
Abstract
Purpose
Biotherapeutics are a large and rapidly growing class of drugs being produced by pharmaceutical companies to treat a diverse range of clinical indications. The overall efficacy and safety of these products can be greatly impacted by their capacity to induce undesired immune responses. This review discusses in vitro cell-based methods used to assess the T cell mediated immunogenicity risk of proteinaceous therapeutic modalities and manufacturing impurities.
Methods
Here, we outline the potential sources and factors that influence immunogenicity. We present patient and product considerations that should be made in designing appropriate in vitro experiments that evaluate T cell epitopes capable of triggering treatment and outcome impacting anti-drug antibody responses and other adverse events.
Results
We present the current in vitro assays used to assess T cell activation towards biotherapeutics and the product impurities. Lastly, we outline the caveats, concerns, and challenges that remain with these cell-based assays.
Conclusions
Data generated from these in vitro antigenicity/immunogenicity assays may be used to derive immunogenicity risk assessments for programs and production processes and provides an opportunity for early selection of candidates or manufacturing impurities with lower likelihood of generating or exacerbating clinical immunogenicity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Protein therapeutics including monoclonal antibodies (mAb), bi- and multi-specific drugs, and enzyme replacement therapies are a rapidly growing class of pharmacological treatments. There is also a booming number of Antibody-Drug Conjugate (ADCs) oncology therapeutics for site-specific drug treatment-each containing a major mAb protein component [1]. Between 2014 and mid-2018, 129 distinct biopharmaceuticals entered the market [2]. The number of product approvals nearly doubled compared to the four years prior, and over half of these first time approvals were mAb products [2]. With the increasing number of these marketed protein therapeutics, delivering the safest and most effective form of these products to patients is essential.
When mAbs or other biotherapeutics are administered to patients, immune responses directed towards those treatments often occur. This negatively affects product safety and efficacy and can lead to a host of harmful side effects [3,4,5]. This immunogenicity most commonly takes the form of anti-drug antibodies (ADAs) that develop in large subsets of patients receiving biotherapeutic treatment over time. ADAs can greatly reduce the clinical efficacy of the therapeutic by neutralizing the product, altering the pharmacokinetics, or potentially result in severe allergic reactions [6,7,8,9]. Clinical development of biotherapeutics can be discontinued due in part to these kinds of adverse immunogenic responses [10, 11]. However, it is also possible for ADA responses to result in non-clinically relevant outcomes [8, 9, 12, 13].
Due to their potential to greatly affect the outcome of patient treatment, much of the guidance set forth by health authorities such as the European Medicines Agency (EMA) and the United States Food and Drug Administration (USFDA) focus on a risk based assessment for ADAs [3, 7, 8, 14]. Currently, regulatory agencies do not require risk assessment for T cell responses during clinical studies. Instead, the guidance focuses primarily on identification, measurement, and characterization of ADAs [7]. However, regulators are increasingly recommending that drug manufacturers evaluate T cell immune responses as a precursor for potential ADA during early drug development, especially during candidate design and selection [3, 8]. If risk assessment is employed during this stage, immunogenic sequences found within drug candidates can be identified, helping direct which candidate should be chosen to continue through to pre-clinical trials as illustrated by the flow chart in Fig. 1. This makes early risk assessment more time and cost effective than assessment at later stages where resources have already been spent on a candidate that might not prove viable.

Flowchart for using predictive antigenicity/immunogenicity assessment tools to design better biotherapeutics. Candidates with low predicted immunogenicity are more desirable for further development. Candidates with higher predicted immunogenicity can be modified to lower their immunogenicity risk and be reassessed prior to further development. AA = Amino Acid
Mechanisms of Immunogenicity
T cell responses are very important to consider in drug design and selection because T cell dependent responses are major drivers for sustained immune memory formation targeting protein therapeutics in the clinic [15,16,17,18]. This is because high affinity antibody development is dependent on the communication between antigen presenting cells (APCs) and CD4+ T cells.
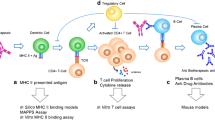
In the context of protein biotherapeutic treatment, APCs such as dendritic cells or antigen specific B cells begin processing the drug first by endocytosis (Fig. 2). The APCs then break the protein down into peptide fragments using internal protein degradation machinery. The populations of peptides that are generated are dependent on the positions of enzymatic cleavage cites within the protein as well as the capacity of a given human leukocyte antigen (HLA) allele to bind the peptide fragment [19]. After they are loaded on to HLAs, these peptides are then presented to circulating T cells [19].

Schematic outlining the events that occur during in vitro T cell assays and the readouts commonly used to assess T cell activation to protein therapeutics. Whole PBMCs or purified APCs such as monocyte derived dendritic cells are pulsed with protein therapeutic or synthesized peptides. The APCs endocytose and process the proteins or synthesized peptides and load peptide fragments onto HLA molecules which are then presented to purified T cells or T cells present within whole PBMCs. After a period of co-culture, T cell activation can be assessed through multiple methods including ELISPOT, immunoassays for secreted cytokines, and T cell proliferation via flow cytometry or [3H]-Thymidine incorporation. Multiple readout methods can be used in conjunction with one another to assess and confirm the immunogenicity potential of biotherapeutics
If a helper T cell recognizes this peptide-HLA complex using its T cell receptor, it becomes activated and provides the necessary signals and cytokines for B cells specific to the biotherapeutic to undergo affinity maturation, proliferate, and differentiate into antibody-secreting plasma cells and memory B cells. These interactions can lead to long term ADA. Efforts to disrupt T cell-B cell interactions concurrently with treatment of a biotherapeutic may provide a means to reduce incidences of ADA, but this is not without potential risks and regulatory hurdles to overcome on its own [20].
