rates of antibody binding to antigen were found, the one corresponding to the same antibody and antigen was chosen when available. The parameters and the extent to which they aligned with the study used for validation are summarized in Supplementary Information, Table S1. Plasma pharmacokinetics for ADCs (2,3,26) were estimated by fitting the reported plasma levels from a single dose of ADC to the parameters in Eq. A (Matlab 2014a, lsqcurvefit, see Supplementary Information, Table S2). The time course of DAR-dependent deconjugation of the payload from the ADC was assumed to be of the form of Eq. 1, and the corresponding parameters were similarly fit from experimental observation (2,3) (see Supplementary Information, Table S3). Parameter values used for bystander effect simulations were chosen each in a range typical of observed values and are shown in Table III.
Measures of Cellular Exposure
The drugs used as payloads for ADCs generally work either by interfering with tubulin dynamics or with DNA integrity or replication. El-Kareh et al. (36,37) analyzed existing in vitro experimental data on cell as a function of drug concentration and exposure time for drugs including doxorubicin (a DNA intercalator) and paclitaxel (which interferes with tubulin dynamics). It was shown that dose-response data for these drugs are inconsistent with models in which cell kill is a function of area under the curve (AUC), defined as \( {\int}_0^{\infty }{C}^{int}(t) dt \). In contrast, models in which cell kill was assumed to depend on the time maximum of intracellular drug concentration showed good agreement with the experimental data. Therefore, in the present study, maximum intracellular concentration, denoted \( {C}_{max}^{int} \), is estimated as a predictor of cellular response.
Results
Spatial Variation of Intratumoral Antibody Distribution
Figure 3 shows the model predictions for the experimental setup in Rhoden et al. (26). In the study, fluorescent intensity of sm3e antibody bound to CEA-positive LS174T xenograft tumors (colorectal carcinoma) was measured as a function of distance to the nearest vessel for a range of doses (5–500 μg). The experiment illustrated the effect of the binding site barrier and its dependence on dose. Figure 3a shows the model prediction of concentration of sm3e at tissue points of increasing distance to the nearest vessel for doses of 5 to 500 μg at 24 h. Results from Rhoden et al. were given in arbitrary units and were scaled by matching the profile for the highest dose with model predictions. High doses resulted in complete saturation of the available antigen, leading to uniform distributions of antibody. Sub-saturating doses lead to gradients in fluorescent intensity, as also predicted by the model. Results are in good agreement with model simulations except for the lowest dose, for which the model underestimates the experimental results. Figure 3b–d illustrate the binding site barrier by showing contours of the predicted antibody distributions for doses 5, 15, and 50 μg.

Intratumoral antibody distributions. a Comparison between results of Rhoden et al. (26) and model simulations of antibody distribution in a solid tumor for doses 2, 15, 50, 100, and 500 μg of the sm3e antibody. Experimental data: relative mean fluorescent intensity (MFI). Model predictions: fractional antigen saturation defined as (total antibody)/(total antigen), where total antibody is the sum of bound antibody and free antibody. b–d Predicted spatial distributions of antibody concentration in a plane through the tissue domain for doses 5, 15, and 50 μg of antibody. Black and gray lines show projections of vessel positions. Vessels whose midpoints are within 50 μm of the contour plane are shown in gray. Parameters values are listed in Table II
Intratumoral Payload Concentration
Figure 4 shows model predictions for the experimental conditions in Alley et al. (2) and Li et al. (20). Alley et al. measured the time course of intratumoral concentration of MMAF released from the h1F6-mcMMAF ADC in CD70-positive renal cell carcinoma xenografts. Li et al. measured the intratumoral concentration of MMAE from two different ADCs (cAC10-vcMMAE, h1F6-vcMMAE) for three doses (0.5, 1, 3 mg/kg) in L82 xenograft models and the time course of intratumoral MMAE concentration from cAC10-vcMMAE in Karpas 299 xenograft models. Figure 4a shows the model prediction for the 10-day time course of intratumoral concentration of MMAF compared with the measurements in Alley et al. The model prediction is in good agreement with the observed accumulation and elimination of MMAF as well as the peak intratumoral concentration. Figure 4b shows the model predictions compared with the observed intratumoral concentrations in the L-82 xenograft models. The predictions for cAC10-vcMMAE are in excellent agreement, whereas the predictions for h1f6-vcMMAE are overestimated by approximately a factor of two. Figure 4c shows model predictions compared with the measurements in Li et al. for the 10-day time course of intratumoral concentration of MMAE in Karpas 299 xenograft models. The predictions have a similar profile of accumulation and elimination of MMAE including peak intratumoral concentration. The model prediction shows a 2-day delay in the predicted time course, however, suggesting that some rates may be underestimated.

