
Abstract
In silico HLA-binding algorithms and in vitro T cell-based assays as predictive tools for human immunogenicity risk have made inroads in the biotherapeutic drug discovery and development process. Currently, these tools are being used only for candidate selection or characterization and not for making a go/no-go decision for further development. A clear limitation for a broader implementation is the lack of correlation between the predicted T cell epitope content/immune reactivity potential of a biotherapeutic and the subsequent ADA-related clinical immunogenicity outcome. The current state of technologies and their pros and cons were discussed as a part of the 2016 AAPS National Biotechnology Conference in a themed session. A review of the advances in the area and the session talks along with the ensuing discussions are summarized in this commentary.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
INTRODUCTION
The rapidly growing number of approved biotherapeutic drug products in the clinic point to the remarkable success of these modalities in many challenging therapeutic areas. A major limitation to the use of biotherapeutics is the development of anti-drug antibodies (ADA) which may decrease the efficacy of the biotherapeutic candidate by neutralizing them or modifying their clearance, and/or impact safety by inducing drug-specific hypersensitivity reactions (1). The ADA may also cross-react with closely related endogenous counterparts of the biotherapeutic thereby interfering with critical physiological functions. Therefore, the early prediction and mitigation of immunogenicity risk during discovery and throughout the course of biopharmaceutical drug development and patient safety is an important consideration.
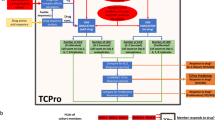
The critical step in the development of an immune response to a biotherapeutic is the activation of CD4+ T cells as a result of its recognition of a cognate peptide MHC class II complex on an antigen-presenting cell. Several immunogenicity risk assessment tools for biotherapeutics such as in silico human leukocyte antigen (HLA)-binding algorithms that identify potential T cell epitopes, LC/MS-based MHC-associated peptide proteomics (MAPPS) assays, in vitro peptide/HLA-binding assays and in vitro human blood-derived cell-based assays based on this critical step have gained prominence over the past decade (2) (Fig. 1). While the MAPPS assay provides a direct assessment and readout of peptides associated with MHC, the human in vitro assays can help understand the heterozygosity of alleles and the T cell repertoire in a healthy population that can potentially react to the presented peptides.

Key processes involved in the development of a humoral, MHC class II-mediated anti-biotherapeutic IgG response and corresponding predictive immunogenicity tools. a Processed peptides from the biotherapeutic are presented as T cell epitopes on MHC class II molecules on an APC like a dendritic cell. Computational tools can be used at this stage to model the affinity of peptides to the different human MHC class II molecules. In vitro HLA-binding assays and MAPPS assays can be used to measure the affinity and stability of peptides binding to MHC class II. b CD4+ helper T cells recognize the T cell epitopes presented by APC’s with their TCRs resulting in activation and proliferation of these T cells. Different assay formats based on diverse donor cells can be utilized to measure T cell activation in response to an antigen in vitro. c Activated T cell interaction with B cells results in the development of plasma cells secreting high affinity antibodies. Transgenic mouse models containing human immune cell repertoires can produce high affinity antibodies to biotherapeutics. d T regulatory cells modulate immune responses to an antigen. Predictive assays and tools that include these cells are therefore more likely to model in vivo immunogenicity. APC antigen-presenting cell, MAPPS liquid chromatography/mass spectrometric (LC/MS)-based MHC-associated peptide proteomics, TCR T cell receptor
The ability of these tools to predict clinical outcomes still needs further validation. This gap hence mandates that such evaluations be instead termed as immunogenicity or antigenicity risk assessments (Fig. 2). In vivo animal models including HLA transgenics or mice with humanized immune systems are being considered by some laboratories to obtain a comparative assessment of the risk for developing an ADA response (for example to compare the marketed drug product to a biosimilar or evaluate multiple formulations or manufacturing lots with variable amounts of process impurity) and breaking of tolerance to the endogenous protein. Currently, rodents and other animal models are not widely believed to be an accurate representation of the immunogenicity risk in humans. However, these models are under development and further refinement and may prove useful in the future. As such, these are not included as a part of this discussion.