Properties relating to both the patient receiving treatment and of the product itself can contribute to the immunogenicity of therapeutic proteins. The aspects that comprise both of these properties must be considered when designing preemptive in silico and in vitro experiments to derive immunogenicity risk assessments.
Contributors to Immunogenicity: Patient Factors
Patient risk factors can be the most difficult property to control when developing biotherapeutics because they cannot be easily manipulated. The immune system is likely to respond differently to a provided therapeutic in older vs. younger patients due to differences in immunological experience and overall immune system efficacy. [21, 22]. The HLA alleles of the patient can also influence rates of immunogenicity towards specific biotherapeutics [15, 23, 24]. For example, treating multiple sclerosis patients of some specific HLA haplotypes was associated with increased immunogenicity towards the administered IFN-β [15].
Patients that require enzyme replacement therapy or gene therapy products typically express a truncated, non-functional form of a protein or no protein at all. When the patient receives treatment, the full length, functional protein might be detected as a foreign protein by the immune system [25, 26]. For example, patients with Pompe disease that still express a portion of lysosomal acid α-glucosidase (GAA) respond better to treatment compared to patients with no GAA expression [23, 27]. This is primarily because GAA− patients mount an immune response to the replacement protein [23, 27]. Similarly, breaking the natural tolerance of endogenous proteins could occur after adding an excess of recombinant biotherapeutics and can cause major long-term consequences [28].
Finally, the disease state of patients also plays a role in the development of an immune response to protein therapeutics. The same biotherapeutic used for the treatment of different conditions or disease indications might result in different rates of ADA formation [29,30,31]. Patients with autoimmune or inflammatory diseases can have a higher propensity to develop ADA responses as opposed to a patient being treated for an oncology indication and these patients may exhibit ADA responses that could be different from those obtained in healthy volunteers [9, 30]. One possible driving factor behind this observation is that the inflammatory state of a patient may change the manner by which APCs acquire, process, and present peptides on their HLA molecules [19, 32]. If this is the case, biotherapeutics that would normally not elicit an immune response would now be capable of triggering an immune reaction due to the appearance of new epitopes. Another possibility is that T regulatory cell mediated suppression and other tolerance mechanisms are so disrupted in autoimmune states that normally non-immunogenic epitopes found in biotherapeutics can trigger an immunogenic response [33, 34].
On the opposite end of the spectrum, immunosuppressed patients may have a stymied immunogenic response compared to healthy volunteers with a fully functioning immune system [35, 36]. To this end, the immunomodulatory medication being used by a patient, the current disease state, and the mechanism of action of the therapeutic are all capable of enhancing or suppressing ADA formation [37, 38]. However, analysis of reports that used mAbs to treat cancer reveals contradictory information on the way each of these characteristics influence ADA formation, indicating these factors may have variable contribution to the overall ADA response outcome [6].
Contributors to Immunogenicity: Product Factors
Unlike patient factors, product risk factors can be easily manipulated to mitigate immunogenicity potential using a multitude of strategies [39]. The primary amino acid sequence of the biologic is a major influence on its immunogenicity. APCs present linear peptides derived from the biologic molecule that can initiate a T cell response if it is recognized as foreign within the patients’ immunological response pathways. In the case of mAbs, even products that are otherwise fully human or are primarily humanized potentially contain foreign amino acid sequences located inside the antibody’s hypervariable regions. Post translational modifications such as glycosylation and modified amino acids as well as other indirect factors including concentration of the dose, frequency of dose, presence of protein aggregates or manufacturing impurities, and the route of administration can result in detectable differences in clinical immunogenicity [40,41,42,43,44,45,46].
Many protein therapeutics have immunomodulatory mechanisms of action which can directly influence their immunogenic potential. Of particular interest, biotherapeutics that enhance immune responses such as the immune checkpoint inhibitors anti-CTLA4 and anti-PD-1 might increase immunogenic responses [31]. Similarly, bi-specific antibodies that have a T cell engaging component or ADCs that carry an immunostimulatory payload might also result in enhanced immunogenicity [31, 47].
In contrast, biotherapeutics that directly engage immune cells in order to deplete or suppress normal immune system function might decrease frequency of immunogenicity. This is exemplified by low occurrence of ADAs in patients treated with B cell depleting anti-CD20 or anti-CD19 mAbs [31]. However, not all biotherapeutics with anti-inflammatory characteristics are less prone to generating immune responses. The anti-IL-21R mAb, ATR-107, is intended to suppress lymphoid cell proliferation and B cell differentiation. Despite this, ADAs occur in 76% of tested healthy donors-in part due to an unintended enhancement of dendritic cell activation and antigen presentation [48, 49].
Contributors to Immunogenicity: Host Cell Proteins and Other Critical Quality Attributes
When biotherapeutic products are produced and purified, host-cell proteins (HCPs) are inevitably co-purified, and can often be detected in small quantities in drug substance/product analyzed by ELISA and mass spectrometry [50, 51]. While a great deal of time and effort are involved in eliminating these from the final deliverable during process development, HCPs that remain are a potential risk factor for immunogenicity of a biotherapeutic when it is administered to patients [52]. For example, during clinical development of a recombinant form of human growth hormone, Somatotropin, ADAs formed in approximately 60% of patients. Reducing HCP impurities in Somatotropin preparations from approximately 1400 ppm to less than 50 ppm brought the observed rate of ADA to less than 3% [53, 54].