Intratumoral payload concentrations. a Model predictions and experimental results from Alley et al. (2) of intratumoral payload time course for a 1.5 mg/kg dose of h1f6-mcMMAF. b Model predictions and experimental results from Li et al. (9) of intratumoral concentration of MMAE from two ADCs (cAC10-vcMMAE, h1F6-vcMMAE) for three doses (0.5, 1, 3 mg/kg) measured at 72 h. c Model predictions and experimental results from Li et al. (9) of intratumoral payload time course for a 2 mg/kg dose of cAC10-vcMMAE. Parameter values are listed in Table II
Spatial Bystander Effect (SBE)
The SBE is analyzed by considering the payload exposure of cells at different distances to the nearest vessel, as functions of payload diffusivity and dose. To provide an index of exposure to payload, \( \overline{C_{max}^{int}} \) is defined as the peak intracellular payload concentration, averaged over intervals of distance from nearest vessel. Figure 5a shows the dependence of \( \overline{C_{max}^{int}} \) on distance to the nearest vessel for several values of the payload diffusivity Dpayload for DAR = 4 and a dose of 1 mg/kg. Steep gradients are evident, showing the effect of the binding site barrier. For distances approximately 50 μm and greater, the payload levels are almost constant and non-zero due to the SBE. In Fig. 5b, a total dose of 10 mg/kg was simulated with 1 mg/kg of DAR = 4 and 9 mg/kg of DAR = 0. These values result in the same total payload dose, but with the lower average DAR value of 0.4. Gradients are less steep for cells near the vessels with overall higher levels of payload for cells distant from the vessel because of direct cell targeting due to the increased penetration distance of the higher dose. With increasing payload diffusivity, overall cellular exposure decreases due to increasing payload washout. Figure 5c shows computed values of payload AUC in blood up to time t. Corresponding results for the 10 mg/kg dose (Fig. 5d) are similar, indicating a weak sensitivity to DAR when the total payload dose is fixed.

Cellular exposure to payload and payload washout. a, b Peak intracellular payload concentration (\( \overline{C_{max}^{int}} \)) averaged over 10-μm intervals of distance from the nearest vessel, for three levels of payload diffusivity (Dpayload = 1, 10, 100 μm2/s). a Dose = 1 mg/kg, DAR = 4. b Dose = 1 mg/kg, DAR = 4 together with 9 mg/kg, DAR = 0, resulting in an effective average DAR = 0.4. c, d Area under the payload concentration curve in plasma at time t, AUC(t), due to payload washout. c Dose = 1 mg/kg, DAR = 4. d Dose = 1 mg/kg, DAR = 4 together with 9 mg/kg, DAR = 0. No DAR-dependent deconjugation was assumed. Parameter values are listed in Table III. In each case, results are also shown for a payload with no bystander effect
Heterogeneous Bystander Effect (HBE)
When a fraction of the cell population is antigen negative (Ag−, i.e., φ– > 0), the conditions that permit the occurrence of the SBE also lead to a HBE. Illustrating the combined effects of the SBE and the HBE, Fig. 6 shows values of \( \overline{C_{max}^{int}} \) averaged over intervals of distance to the nearest vessels in Ag+ cells and Ag– cells, for different values of φ– (10%, 50%, 70%) and different doses (1, 5, 10 mg/kg). In all cases, the number of receptors per cell remains fixed. In Fig. 6a–c, values of \( \overline{C_{max}^{int}} \)in Ag+ cells decrease with increasing distance to the nearest vessel, as a consequence of the binding site barrier. For lower percentages of Ag+ cells, the same quantity of ADC binds to fewer Ag+ cells, resulting in higher concentrations of intracellular payload. The fractional antigen saturation was computed for cells closest to the vessel for each percentage of Ag+ cells and dose (see Fig. 6 caption). As a result of partial saturation, values of \( \overline{C_{max}^{int}} \)at tissue points close to vessels do not increase in proportion to dose.