In silico and in vitro cell-based tools model and measure critical steps in an antibody-mediated immune response. Analytical, functional, and clinical correlation as well as standardization of these tools remains a challenge and an obstacle for greater utility of these tools in the drug development. HLA human leukocyte antigen
Most of these tools are primarily employed in the discovery phase of biotherapeutics development to identify T cell epitope content on various variants of a protein sequence and support candidate selection based on the sequence with the least antigenic property in conjunction with other selection parameters such as efficacy, off/on target effects, manufacturability, and pharmacokinetics (3,4). The information from these tools enables optimization of the sequences by rational design or rank ordering sequences (if multiple variants with similar developability parameters are available) to reduce the likelihood of ADA in the clinic (5,6,7). Additionally, the algorithm-based readouts and their association with specific HLA-DR alleles can be confirmed using human immune cell-based assays (6). These assays can also be used to identify and derisk attributes driven by post translational changes to the biotherapeutics that are not T cell epitope driven (8,9,10) and are instead related to process and manufacturing related changes (8,9,11).
Nevertheless, the utilization of these preclinical tools is hindered by the lack of common assay standards and independent clinically validated data. An American Association of Pharmaceutical Sciences (AAPS) survey in 2014 indicates (manuscript submitted) that a wide cross section of labs across industry and academic institutions that evaluate immunogenicity to biotherapeutics have begun adopting these methods. However, the use of standardized methods and outputs among these peers needs further effort in order to compare risks due to the sequence and other attributes across biologics with similar engineered sequences (e.g., biosimilars), analytical performance of the assays used, and clinical correlation. Another limitation is the availability of clinical studies where a biotherapeutic was administered before and after engineering out the non-self T cell epitopes from the sequence. Results from such a study would clearly indicate if removal of T cell epitopes led to decreased incidence of immunogenicity in clinic.
At the 2016 AAPS National Biotechnology Congress, a “Topics in Transition” session entitled, “Approaches to Evaluate Ex Vivo Immunogenicity Risks with Observed Clinical Outcome” highlighted these promising tools and methods and the associated challenges. The opening talk of the session entitled “Correlation of Preclinical Prediction Outputs with Clinical Outcomes” by Vibha Jawa described evaluations using in silico algorithm and immune cell-based predictive tools and success at correlating to an ADA response in clinic (Jawa et al. manuscript in preparation). Through case studies using unpublished sequences and commercially approved biotherapeutics, the talk showed that output from sequence-based algorithms and in vitro assays, even with their limitations, can improve the prediction accuracy for sequence based risk identification and rank ordering of candidates to achieve the least clinical immunogenicity outcome. The next talk was delivered by Nikolai Schwabe and titled “Tools & Technologies for Managing Immunogenicity Risk - where does the rubber meet the road?”. The presentation focused on the ability of using new and established in vitro cell-based activation as well as HLA-binding epitope analysis methods that can provide a broad correlation of T cell activation potential or epitope content with clinical ADA incidences. While providing examples of success stories where such associations are observed using these methods, he also elaborated on how the B cell expansion in the germinal centers and patient specific factors such as disease, prior exposure and state of the immune system cannot yet be assessed in these preclinical methods and thereby limits the ability of these in vitro T cell activation readouts to assess clinical immunogenicity risk. This commentary provides highlights of the presentations and the ensuing discussions on the challenges and resolutions suggested by the panel members and the audience.
What Do In Silico Tools Predict and Is that Output Relevant for ADA Formation?
Computational algorithm-based in silico tools rely on the ability of the linear peptide sequences parsed into overlapping peptides up to 15 amino acids long to bind to the HLA allele pocket. The size, charge, and polarity of each amino acid at each position collectively determine how well a protein fragment will bind to a given HLA allele. A large variety of algorithms are available for performing the predictive evaluations, with each one addressing a specific element related to binding of the sequence at the MHC pocket (12). Currently, the most utilized application of the predictive algorithms is rank ordering the variants and short listing/selecting the least risky molecule in terms of T cell epitope content as the candidate for further development. Additionally, by utilizing the T helper epitope cluster analysis, the hot spots in the sequence that bind promiscuously to the large number of HLA, alleles (DR, DP, and DQ) can be identified. Such information can be used at early stages of discovery for lead optimization and selection. Although immunogenicity concerns are most related to development of antibodies to a biotherapeutic, these in silico algorithms almost exclusively center on the prediction of potential T helper cell epitopes, which would presumably facilitate the maturation of B cells into ADA-producing plasma cells. However, these in silico tools are not currently designed to assess the impact of non-natural amino acids, nucleic acids, or post translational modifications, which may be studied using in vitro cell-based T cell activation assays.