However, not all HCPs have the same potential to be immunogenic, and overall abundance of the HCP appears to play a role. One report has determined a threshold of <4000 ppm total HCP content in the final drug product of mAbs as a limit before HCP immunogenicity becomes a concern [55]. Additionally, there is an growing trend in the use of mammalian expression systems such as Chinese Hamster Ovary (CHO) cells over Escherichia coli or Saccharomyces cerevisiae based systems for product manufacturing since 2014 [2]. This unsurprisingly correlates with an increase in the number of mAbs in development and the overall importance of post translational modifications such as glycosylation for protein stability [2]. Because of this, many observed HCPs share high sequence homology with naturally occurring human proteins and are less likely to be immunogenic as compared to microbial impurities. However, data proving that individual, non-human HCPs with high in silico predicted risk are actually more immunogenic in vitro and in vivo remains to be obtained. Until this is established, manufacturing process development workflows are required to keep the levels of HCPs below detection or as low as possible.
Along with HCPs, protein aggregates are of major concern in biotherapeutics [44, 45]. For example, immunogenicity differences observed between different formulations of rhIFN-β products can be attributed to the presence or absence of aggregates [43]. These kinds of aggregates can form for a multitude of reasons including naturally over time during storage, or upon contact with silicone oils or metals found in the syringes used to deliver the biotherapeutic products and formulations [45]. A major consequence of protein aggregates is their capability of triggering innate immune responses which enhance the formation of adaptive responses [56,57,58]. However, the exact factors that influence the immune responses towards aggregates are still being investigated.
Similarly, other critical quality attributes such as residual host DNA, RNA, endotoxins, and inorganic compounds might still be present in drug substance after purification. Similar to the consequence of protein aggregates, these compounds can contribute to a breakdown of tolerance to a biotherapeutic by initiating innate immune responses through engagement of pattern recognition receptors such as toll like receptors [59,60,61]. In vitro cell-based assays used to asses these types of product impurities are beyond the scope of this review, but additional information can be found in recent publications [59,60,61].
Pre-emptive risk assessment on HCPs, protein aggregates, and other critical quality attributes that arise during manufacturing of one product can be beneficial for the future production of similar compounds [60]. This is especially true when biotherapeutics such as mAbs with the same IgG isotype are manufactured and purified via similar methods. Determining if an HCP that regularly appears in these processes is immunogenic could help established methods to completely remove these from current and future products.
In Silico Prediction of T Cell Epitopes
Immunogenicity risk assessment typically begins early in product development and remains relevant through each stage of the drug development process. The first assessment usually performed is T cell epitope prediction using in silico models that search for peptides sequences within the protein likely to bind to HLA molecules with high affinity.
Generally, proteins with a large number of high affinity HLA binding epitopes receive a comparatively higher ranking score and would thus be considered higher risk. T cell epitopes are the focus for these assessments because using in silico tools to predict a B cell epitope is complicated by B cells’ capacity to recognize structural folds of a protein in addition to specific amino acid sequences that make up the protein [62, 63]. Predicting T cell epitopes is comparatively easier due to the linear nature of processed peptides that are presented in the context of an MHC molecule. Additionally, these tools and others can be trained and tested using the large source of publically available data that can be found on the Immune Epitope Database (IEDB) online [64].
There are a multitude of HLA II peptide binding prediction algorithms such as the proprietary EpiMatrix [65, 66] or the publically available IEDB consensus approaches [64, 67]. Data from the clinical trials of prior approved drugs can be used in conjunction with results from these in silico analyses to create a scale of relative immunogenicity risk. However, these in silico tools are widely considered to be over-predictive because they often return false-positive epitopes due to the fact that these algorithms only consider a small fraction of the factors that influence an immune response [65, 68]. Despite this, these tools have been used by vaccine developers for some time, and are increasingly being used by pharmaceutical companies for early risk assessment during drug design and low immunogenicity risk candidate selection.
One additional factor to consider with in silico prediction tools is that DP and DQ HLA-II alleles are typically omitted during the analysis. This may be because the peptide binding motifs are less well defined compared to HLA-DR alleles, but binding affinity prediction algorithms are improving [64, 68]. The omission of DQ and DP alleles in the EpiMatrix T cell epitope prediction algorithm is due to the observation that there was no correlation between the presence of DQ or DP and the resulting immunogenicity in a portion of mAbs evaluated in clinical trials [69]. Regardless, these omissions might still leave potential gaps in the predicted immunogenic potential of a therapeutic.
In Vitro Binding Assays
One way to bypass pitfalls of in silico prediction is to use MHC-associated Peptide Proteomics (MAPPs) assays [70]. MAPPs utilizes mass spectrometry to provided unambiguous and definitive identification of peptide epitopes that are eluted from HLA molecules [71]. For example, peptides determined by MAPPS assays were used to stimulate peripheral blood mononuclear cells (PBMCs) from donors that have ADA towards Rituximab and Infliximab and were sufficient to show T cell memory responses [16]. Similar experiments performed on the low immunogenic mAb, Secukinumab, showed a dearth of immunogenic T cell epitopes [72]. This technique was also used to identify neoepitopes in bioengineered recombinant factor VIIa and recombinant factor VIII while also testing the predictive performance of the NetMHCIIpan in silico tool [11, 73].