Dependence of cellular exposure to payload on distance to the nearest vessel, percentage of Ag+ cells, and dose. Values of \( \overline{C_{max}^{int}} \) are averaged over cells within 10-μm intervals of distance from the nearest vessel, for 30%, 50%, and 90% Ag+ cells and correspondingly 70%, 50%, and 10% Ag− cells, and for doses of 1, 5, and 10 mg/kg. Bars represent one standard deviation above the mean. The fractional antigen saturation as defined in Fig. 3 caption was calculated for cells within 10 μm to vessels: 0.15, 0.116, and 0.088 for 30%, 50%, and 90% Ag+ cells at 1 mg/kg, respectively, 0.551, 0.448, and 0.36, for 30%, 50%, and 90% Ag+ cells at 5 mg/kg, respectively, and 0.806, 0.681, and 0.574 for 30%, 50%, and 90% Ag+ cells at 10 mg/kg, respectively. Values of \( \overline{C_{max}^{int}} \)for 70%, 50%, and 10% Ag− cells are emphasized on the axes. Parameter values are listed in Table III
In Fig. 6a–c, intracellular payload in Ag– cells is seen to be independent of the percentage of antigen expression. This occurs because antigen saturation is incomplete, and all available ADC is catabolized independent of the percentage of Ag+ cells, as is the amount of free payload released into the extracellular space (results not shown). The free payload diffuses readily in the extracellular space and results in essentially uniform uptake by Ag+ and Ag– cells at different distances to the nearest vessel. For 90% Ag+ cells and all doses, the payload reaching cells approximately 75 μm or more from the nearest vessel is a consequence of the SBE. For lower percentages of Ag+ cells, payload reaches this distance from combined effects of the SBE and ADC binding, due to the greater penetration distance of free ADC. With higher doses (Fig. 6b, c), gradients in \( \overline{C_{max}^{int}} \) are less prominent because ADC binding to Ag+ cells near the vessels saturates, allowing more ADC to diffuse to distant cells. In Ag– cells, \( \overline{C_{max}^{int}} \) increases linearly with dose because cellular efflux and uptake of free payload are assumed to be first order.
With regard to therapeutic effectiveness, it is relevant to consider the minimum value of \( \overline{C_{max}^{int}} \) attained within the tissue domain, since cells receiving the lowest exposure are more likely to survive treatment. Figure 7 shows the spatial minimum values of \( \overline{C_{max}^{int}} \) in Ag+ and Ag– cells for doses of 1, 5, and 10 mg/kg. These values increase linearly with dose in Ag– cells, but the increase is non-linear for 30% Ag+ cells, because more ADC is available to distant cells due to saturation of surface antigen binding. For higher percentages of Ag+ cells, the dependence on dose is almost linear and similar to that predicted for Ag– cells.