An improvement in prediction accuracy can be attempted by utilizing multiple algorithms that can each provide their own specific readouts (Jawa et al., manuscript in preparation). The outputs from multiple algorithms not only predict the binding of amino acids to the HLA pocket but also provide an understanding of the cross reactivity and tolerogenicity of the sequences due to homology to human genome and microbiome which educates the T cell repertoire (13). Additionally, the overall prediction score was also normalized by taking into account the presence of T regulatory epitopes in addition to effector CD4 T cell epitopes (14). An additional in silico algorithm prediction method that follows the consensus approach that combines outputs from multiple prediction approaches and is sourced by in vitro peptide binding to HLA-DR pockets was also used as an orthogonal means to identify the top 1% binders to prevalent HLA DRB1 alleles in the worldwide population as well as less frequently reported DRB3 and DP/DQ alleles (15). Most of the HLA-binding datasets in the databases used for the in silico algorithms are derived from HLA DRB1 binding machine learning or experimental observations; hence, these are most frequently used to power the in silico peptide-HLA binding predictions. Overall, the concept was to strengthen the predictability by layering the features from each tool.
What Do In Vitro T Cell Activation and HLA-Binding Assays Predict and Is that Data Relevant for ADA Formation in the Clinic?
In vitro HLA-binding immunoassays or human PBMC and dendritic cell (DC) assays (14) provide additional risk assessment and represent a means to confirm as well as probe for any post sequence quality attributes and impurities that could become an immunogenicity risk during the manufacturing and formulation process. These methods are based on measuring immune cell activation following exposure to the intact biotherapeutic or overlapping small peptides derived from it, and have been extensively discussed in the recent published literature (3,4,9,10,16,17,18,19,20). It was very clear from the presentations that these human immune cell-based or HLA-binding assays are most beneficial when powered with diverse donor pool and HLA genotypes representative of the world population. These assays are useful not only to measure the risk due to the foreign epitope content of the biologic but can also assess the immune activation danger signals due to product attributes like aggregation (10,21), host cell proteins (8), degraded protein fragments, leachates like silicon oil, tungsten, and other metals.
In vitro HLA-peptide binding immunoassays have traditionally been used to provide the datasets that are used to build the in silico algorithms (22,23). Recently, in vitro methods such as MAPPS have made significant inroads in preclinical immunogenicity by mapping the naturally processed and presented T cell epitopes on biotherapeutics (19,24). The DCs naturally process proteins and present the derived peptides in the context of HLA-class II. HLA-DR-associated biotherapeutic-derived peptides, representing potential T cell epitopes, are identified in the MAPPS assay and appear to represent the naturally presented T cell engaging epitopes. These assays indicate that natural antigen processing plays an important role in identifying potential T cell epitopes as much smaller numbers of peptides are eluted from the HLA pocket than what is predicted by the HLA-binding assays and in silico algorithm-based outputs. Hence, data from these MAPPS assays help to address the limitation of “over prediction” of in silico algorithms and can also help in the identification of “dominant” T cell epitopes.
Schwabe et al. showed a good correlation of a biotherapeutic protein with a low immunogenicity incidence in clinic with a low number of T cell epitopes identified by MAPPS assay and a low T cell activation in the in vitro PBMC assay. Similarly, biotherapeutics with relatively higher clinical immunogenicity incidences also showed increased numbers of potential T cell epitopes in the MAPPS assay and increased T cell response rates in T cell activation assays.
Another key application of MAPPS assay was the ability to correlate T cell epitopes from a biotherapeutic identified using in silico algorithm with the MAPPS assay output. The results from the HLA-binding study confirmed that the peptides eluted from specific HLA DR alleles of antigen-presenting cells of patients dosed with a biotherapeutic were also predicted as potential high binders by the in silico algorithm (3,25). A comprehensive evaluation of the correlation of preclinically identified T cell epitopes and clinical T cell activation is ongoing as part of the European consortium, Anti-Biopharmaceutical Immunization: prediction and analysis of clinical relevance to minimize the RISK (ABIRISK) initiative and data from these studies should become available in the near future. The outcomes from such studies seem to indicate that layering in silico and in vitro data could provide a higher degree of confidence in the immunogenicity assessment.