However, MAPPs assays do have some drawbacks and technical limitations depending on the approach taken [74]. Some methods can be time and resource intensive. Multiple HLA alleles must also be accounted for in order to learn the breadth of presented peptides. The peptides identified by MAPPS assays depend on the signal to noise threshold used to determine positive binding, leading to the possibility of over-predicting positive responses or not detecting low abundance peptides. In many cases, MAPPs may actually under-represent the number of peptides bound by MHC II [70]. Additionally, the epitopes determined by these assays still need to be tested in vitro to determine if they have the potential to activate T cells.
It is also possible to perform epitope mapping by measuring T cell activation in response to synthesized, pools of overlapping peptide fragments that span the entire protein of interest [15, 16, 75,76,77,78]. However, using this method alone can be much more time consuming for large proteins and the predicted HLA-peptide binders predicted in this method face the similar requirement for different combinations of peptide-HLA II alleles as those determined by MAPPs assays. Ultimately, the data gained from in vitro binding assays can also be used to improve the accuracy of in silico prediction algorithms [79].
Epitopes that bind more strongly to HLA molecules may be more likely to elicit a T cell response [16, 72, 80,81,82]. There is also growing evidence that the overall stability of peptide binding, a component not always considered in most current in silico prediction algorithms, plays a major role in determining the antigenicity of a peptide and could improve predictive methods [83]. Despite this, there is also plenty of evidence demonstrating that not every peptide that binds strongly to HLA molecules is equally likely to lead to T cell activation [16, 80, 84, 85].
For a peptide to be immunogenic, there must be a corresponding naïve or memory effector T cell that recognizes it. The presented peptide could resemble a self or commensal peptide which developing T cells are either eliminated or tolerized towards by central or peripheral tolerance mechanisms respectively [86]. Hence it is advisable that in silico models and MAPPs assays be used in conjunction with other methods such as additional in vitro cell-based assays for immunogenicity prediction or in vivo animal models where appropriate [87, 88]. Some groups are developing in silico tools to try and predict the immunogenicity of a peptide based on the energetic potential between the amino acid sequence of a peptide and the amino acids on the CDR3β loops T cell receptor [89]. However, correlation between the output of these methods in terms of predicted immunogenic sequences and observed incidence of clinical immunogenicity is pending additional validation.
Immunogenic epitopes identified by in silico or in vitro binding assays and then subsequently confirmed using in vitro cell-based assays can be modified by mutating amino acids before the biotherapeutic moves to large scale production as long as the changes do not alter the product’s intended activity [18, 80]. This approach was utilized in the case recombinant human erythropoietin [80]. Modifying the erythropoietin protein reduced the binding affinity of immunodominant epitopes without altering functionality and resulted in reduced immunogenicity [80]. The immunotoxin, α-sarcin, was also successfully deimmunized after using an in vitro binding assay to determine T cell epitopes [78].
In Vitro Cell-Based Assays for Risk Assessment
In vitro cell-based assays are increasingly being used to assess the immunogenicity of biotherapeutics. Several iterations of these methods have been implemented across various types of biotherapeutic development workflows to identify risk and design candidates with lower clinical immunogenicity liability, Fig. 2. The PBMC and DC:T cell assays detailed below may be developed in house to tailor to the biotherapeutic in question or outsourced to contract research organizations (CROs) such as ProImmune Ltd. (Reveal®), VaxDesign (MIMIC®), Abzena (EpiScreen™), Lonza (EpiBase™), ImmuneXperts, and EpiVax Inc. [90]. Tables 1 and 2 contain a non-exhaustive selection of the cell-based assays that were performed to characterize the immunogenicity of different biotherapeutics in the past few years. Table 1 details reports that focus primarily on assessing the biotherapeutic or synthesized peptides within the biotherapeutic. Table 2 details reports that delve into the biotherapeutics’ formulation, protein aggregates, and any HCPs that co-purify along with the biotherapeutic.
While these in vitro methods perform well for comparing relative immunogenicity between similar biotherapeutics, they are limited in their capacity to fully predict the clinical incidence of immunogenicity. Contributing to this limitation is the potential differences in the overall distribution and tissue localization of a tested biological in vivo as well as the presence of other cell and tissue types at relevant biological ratios. Nonetheless, there are studies where in vitro observations correspond to observed clinical immunogenicity, but more studies are needed to better understand the correlation between in vitro and clinical observations [49, 72, 91,92,93].
In general, assays with fresh or frozen whole PBMCs are comparatively high throughput and can assess a variety of HLA types whereas specialized assays such as monocyte derived dendritic cell (moDC)-T cell co-cultures enhance the likelihood of antigen presentation interactions without confounding factors in whole blood. These moDC-T cell assays can also more easily assess therapeutics with T cell activating or engaging characteristics. Currently, moDC-T cell assays are utilized more frequently for evaluating both biotherapeutics and CQAs as indicated in Tables 1 and 2.
Despite a steady stream of information coming in from publications from industry, academia, and consortium based efforts to rank these methods, there is no current consensus on the most sensitive or preferred method. Often times, the preference of one assay over the other is determined by the type of molecule, stage of program under development, and overall risk predicted by in silico assessments or MAPPs for the molecule. However, head to head comparisons between the power of assays is still pending and investigators are encouraged to weigh the advantages and limitations of each assay type before selecting one for their risk assessments.