Spatial minimum values of cellular exposure in tissue region for varying doses and percentages of Ag+ and Ag− cells. Minimum values of \( \overline{C_{max}^{int}} \) for doses of 1, 5, and 10 mg/kg are shown for 30%, 50%, and 90% Ag+ cells and correspondingly 70%, 50%, and 10% Ag− cells. Parameter values are listed in Table III
Discussion
ADCs have shown strong potential for selective killing of tumor cells both in vitro and pre-clinically in vivo, but success in demonstrating clinical efficacy against solid tumors has been limited. As of 2017, over 60 clinical trials using ADCs for the treatment of solid tumors were reported, but nearly half have been stopped and few are in phase III (38). Currently, only one ADC is approved for the treatment of solid tumors (trastuzumab emtansine). The complex transport and kinetic properties of ADCs within solid tumors present significant challenges for achieving efficacy. Notably, limited penetration of ADCs from vessels into tumor tissue, i.e., the binding site barrier, results in heterogeneous distributions of ADC within the tumor. Some ADCs can induce a bystander effect, in which payload released from a degraded ADC within a cell is taken up by neighboring cells. Bystander effects have potential therapeutic benefits, as they can cause exposure to toxic payload of cells that are beyond the binding site barrier (the spatial bystander effect) or that do not express the target antigen (the heterogeneous bystander effect), but their effects are difficult to predict in vivo. It remains unclear whether bystander effects are even desirable for ADCs and, if so, which design options should be chosen in combination to create the maximum therapeutic index.
In view of the challenges mentioned above, a systems pharmacology model based on reaction diffusion processes is presented here as a tool to support preclinical ADC drug development by predicting cellular exposure to payload in solid tumors. The model simulates the transport and kinetics of ADCs in a tumor region with realistic vessel network geometry and accounts for convective delivery of ADC to the tumor, ADC diffusion within the tumor, ADC-antigen binding and internalization, intracellular degradation of the ADC, cellular release of payload, uptake of payload by neighboring cells, and payload washout by blood vessels. The model utilizes parameters from the literature and uses fitted parameters only for the time course of ADC in plasma. The model was validated by showing agreement between model predictions and experimental observations from previous studies that examined the binding site barrier and intratumoral payload concentrations. Simulations were performed to analyze the bystander effects and their dependence on payload and tumor properties. The model provides a general framework that can be customized to the properties of a specific ADC and tumor. More precise parameterization can be achieved by designing experiments for the purpose of quantifying unknown transport and/or kinetic parameters (18,35).
Model predictions simulating the experimental conditions of Rhoden et al., Alley et al., and Li et al. (2,9,26) showed good agreement in predicting various spatial and temporal ADC-related quantities. In Rhoden et al., the relative magnitudes between the experimental fluorescent intensity of the sm3e antibody and the model prediction of antibody concentration relative to distance to the nearest vessel are in close agreement. The model quantitatively distinguishes between saturating and sub-saturating doses and accurately predicts gradients in concentration as compared with fluorescent intensity. The discrepancy in the predictions for the lowest dose can be possibly explained by a background level of fluorescence which influences the relative difference in the fluorescent signals. Model predictions for Li et al. show excellent agreement for intratumoral concentration of payload using cAC10-vcMMAE, but poorer agreement for h1F6-vcMMAE with model predictions approximately twofold higher than observations (Fig. 4b). In this case, the values of kon and kint used for this prediction were measured for CD70 but not h1f6 and may not be reasonable surrogates for the kinetics of h1f6-CD70 complex. In contrast, these parameters were measured for cAC10-CD30 which likely correlates with the more accurate model prediction for cAC10-vcMMAE. Predictions for the time course of intratumoral concentration (Fig. 4a, c) are also in close agreement with experimental observations of Li et al. and Alley et al. Both model predictions show a slightly delayed accumulation of intratumoral payload, suggesting that one or more of the binding, internalization, or antibody degradation rates are underestimated.
The model was used to analyze bystander effects, including the SBE, defined here as the exposure of cells distant from the vessels to freely diffusing payload, and the HBE, defined here as exposure of Ag– cells to freely diffusing payload. The SBE was investigated by predicting the dependence of average peak intracellular concentration in cells on distance to the nearest vessel, for various payload diffusivities and doses, with total payload remaining fixed (Fig. 5). The HBE was investigated by predicting exposure of both Ag+ and Ag– cells to payload, for various Ag– cell fractions and doses (Fig. 6).