Gaps and Challenges in Antigenicity Assessments and the Path Forward
In 2014, the Immunogenicity Prediction Action Program Area (IPAPA) within the AAPS Therapeutic Product Immunogenicity Focus Group (TPIFG) systemically surveyed industry, academic and regulatory agencies stakeholders on the use, impact, applicability and limitations of the in silico and in vitro HLA-binding or cell-based T cell epitope identification tools and collated the responses. In these surveys, it was noticed that some of the shortcomings of these methods were the limited correlation (both analytical and clinical) of the prediction output and lack of commonly established parameters to define the validation criteria. Additionally, none of these methods consider the numerous exacerbating patient and product related factors that promote ADA responses. A summary of gaps and potential mitigation strategies are provided in Table I. Koren et al. (26) have attempted to address these shortcomings by comparing the T cell responses and their memory to the administered protein by probing for a recall response with immune dominant epitopes from T cells derived antibody positive subjects. More studies are needed to show such correlations using the cells from dosed donors.
It appears that the strength and gaps from such readouts when pieced together can be used to gauge the overall value even though the correlations might not be perfect. Such instances of comparative antigenicity assessments are unfortunately frequently not published, presumably, due to foreseeable impact to internal R&D programs and lack of concrete requirements or recommendation guidance from regulatory agencies.
The biggest challenge remains with the predictive accuracy and correlations with clinical immunogenicity incidence. Some ways to improve the predictive accuracy are by (1) utilizing the multi-functionality of algorithms and confirming the top 1–10% binders with the strongest binding affinity in a sequence, (2) incorporating a diverse HLA allele coverage to encompass global diversity and mining of pharmacogenomics data from clinic and obtaining associations of HLA alleles with immunogenicity incidences, (3) assessing the binding of peptide MHC complex to the variable T cell receptor repertoire of healthy and diseased subjects, (4) accounting for the regulatory epitopes and differentiating between T cell epitopes conserved between human genome as well as environmental pathogens, and (5) using in vitro T cell activation and HLA-binding assay outputs to confirm the peptides identified as potential epitopes. Additionally, by assessing for recall response in diseased subjects challenged with the predicted peptides, more confidence can be generated around the predictive accuracy of such tools.
Some obvious caveats in performing correlations of T cell derived outputs to ADA incidences are comparing the fate of linear sequences from a biotherapeutic that could drive a T cell functional readout in vitro and linking to a B cell-derived antibody response-based outcome in patients. Such relationships can at the most be considered associative and require underlying assumptions that linear epitopes that drive T cell response in vitro can potentially drive the B cell-driven antibody responses in vivo. The kinetics of such an ADA generating response and other factors like disease indications, technical variability in the detection of such antibodies and the role of standard of care immune suppressive medications in influencing the overall outcome have to be taken into account and need further evaluation systematically. Nevertheless, these methods are close approximations of the human antigen presentation and T cell activation steps that precede B cell maturation and ADA secretion. It was evident in the discussions that B cell immunogenic epitopes can be incorporated into immunogenicity prediction models. Although several in silico tools for predicting B cell epitopes have been developed to predict both linear or conformational epitopes, these are not yet being utilized widely or systematically to predict immunogenicity to biotherapeutics. Finally, during the break out session, the panel and the audience encompassing academic, industry and regulatory agency partners discussed the best practices within the current state of technology and understanding of immune systems and agreed that at present, in vitro HLA-binding and in vitro cell-based proliferation or cytokine activation assays provide an additional level of accuracy/reliability, as well as an opportunity to corroborate the in silico prediction outcomes. No approach or strategy yet approaches 100% accuracy when it comes to predicting a clinical outcome and there are challenges including but not limited to (1) inability of all relevant players of the immune system that are present in vivo to be a part of in vitro assays, (2) cells and their functional outputs in long-term cultures can be rate limiting, (3) lack of clinical studies where a biotherapeutic was administered before and after optimization/rational design for engineering out potential immunogenic epitopes, and (4) the fate of the biotherapeutic in an in vivo environment with interplay of tissue architecture like extracellular matrix (ECM), interstitial space and vasculature cannot be reproduced. Some effort has been made by development of artificial lymph node systems which need further validation (27).
It appears that the abovementioned gaps and the mitigation strategies when pieced together can be used to gauge the overall value even though the correlations might not be perfect. Such instances of comparative antigenicity assessments are unfortunately frequently not published by industry peers, presumably, due to foreseeable impact to internal R&D programs and lack of concrete requirements or recommendation guidance from regulatory agencies.