Considerations Prior to Performing In Vitro Immunogenicity Analysis
When designing an in vitro assay to assess immunogenicity risk, it is important to consider the patient population that will receive the therapeutic. The vast diversity of HLA alleles makes it difficult to perfectly predict the immunogenicity of a therapeutic with respect to the worldwide human population because individuals’ APCs can present different peptides on their HLA class I and II molecules [94,95,96]. Hence, most in vitro risk assessments should try to utilize a population of donors with diverse HLA alleles while keeping in mind the frequencies with which those alleles appear in the target population. It is recommended that the analysis include at risk HLA alleles-especially if they are correlated with disease indication and treatment outcome.
Some autoimmune and inflammatory diseases such as Type I Diabetes, Celiac Disease, Multiple Sclerosis, and Rheumatoid Arthritis are associated with enhanced or protective properties associated with specific HLA alleles [97,98,99]. If a biotherapeutic is intended for the treatment of these diseases, it would be preferable to skew the source of cells used for in vitro experiments towards donors that express relevant disease related alleles. Similarly, in silico algorithms or MAPPs assays can also be narrowed to focus on disease specific alleles to reduce the number of peptides requiring in vitro testing.
Most studies typically use healthy donors as their cell source for in vitro assays. Obtaining large amounts of cells from healthy donors is more reliable, easier, and cheaper than obtaining cells from donors with different disease states. Cells from healthy donors are considered antigen inexperienced because they have not received biotherapeutic treatments that could potentially have established pre-existing populations of therapeutic-specific memory T and B cells. However, the results collected from experiments using healthy donors come with a caveat because immune responses from healthy donors might not accurately predict the risk of immunogenicity towards a biotherapeutic because of the lack of influence from a disease state.
Because of this, immunogenicity risk assessment should ideally also include cells isolated from donors experiencing a relevant disease state. However, this is not without its own disadvantages. The availability of donors with relevant disease states as well as the overall viability of the cells reduces the feasibility of including samples from sick patients. Additionally, the current medication patients receive to treat their disease might complicate analysis and interpretation of data generated using their cells by interfering with the readout of the experiment [55].
Cell-Based Methods Used for Pre-Emptive Immunogenicity Profiling/Prediction
Fresh Whole Blood
Long term experiments using fresh, whole blood from donors for immunogenicity risk assessment are not commonly preferred. This is because extended culturing of fresh, whole blood is problematic in assessing naïve donors because high background and increased cell death after 48 h. Despite this, whole blood is the most high throughput in vitro cell-based method and is very easy to execute, requiring no additional cell isolation after blood collection. It is also easy to include a wide range of HLA types or bias the collection of blood to HLA types relevant to a specific disease or patient population.
Re-stimulation of whole blood to measure the T cell recall responses of antigen experienced donors has been used to confirm immunogenic epitopes identified by in silico methods or known antigens derived from pathogens with varying levels of efficacy [100, 101]. These methods were primarily developed for identifying potential options for vaccine components but also could be applied for understanding immunogenicity generated by biotherapeutics.
Single and Multiple Challenge PBMC Assays
PBMCs isolated from whole blood are the typical go-to source for most current in vitro cell-based assays for immunogenicity prediction [93, 102]. The PBMCs used in experiments can be freshly isolated from a donor or thawed from a cryopreserved stock. One of the benefits of building a bank of cryopreserved cells is the ability to consistently return to the same donor for autologous cells depending on the type of experiment being performed. This provides the capability to select specific HLA types depending on the needs of the experiment and therapeutic being evaluated. However, establishing a large bank of donors can be expensive and requires a lot of preparation and space. Donor PBMCs also need to be tested using positive and negative stimulatory controls to show they display consistent immune responses. Control PBMCs from donors that display these consistent responses also need to be maintained so that they can serve as reference points for assay performance.
Due to the comparatively lower cost and the ease of execution, the most commonly performed in vitro cell-based assay is directly stimulating isolated donor whole PBMCs, or CD8+ T cell depleted PBMCs with whole biotherapeutic and measuring the response of activated T cells (See Assay column, Tables 1 and 2). Whole PBMC stimulation requires less hands on work, but has the disadvantage of not being able to easily control the ratio of antigen presenting cells to T cells--a property that can vary from donor to donor [22, 93].
Host cell protein contaminants, protein aggregates, synthesized peptide fragments, and whole biotherapeutic can simply be added to these kinds of PBMC cultures at the desired concentration. APCs within the whole PBMCs will process and present immunogenic antigens to T cells. Multiple rounds of stimulation can be performed by replacing cell supernatants with fresh media spiked with the desired stimulant during extended culturing in order to expand populations of antigen specific T cells for further characterization [11, 80, 93]. Multiple challenges during long culture conditions provide more time and opportunities for naïve T cells to expand. However, multiple challenges require more time, maintenance, and the addition of cytokines such as IL-2 and IL-7 to sustain the T cells in culture [93].