Several notable findings arise from the simulations: (i) Payload diffusivity strongly influences the extent to which the SBE results in increased payload exposure to distant cells. Relative to low payload diffusivities (< 10 μm2/s), higher diffusivities (> 10 μm2/s) result in lower payload levels in cells distant from the vessels, as a result of payload washout in blood. These results highlight the importance of considering the diffusive properties of the payload in the interstitial space, as described in Khera et al. (22). In that study, a potential optimal diffusivity was evidenced. No such optimum arises in this work, possibly because of the different vascular geometry assumed here or because of different lipid/aqueous partition coefficients of the payloads considered. (ii) Co-administration of antibody without payload together with ADC increases the number of cells receiving payload, for a given total payload dose. This result is consistent with Cilliers et al. (20) where T-DM1 was co-administered with increasing doses of trastuzumab to increase tumor penetration. (iii) The payload washout when antibody without payload is co-administered is nearly identical to that without co-administration, suggesting that the therapeutic window can be increased since potential toxicity from free payload washout remains unchanged with co-administration. (iv) Exposure of Ag– cells to payload is insensitive to the percentage of Ag– cells. If the ADC dose is sufficiently low such that saturation is not complete, then all available ADC binds to target antigens and the same amount of payload is expected to be released for passive uptake into Ag– cells. This finding is consistent with previous studies showing approximately the same reduction in tumor size for a range of percentages of Ag– cells (9,10).
Several considerations should be taken into account when interpreting the results. First, the simulations presented here are based on a single vascular network geometry derived from observations of a mammary carcinoma implanted in a rat. The tumor is assumed to be well perfused, which is reasonable for small tumors, but may not apply to larger ones, which often contain necrotic cores. Relative to previous models using compartmental models or a Krogh cylinder, the present model has the advantage of providing a realistic representation of the heterogeneity of vascular structures in tumors, and the resulting wide distribution of distances of tissue points to the nearest vessel (Fig. 2b). While it does not represent the full range of variability in tumor vascular structure and perfusion, the conclusions of this study are not dependent on the specific network simulated here, and similar results would be expected for other network geometries and flow distributions. Second, the model uses the metric \( {C}_{max}^{int} \) while in reality, the actual metric is likely a combination of an intracellular AUC and \( {C}_{max}^{int} \), i.e., exposure to some minimum concentration for some minimum duration of time. The use of \( {C}_{max}^{int} \)simplifies the analyses, but a more precise metric could be defined since concentrations are known at all tissue points and time. Third, the model used here neglects the effects of cell kill on the kinetics of payload release and on the resulting bystander effects. In reality, tumor cells may undergo apoptosis within the period considered in the simulations. This would be especially important if there are relatively few Ag+ cells. In this case, Ag+ cells would likely accumulate higher concentrations of payload, which may decrease the time to cell kill. After apoptosis, such cells would cease internalizing and processing ADCs and therefore affect overall cellular exposure to payload. In order to predict these effects, the model could be combined with a spatially dependent model for cellular pharmacodynamics to predict overall tumor volume, since the predicted time course of intracellular payload concentration at any given point in the tissue is available. Lastly, the results are strongly dependent on the assumed parameter values. The wide range of potential kinetics for ADCs makes it difficult to extrapolate parameters measured for a particular ADC or ADC component to others. In this work, care was taken to identify parameter values that matched the experimental conditions of the study whenever possible.
The present work seeks to provide insights regarding the potential clinical benefits of ADC bystander effects. The choice whether the design of ADCs should include bystander capabilities is a complex balance between efficacy and toxicity. Mechanisms of toxicity may include free payload washout, deconjugation of payload from the antibody in circulation, non-specific ADC targeting, and hepatic clearance. To build evidence for or against the use of bystander payloads, these mechanisms should be quantified alongside cellular exposure, which is itself highly dependent on the characteristics of the ADC and the tumor. In general, experiments providing estimates of the kinetic parameters for a particular antibody, linker, payload, and cell line would be of great benefit in the application of the present model in support of decision making for design of ADCs.