CONCLUSIONS
Some of the key takeaways from these sessions were as follows. The participants and the audience agreed that a robust immunogenicity risk assessment would require multiple orthogonal tools to confirm and complement the final outcome. It was clear that the drug type and the stage of the research and development program would be the guiding points for the selection of the in silico or in vitro assay platforms. Additionally, it would be beneficial to employ layering of algorithms or use orthogonal methods to increase prediction accuracy since no recommendation on the most beneficial tool is currently available.
In cognizance of the gaps in correlation of predicted immunogenicity with clinical immunogenicity, ongoing efforts to standardize these technologies both within US through the AAPS focus groups and recent publications by the European ABIRISK consortium to validate the methods and correlate the predictive platforms to ADA and clinical immunogenicity outcomes have been encouraging (5,28,29,30).
Furthermore, in order to achieve the necessary clinical validation, drug development process could compare T cell repertoire from both naïve and dosed clinical samples to correlate the T cell epitope profile with ADA risk. Systematic collection of clinical HLA data and whole blood-derived PBMC samples pre and post dosing with biotherapeutic through informed consent in clinical protocols could also add to further understanding of the association of HLA of individuals to the risk of immunogenicity in clinic. Also, such efforts are likely to succeed if a side-by-side comparison of two drug sequences can be performed. This might be feasible in early discovery where candidates before and after optimization can be expressed in small scale and tested in in vitro systems. Overall, the participants agreed that pending analytical and clinical validation; preclinical tools can be used to derisk/deimmunize the non-self epitopes to reduce the immunogenicity risk.
References
Shankar G, Arkin S, Cocea L, Devanarayan V, Kirshner S, Kromminga A, et al. Assessment and reporting of the clinical immunogenicity of therapeutic proteins and peptides-harmonized terminology and tactical recommendations. AAPS J. 2014;16(4):658–73.
Jawa V, Cousens LP, Awwad M, Wakshull E, Kropshofer H, De Groot AS. T cell dependent immunogenicity of protein therapeutics: preclinical assessment and mitigation. Clin Immunol. 2013;149(3):534–55.
Karle A, Spindeldreher S, Kolbinger F. Secukinumab, a novel anti-IL-17A antibody, shows low immunogenicity potential in human in vitro assays comparable to other marketed biotherapeutics with low clinical immunogenicity. MAbs. 2016;8(3):536–50.
Xue L, Hickling T, Song R, Nowak J, Rup B. Contribution of enhanced engagement of antigen presentation machinery to the clinical immunogenicity of a human interleukin (IL)-21 receptor-blocking therapeutic antibody. Clin Exp Immunol. 2016;183(1):102–13.
Lamberth K, Reedtz-Runge SL, Simon J, Klementyeva K, Pandey GS, Padkjaer SB, et al. Post hoc assessment of the immunogenicity of bioengineered factor VIIa demonstrates the use of preclinical tools. Sci Transl Med. 2017;9(372):eaag1286.
Mazor R, Onda M, Pastan I. Immunogenicity of therapeutic recombinant immunotoxins. Immunol Rev. 2016;270(1):152–64.
Mazor R, Zhang J, Xiang L, Addissie S, Awuah P, Beers R, et al. Recombinant immunotoxin with T cell epitope mutations that greatly reduce immunogenicity for treatment of mesothelin-expressing tumors. Mol Cancer Ther. 2015;14(12):2789–96.
Jawa V, Joubert MK, Zhang Q, Deshpande M, Hapuarachchi S, Hall MP, et al. Evaluating immunogenicity risk due to host cell protein impurities in antibody-based biotherapeutics. AAPS J. 2016;18(6):1439–52.
Joubert MK, Deshpande M, Yang J, Reynolds H, Bryson C, Fogg M, et al. Use of in vitro assays to assess immunogenicity risk of antibody-based biotherapeutics. PLoS One. 2016;11(8):e0159328.
Moussa EM, Panchal JP, Moorthy BS, Blum JS, Joubert MK, Narhi LO, et al. Immunogenicity of therapeutic protein aggregates. J Pharm Sci. 2016;105(2):417–30.
Jiskoot W, Kijanka G, Randolph TW, Carpenter JF, Koulov AV, Mahler HC, et al. Mouse models for assessing protein immunogenicity: lessons and challenges. J Pharm Sci. 2016;105(5):1567–75.
De Groot AS, Martin W. Reducing risk, improving outcomes: bioengineering less immunogenic protein therapeutics. Clin Immunol. 2009;131(2):189–201.