A novel PBMC cell-based assay was recently developed that enriches the number of CD4+ T cells prior to co-culture with irradiated syngeneic PBMCs in an effort to increase throughput and sensitivity [90]. This method showed higher sensitivity compared to CD8+ T cell depleted PBMCs and was able to demonstrate high in vitro immunogenicity towards biotherapeutics that are reported to have high clinical immunogenicity [90].
moDC-T Cell Co-Culture
Another commonly performed cell-based assay is co-culturing moDCs and autologous CD4+ T cells (See Assay column, Tables 1 and 2). This moDC-T cell co-culture method pares the system down to the basic initiating components of cell mediated immunity: CD4+ T cells interacting with an APC with the added benefit that cell ratios can be actively controlled by the investigator. Controlling cell ratios allows the system to be more sensitive as the total number of potential responder cells is much greater than the whole PBMC milieu. However, co-culturing is a more time consuming method that requires isolation and differentiation of monocytes into dendritic cells followed by an antigen loading/pulsing step.
The way cells are isolated from PBMCs for co-cultures vary from study to study. Some opt to take plastic adherent cells from plated PBMCs to differentiate into DCs, while others use magnetic bead separation to isolate specific populations of monocytes from PBMCs. The monocytes are then cultured for up to 5 days in media containing IL-4 and GM-CSF. These cytokines can be added individually into the desired cell culture media, or differentiation media created specifically for immature moDC generation can be readily purchased and used. Maturation of moDCs is then performed through adding exogenous factors such as TNFα or LPS [46] concurrently with addition of the desired biotherapeutic, peptide fragments, or aggregates. The moDCs are incubated for 1–2 days to allow for maturation, antigen processing, and antigen presentation. The matured, pulsed DCs are then typically combined in a co-culture with autologous, purified CD4+ T cells, generally at a 1:10 ratio of moDC:T Cells. However, this ratio is not always followed as can be seen in the # Cells column of Tables 1 and 2.
Multiple rounds of stimulation can be performed for long term moDC-T cell cultures in order to expand antigen specific T cell populations [16, 103,104,105]. However, this can be resource intensive compared to whole PBMC restimulation because it requires differentiation, maturation and pulsing of moDCs prior to each restimulation. This also requires returning to dedicated large stock of autologous PBMCs so that cells from the same donor are always used in co-culture.
An advanced variation of the moDC-T cell system is the Modular Immune In vitro Construct (MIMIC®) model which is capable of reproducibly generating antigen-specific innate and adaptive immune responses against vaccine targets and monoclonal antibodies and is sensitive to donor immunophysiology [106,107,108]. This system adds additional complexity through recreating the order of events that would occur in vivo by supplying dendritic cells, T cells and then B cells sequentially in order to simulate an adaptive immune response [106]. Because of this, assessment of both T cell and B cell responses through flow cytometry and antibody forensics is possible [108].
Assay Readouts
Typically, the total duration of co-culture systems depends on the readout chosen for the experiment, but even then there is variability between the methods performed by different studies (Tables 1 and 2, co-culture duration column). There is an ample selection of readouts for T cell activation that can be used for each of these assays, and groups often opt to use more than one (Fig. 2). ELISPOT assays are commonly used to quantitatively determine the frequency of antigen specific, IFN-γ or IL-2 secreting T cells responding to biotherapeutics (Tables 1 and 2, readout column).
Some studies opt to directly measure T cell proliferation by one of two well-established methods: through flow cytometry by measuring dilution of fluorescent dyes such as CFSE used to label cells [48, 109], or through the incorporation of radiolabeled thymidine pulsed into the cell culture. Most reported immunogenicity assessment studies measure proliferation by thymidine incorporation (See Readout Column, Tables 1 and 2), but flow cytometry can provide additional information because it is possible to characterize and differentiate between T effector subsets based on the surface marker expression with the proper staining panels [93]. This characterization increases the accuracy of the risk assessment because it can be used to identify T regulatory cells which are capable of suppressing immune responses [110].
ELISA and Multiplex fluorescence based analysis of cell supernatants for qualitative assessment of T cell activation related cytokines can also be performed simultaneously along with the measurement of T cell proliferation. Typically, supernatant is harvested and stored until the desired method of analysis is performed. Some studies opt to run ELISAs on the supernatants to analyze only a few cytokines of interest such as IL-2, ILl-4 or IFN-γ. However, multiplex cytokine assay methods now allow for the screening of many different cytokines concurrently. For the purposes of immunogenicity risk assessment, the levels of pro-inflammatory innate and T cell activation related cytokines such IL-1β, IL-2, IL-4, IL-5, IL-6, IL-8, IL-10, IL-13, IL-17, IFN-γ, and TNF-α can all be assessed at the same time with commercially available single or multiplexed cytokine detection kits.
Emerging 3D In Vitro Cell-Based Assays
While stimulation of PBMCs or moDC-T cell assays are most commonly used, different in vitro organ-on-chip methods are being utilized to better mimic the interface between innate and adaptive immunity that occurs in vivo [111, 112]. Groell et al. provide an in-depth look at many 3D in vitro assays that could be adapted for immunogenicity prediction [113]. However, pending further validation of the prediction outcomes from these models systems, at this time, 3D artificial lymph nodes are one of the most promising models for immunogenicity risk assessment.
One such assay utilizes a bioreactor seeded with 3D human artificial lymph nodes made out of a RGD-dextran hydrogel matrix [109]. Using this system, the cytokine profile of the culture was monitored over many time points to track cell activation upon stimulation with mAb aggregates [109]. Another artificial 3D lymph node system using a microfluidic chip may have the potential to simulate mature DC migration and T cell activation, but has not been adapted to determine immunogenicity of biotherapeutics after extended co-culturing [114]. Due to their complexity, these types of assays have not been taken up for risk assessments. This is also due in part because it remains unclear if these methods will serve as better predictors of clinical immunogenicity compared to standard PBMC or DC-T cell assays until additional development and more rigorous comparisons are made.