Conclusion
ADCs have a complex design space based on the choice of antibody, linker, and payload, and improved quantitative tools for drug development are needed. Predicting the effectiveness of ADCs in solid vascularized tumors requires consideration of spatial gradients in concentration around vessels. The model presented here provides a novel approach for predicting the distribution of ADCs and payloads in tumor tissues including bystander effects, taking into account the heterogeneous structure of the tumor vasculature and the kinetics of the ADC and payload. The model has potential application in the development of more effective ADCs for use in treating solid tumors.
References
Chari RVJ, Miller ML, Widdison WC. Antibody–drug conjugates: an emerging concept in cancer therapy. Angew Chem Int Ed. 2014;53(15):3796–827. https://doi.org/10.1002/anie.201307628.
Alley SC, Zhang X, Okeley NM, Anderson M, Law C-L, Senter PD, et al. The pharmacologic basis for antibody-auristatin conjugate activity. J Pharmacol Exp Ther. 2009;330(3):932–8. https://doi.org/10.1124/jpet.109.155549.
Sanderson RJ, Hering MA, James SF, Sun MMC, Doronina SO, Siadak AW, et al. In vivo drug-linker stability of an anti-CD30 dipeptide-linked auristatin immunoconjugate. Clin Cancer Res. 2005;11:843–52.
Hamblett KJ, Senter PD, Chace DF, Sun MMC, Lenox J, Cerveny CG, et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin Cancer Res. 2004;10(20):7063–70. https://doi.org/10.1158/1078-0432.ccr-04-0789.
Bakhtiar R. Antibody drug conjugates. Biotechnol Lett. 2016;38(10):1655–64. https://doi.org/10.1007/s10529-016-2160-x.
Liu C, Tadayoni BM, Bourret LA, Mattocks KM, Derr SM, Widdison WC, et al. Eradication of large colon tumor xenografts by targeted delivery of maytansinoids. Proc Natl Acad Sci. 1996;93(16):8618–23.
Kovtun YV, Audette CA, Ye Y, Xie H, Ruberti MF, Phinney SJ, et al. Antibody-drug conjugates designed to eradicate tumors with homogeneous and heterogeneous expression of the target antigen. Cancer Res. 2006;66(6):3214–21. https://doi.org/10.1158/0008-5472.can-05-3973.
Okeley NM, Miyamoto JB, Zhang X, Sanderson RJ, Benjamin DR, Sievers EL, et al. Intracellular activation of SGN-35, a potent anti-CD30 antibody-drug conjugate. Clin Cancer Res. 2010;16(3):888–97. https://doi.org/10.1158/1078-0432.ccr-09-2069.
Li F, Emmerton KK, Jonas M, Zhang X, Miyamoto JB, Setter JR, et al. Intracellular released payload influences potency and bystander-killing effects of antibody-drug conjugates in preclinical models. Cancer Res. 2016;76(9):2710–9. https://doi.org/10.1158/0008-5472.can-15-1795.
Golfier S, Kopitz C, Kahnert A, Heisler I, Schatz CA, Stelte-Ludwig B, et al. Anetumab ravtansine: a novel mesothelin-targeting antibody–drug conjugate cures tumors with heterogeneous target expression favored by bystander effect. Mol Cancer Ther. 2014;13(6):1537–48. https://doi.org/10.1158/1535-7163.mct-13-0926.
Erickson HK, Park PU, Widdison WC, Kovtun YV, Garrett LM, Hoffman K, et al. Antibody-maytansinoid conjugates are activated in targeted cancer cells by lysosomal degradation and linker-dependent intracellular processing. Cancer Res. 2006;66(8):4426–33. https://doi.org/10.1158/0008-5472.can-05-4489.
Chauhan VP, Jain RK. Strategies for advancing cancer nanomedicine. Nat Mater. 2013;12(11):958–62. https://doi.org/10.1038/nmat3792.
Juweid M, Neumann R, Paik C, Perez-Bacete MJ, Sato J, van Osdol W, et al. Micropharmacology of monoclonal antibodies in solid tumors: direct experimental evidence for a binding site barrier. Cancer Res. 1992;52(19):5144–53.