Moise L, Gutierrez AH, Bailey-Kellogg C, Terry F, Leng Q, Abdel Hady KM, et al. The two-faced T cell epitope: examining the host-microbe interface with JanusMatrix. Hum Vaccin Immunother. 2013;9(7):1577–86.
De Groot AS, Moise L, McMurry JA, Wambre E, Van Overtvelt L, Moingeon P, et al. Activation of natural regulatory T cells by IgG Fc-derived peptide “Tregitopes”. Blood. 2008;112(8):3303–11.
Kim Y, Sette A, Peters B. Applications for T cell epitope queries and tools in the immune epitope database and analysis resource. J Immunol Methods. 2011;374(1–2):62–9.
Haile LA, Puig M, Kelley-Baker L, Verthelyi D. Detection of innate immune response modulating impurities in therapeutic proteins. PLoS One. 2015;10(4):e0125078.
Rosenberg AS, Verthelyi D, Cherney BW. Managing uncertainty: a perspective on risk pertaining to product quality attributes as they bear on immunogenicity of therapeutic proteins. J Pharm Sci. 2012;101(10):3560–7.
Verthelyi D, Wang V. Trace levels of innate immune response modulating impurities (IIRMIs) synergize to break tolerance to therapeutic proteins. PLoS One. 2010;5(12):e15252.
Baker MP, Reynolds HM, Lumicisi B, Bryson CJ. Immunogenicity of protein therapeutics: the key causes, consequences and challenges. Self Nonself. 2010;1(4):314–22.
Harding FA, Stickler MM, Razo J, DuBridge RB. The immunogenicity of humanized and fully human antibodies: residual immunogenicity resides in the CDR regions. MAbs. 2010;2(3):256–65.
Joubert MK, Hokom M, Eakin C, Zhou L, Deshpande M, Baker MP, et al. Highly aggregated antibody therapeutics can enhance the in vitro innate and late-stage T cell immune responses. J Biol Chem. 2012;287(30):25266–79.
Davies V, Vaughan K, Damle R, Peters B, Sette A. Classification of the universe of immune epitope literature: representation and knowledge gaps. PLoS One. 2009;4(9):e6948.
Paul S, Lindestam Arlehamn CS, Scriba TJ, Dillon MB, Oseroff C, Hinz D, et al. Development and validation of a broad scheme for prediction of HLA class II restricted T cell epitopes. J Immunol Methods. 2015;422:28–34.
Bryson CJ, Jones TD, Baker MP. Prediction of immunogenicity of therapeutic proteins: validity of computational tools. BioDrugs. 2010;24(1):1–8.
Rombach-Riegraf V, Karle AC, Wolf B, Sorde L, Koepke S, Gottlieb S, et al. Aggregation of human recombinant monoclonal antibodies influences the capacity of dendritic cells to stimulate adaptive T cell responses in vitro. PLoS One. 2014;9(1):e86322.
Koren E, De Groot AS, Jawa V, Beck KD, Boone T, Rivera D, et al. Clinical validation of the “in silico” prediction of immunogenicity of a human recombinant therapeutic protein. Clin Immunol. 2007;124(1):26–32.
Dhir V, Fort M, Mahmood A, Higbee R, Warren W, Narayanan P, et al. A predictive biomimetic model of cytokine release induced by TGN1412 and other therapeutic monoclonal antibodies. J Immunotoxicol. 2012;9(1):34–42.
Hamze M, Meunier S, Karle A, Gdoura A, Goudet A, Szely N, et al. Characterization of CD4 T cell epitopes of infliximab and rituximab identified from healthy donors. Front Immunol. 2017;8:500.
Schultz HS, Reedtz-Runge SL, Backstrom BT, Lamberth K, Pedersen CR, Kvarnhammar AM, et al. Quantitative analysis of the CD4+ T cell response to therapeutic antibodies in healthy donors using a novel T cell: PBMC assay. PLoS One. 2017;12(5):e0178544.
Ing M, Gupta N, Teyssandier M, Maillere B, Pallardy M, Delignat S, et al. Immunogenicity of long-lasting recombinant factor VIII products. Cell Immunol. 2016;301:40–8.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Gokemeijer, J., Jawa, V. & Mitra-Kaushik, S. How Close Are We to Profiling Immunogenicity Risk Using In Silico Algorithms and In Vitro Methods?: an Industry Perspective. AAPS J 19, 1587–1592 (2017). https://doi.org/10.1208/s12248-017-0143-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1208/s12248-017-0143-z