Caveats
It is important to keep in mind that the mechanism of action for a given therapeutic may also greatly alter the methods needed to perform immunogenicity prediction as well as the outcome of these assays. ADCs have the potential to be more or less immunogenic depending on the sequence of the linker built attaching the payload to the mAb as well as the actual effects and identity of the payload they deliver [47]. Some mAbs can stymie the maturation and survival of APCs, alter cytokine expressions signatures, and inhibit T cell proliferation in vitro [115,116,117]. This may lead to underestimating the immunogenicity of the mAb due to weaker observed in vitro responses.
Many mAbs currently in development for treating malignancies are immune checkpoint inhibitors that boost immune system activation. This is exemplified by CTLA-4 or PD1 inhibitors that cause enhanced T cell responses and increased risk [31]. If the biotherapeutic has properties that bind to and directly interact with T cells to enhance or suppress their activation, assays using whole PBMCs should not be used without careful considering the magnitude of the biotherapeutic’s effect. This is because it would be difficult to determine if the measured T cell responses are either due to the presence of immunogenic epitopes within the protein, or if their response was due to the biotherapeutic’s mechanism of action. This can be circumvented by using peptide fragments determined by in silico methods or MAPPs.
Developing an assay to test whole biotherapeutic that works on a case by case basis may not be practical or as effective as using one that could consistently perform better at assessing biotherapeutic molecules in a similar class. This makes the moDC-T cell assay a great option for testing T cell engaging biotherapeutics because moDC’s can be pulsed with whole protein and washed prior to their co-culture with isolated CD4+ T cells in order to remove excess drug product. However, this method makes long term culturing difficult because the stability of the peptide-MHC complex is unknown, and the relative abundance of the peptide fragment will decline once the antigen source is removed from the moDCs. Because the MOA of the biotherapeutic prevents re-challenge, the time consuming step of introducing more, freshly loaded moDCs to the co-culture would be necessary in order to provide the optimal duration of T cell-APC interaction for naïve T cell activation and proliferation [118].
Another consideration that often goes overlooked in cell-based in vitro assays is the total number of T cells used to screen for immunogenicity. There may be up to ~3 × 1011 total T cells maintained in a human [119]. An estimated ~1 × 108 unique T cell receptors are expressed within this population of naïve T cells [120, 121]. The cross reactive properties of T cell receptors would suggest each of these are capable of recognizing multiple distinct peptide-HLA complexes with overlapping recognition, however accurately measuring this cross reactivity has proven difficult [122, 123]. Despite this, only a tiny fraction of CD4+ T cells are typically ever examined in vitro. Many assays ultimately use less than 1–3 × 106 total CD4+ T cells per condition, and there has been no general consensus as to the total number of T cells needed to accurately predict the immunogenicity of a therapeutic (See # cells column in Tables 1 and 2). Given this observation, most current studies risk under-predicting rates of immunogenicity with their current in vitro assay methods, although some report relatively high sensitivity to detect pre-existing T cells through long term culturing DC-T cell co-culturing methods [104, 105].
Conclusions
Clinical immunogenicity is a major concern for the development and approval of new biotherapeutics. Given the rapid influx of mAb and ADCs in clinical pipelines, effective and accurate immunogenicity risk assessment tools are needed now more than ever. Risk assessment can and should be performed at multiple steps along the drug development pathway, from initial protein design to assessment of quality product attributes in manufacturing. While they are not yet required, the USFDA and EMA are increasingly suggesting pharmaceutical companies to assess T cell activation in response to protein therapeutics [7, 8]. Designing and selecting protein sequences with a lower risk of immunogenicity using qualified tools is a promising alternative to elaborate monitoring of ADA responses in the clinical trials. Presently, a single platform or process cannot be recommended based on the available data. However, as more information on the use and correlative applicability of these assays is made available by drug developers in the public domain, a harmonized and rational approach to design a candidate with lowest predicted immunogenicity risk will become possible in the near future.
The cell-based in vitro immunogenicity assessments outlined here are an essential component of the risk assessment tool kit. We have provided details on the patient and product related factors that need to be considered before designing an effect in vitro experiment to perform. While each of these assays can provide some insight into the potential for clinical immunogenicity, none are completely validated to predict it. In vitro assays cannot completely capture the entire picture of what occurs in an organism. Cell types as well as tissue specific distribution of protein processing cathepsins and cystatins might also influence the peptides that are loaded and presented on MHC molecules [19]. While the immediate and long-term impact of the output of these assays cannot be over emphasized, industry wide standardization of in vitro assays may be necessary, and more work is still needed to better understand how in vitro T cell activation correlates with observed clinical immunogenicity of current and future biotherapeutics.
References
Beck A, Goetsch L, Dumontet C, Corvaïa N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat Rev Drug Discov. 2017;16(5):315–37.
Walsh G. Biopharmaceutical benchmarks 2018. Nat Biotechnol. 2018;36(12):1136–45.
FDA, U., Immunogenicity Assessment for Therapeutic Protein Products, H.a.H. Services, Editor. 2014, Guidance for industry: 10903 New Hampshire avenue, WO 71, room 3128 silver spring, MD 20993-0002.