Widdison WC, Ponte JF, Coccia JA, Lanieri L, Setiady Y, Dong L, et al. Development of anilino-maytansinoid ADCs that efficiently release cytotoxic metabolites in cancer cells and induce high levels of bystander killing. Bioconjug Chem. 2015;26(11):2261–78. https://doi.org/10.1021/acs.bioconjchem.5b00430.
Cormier JN, Hijazi YM, Abati A, Fetsch P, Bettinotti M, Steinberg SM, et al. Heterogeneous expression of melanoma-associated antigens and HLA-A2 in metastatic melanoma in vivo. Int J Cancer. 1998;75(4):517–24. https://doi.org/10.1002/(SICI)1097-0215(19980209)75:4<517::AID-IJC5>3.0.CO;2-W.
Smith LM, Nesterova A, Ryan MC, Duniho S, Jonas M, Anderson M, et al. CD133/prominin-1 is a potential therapeutic target for antibody-drug conjugates in hepatocellular and gastric cancers. Br J Cancer. 2008;99(1):100–9.
Shah DK, Haddish-Berhane N, Betts A. Bench to bedside translation of antibody drug conjugates using a multiscale mechanistic PK/PD model: a case study with brentuximab-vedotin. J Pharmacokinet Pharmacodyn. 2012;39(6):643–59. https://doi.org/10.1007/s10928-012-9276-y.
Singh AP, Shah DK. Measurement and mathematical characterization of cell-level pharmacokinetics of antibody-drug conjugates: a case study with trastuzumab-vc-MMAE. Drug Metab Dispos. 2017;45:1120–32.
Singh AP, Shin YG, Shah DK. Application of pharmacokinetic-pharmacodynamic modeling and simulation for antibody-drug conjugate development. Pharm Res. 2015;32(11):3508–25. https://doi.org/10.1007/s11095-015-1626-1.
Cilliers C, Guo H, Liao J, Christodolu N, Thurber GM. Multiscale modeling of antibody-drug conjugates: connecting tissue and cellular distribution to whole animal pharmacokinetics and potential implications for efficacy. AAPS J. 2016;18(5):1117–30. https://doi.org/10.1208/s12248-016-9940-z.
Vasalou C, Helmlinger G, Gomes B. A mechanistic tumor penetration model to guide antibody drug conjugate design. PLoS One. 2015;10(3):e0118977. https://doi.org/10.1371/journal.pone.0118977.
Khera E, Cilliers C, Bhatnagar S, Thurber GM. Computational transport analysis of antibody-drug conjugate bystander effects and payload tumoral distribution: implications for therapy. Mol Syst Des Eng. 2018;3(1):73–88. https://doi.org/10.1039/C7ME00093F.
Secomb TW, Hsu R, Dewhirst MW, Klitzman B, Gross JF. Analysis of oxygen transport to tumor tissue by microvascular networks. Int J Radiat Oncol Biol Phys. 1993;25(3):481–9. https://doi.org/10.1016/0360-3016(93)90070-C.
Hicks KO, Pruijn FB, Secomb TW, Hay MP, Hsu R, Brown JM, et al. Use of three-dimensional tissue cultures to model extravascular transport and predict in vivo activity of hypoxia-targeted anticancer drugs. J Natl Cancer Inst. 2006;98(16):1118–28. https://doi.org/10.1093/jnci/djj306.
Secomb TW. A Green’s function method for simulation of time-dependent solute transport and reaction in realistic microvascular geometries. Math Med Biol. 2016;33(4):475–94. https://doi.org/10.1093/imammb/dqv031.
Rhoden JJ, Wittrup KD. Dose dependence of intratumoral perivascular distribution of monoclonal antibodies. J Pharm Sci. 2012;101(2):860–7. https://doi.org/10.1002/jps.22801.
Secomb TW, Hsu R, Braun RD, Ross JR, Gross JF, Dewhirst MW. Theoretical simulation of oxygen transport to tumors by three-dimensional networks of microvessels. In: Hudetz AG, Bruley DF, editors. Oxygen Transport to Tissue XX. Boston: Springer US; 1998. p. 629–34.
Jacobsen ED, Sharman JP, Oki Y, Advani RH, Winter JN, Bello CM, et al. Brentuximab vedotin demonstrates objective responses in a phase 2 study of relapsed/refractory DLBCL with variable CD30 expression. Blood. 2015;125(9):1394–402. https://doi.org/10.1182/blood-2014-09-598763.
Ogitani Y, Hagihara K, Oitate M, Naito H, Agatsuma T. Bystander killing effect of DS-8201a, a novel anti-human epidermal growth factor receptor 2 antibody–drug conjugate, in tumors with human epidermal growth factor receptor 2 heterogeneity. Cancer Sci. 2016;107(7):1039–46. https://doi.org/10.1111/cas.12966.
Silence K, Dreier T, Moshir M, Ulrichts P, Gabriels SME, Saunders M, et al. ARGX-110, a highly potent antibody targeting CD70, eliminates tumors via both enhanced ADCC and immune checkpoint blockade. mAbs. 2014;6(2):523–32. https://doi.org/10.4161/mabs.27398.
Wang R, Li L, Zhang S, Li Y, Wang X, Miao Q, et al. A novel enediyne-integrated antibody–drug conjugate shows promising antitumor efficacy against CD30+ lymphomas. Mol Oncol. 2018;12(3):339–55. https://doi.org/10.1002/1878-0261.12166.
Schmidt MM, Thurber GM, Wittrup KD. Kinetics of anti-carcinoembryonic antigen antibody internalization: effects of affinity, bivalency, and stability. Cancer Immunol Immunother. 2008;57(12):1879–90. https://doi.org/10.1007/s00262-008-0518-1.
Adam PJ, Terrett JA, Steers G, Stockwin L, Loader JA, Fletcher GC, et al. CD70 (TNFSF7) is expressed at high prevalence in renal cell carcinomas and is rapidly internalised on antibody binding. Br J Cancer. 2006;95:298–306. https://doi.org/10.1038/sj.bjc.6603222.
Sutherland MSK, Sanderson RJ, Gordon KA, Andreyka J, Cerveny CG, Yu C, et al. Lysosomal trafficking and cysteine protease metabolism confer target-specific cytotoxicity by peptide-linked anti-CD30-auristatin conjugates. J Biol Chem. 2006;281(15):10540–7. https://doi.org/10.1074/jbc.M510026200.
Maass KF, Kulkarni C, Betts AM, Wittrup KD. Determination of cellular processing rates for a trastuzumab-maytansinoid antibody-drug conjugate (ADC) highlights key parameters for ADC design. AAPS J. 2016;18(3):635–46. https://doi.org/10.1208/s12248-016-9892-3.
El-Kareh AW, Secomb TW. Two-mechanism peak concentration model for cellular pharmacodynamics of doxorubicin. Neoplasia. 2005;7(7):705–13. https://doi.org/10.1593/neo.05118.
El-Kareh AW, Labes RE, Secomb TW. Cell cycle checkpoint models for cellular pharmacology of paclitaxel and platinum drugs. AAPS J. 2008;10(1):15–34. https://doi.org/10.1208/s12248-007-9003-6.
Beck A, Goetsch L, Dumontet C, Corvaia N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat Rev Drug Discov. 2017;16(5):315–37. https://doi.org/10.1038/nrd.2016.268.
Funding
This work was supported by the NIH National Heart, Lung and Blood Institute, grant HL034555.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(PDF 316 kb)
Rights and permissions
About this article

Cite this article
Burton, J.K., Bottino, D. & Secomb, T.W. A Systems Pharmacology Model for Drug Delivery to Solid Tumors by Antibody-Drug Conjugates: Implications for Bystander Effects. AAPS J 22, 12 (2020). https://doi.org/10.1208/s12248-019-0390-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1208/s12248-019-0390-2