Vultaggio A, Nencini F, Pratesi S, Petroni G, Maggi E, Matucci A. Manifestations of antidrug antibodies response: hypersensitivity and infusion reactions. J Interf Cytokine Res. 2014;34(12):946–52.
Vanderlugt CL, Miller SD. Epitope spreading in immune-mediated diseases: implications for immunotherapy. Nat Rev Immunol. 2002;2(2):85–95.
van Brummelen EM, et al. Antidrug antibody formation in oncology: clinical relevance and challenges. Oncologist. 2016;21(10):1260–8.
FDA, U., Immunogenicity Testing of Therapeutic Protein Products — Developing and Validating Assays for Anti-Drug Antibody Detection, H.a.H. Services, Editor. 2019: 10001 New Hampshire Ave., Hillandale Bldg., 4th Floor Silver Spring, MD 20993–0002.
EMA. Guideline on Immunogenicity Assessment of Therapeutic Proteins. 2017; Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-immunogenicity-assessment-therapeutic-proteins-revision-1_en.pdf.
van Schouwenburg PA, Rispens T, Wolbink GJ. Immunogenicity of anti-TNF biologic therapies for rheumatoid arthritis. Nat Rev Rheumatol. 2013;9(3):164–72.
Ridker PM, Tardif JC, Amarenco P, Duggan W, Glynn RJ, Jukema JW, et al. Lipid-reduction variability and antidrug-antibody formation with Bococizumab. N Engl J Med. 2017;376(16):1517–26.
Lamberth, K., et al., Post hoc assessment of the immunogenicity of bioengineered factor VIIa demonstrates the use of preclinical tools. Sci Transl Med, 2017. 9(372).
Agrawal S, Statkevich P, Bajaj G, Feng Y, Saeger S, Desai DD, et al. Evaluation of immunogenicity of Nivolumab Monotherapy and its clinical relevance in patients with metastatic solid tumors. J Clin Pharmacol. 2017;57(3):394–400.
Pineda C, Castañeda Hernández G, Jacobs IA, Alvarez DF, Carini C. Assessing the immunogenicity of biopharmaceuticals. BioDrugs. 2016;30(3):195–206.
Kurki, P., Compatibility of immunogenicity guidance by the EMA and the US FDA. Bioanalysis, 2019. 0(0): p. null.
Barbosa MD, Vielmetter J, Chu S, Smith DD, Jacinto J. Clinical link between MHC class II haplotype and interferon-beta (IFN-beta) immunogenicity. Clin Immunol. 2006;118(1):42–50.
Hamze M, et al. Characterization of CD4 T cell epitopes of infliximab and rituximab identified from healthy donors. Front Immunol. 2017;8:500.
Vultaggio A, Petroni G, Pratesi S, Nencini F, Cammelli D, Milla M, et al. Circulating T cells to infliximab are detectable mainly in treated patients developing anti-drug antibodies and hypersensitivity reactions. Clin Exp Immunol. 2016;186(3):364–72.
Mazor R, Eberle JA, Hu X, Vassall AN, Onda M, Beers R, et al. Recombinant immunotoxin for cancer treatment with low immunogenicity by identification and silencing of human T-cell epitopes. Proc Natl Acad Sci U S A. 2014;111(23):8571–6.
Unanue ER, Turk V, Neefjes J. Variations in MHC class II antigen processing and presentation in health and disease. Annu Rev Immunol. 2016;34:265–97.
Lee JI, Choi YJ, Park HJ, Jung KC, Park SH. RD-05, a novel anti-CD154 antibody, efficiently inhibits generation of anti-drug antibody without the risk of thrombus formation in non-human primates. Biochem Biophys Res Commun. 2018;498(4):996–1001.
Bachelet D, Hässler S, Mbogning C, Link J, Ryner M, Ramanujam R, et al. Occurrence of anti-drug antibodies against interferon-Beta and Natalizumab in multiple sclerosis: a collaborative cohort analysis. PLoS One. 2016;11(11):e0162752.
Brodin P, Davis MM. Human immune system variation. Nat Rev Immunol. 2017;17(1):21–9.
De Groot AS, et al. HLA- and genotype-based risk assessment model to identify infantile onset pompe disease patients at high-risk of developing significant anti-drug antibodies (ADA). Clin Immunol. 2019;200:66–70.
Zota V, et al. HLA-DR alleles in amyloid beta-peptide autoimmunity: a highly immunogenic role for the DRB1*1501 allele. J Immunol. 2009;183(5):3522–30.
Zarate YA, Hopkin RJ. Fabry's disease. Lancet. 2008;372(9647):1427–35.
Harmatz, P., et al., Enzyme replacement therapy for mucopolysaccharidosis VI: A phase 3, randomized, double-blind, placebo-controlled, multinational study of recombinant human N-acetylgalactosamine 4-sulfatase (recombinant human arylsulfatase B or rhASB) and follow-on, open-label extension study. The Journal of Pediatrics, 2006. 148(4): p. 533–539.e6.
Berrier KL, Kazi ZB, Prater SN, Bali DS, Goldstein J, Stefanescu MC, et al. CRIM-negative infantile Pompe disease: characterization of immune responses in patients treated with ERT monotherapy. Genet Med. 2015;17(11):912–8.
Casadevall N, Nataf J, Viron B, Kolta A, Kiladjian JJ, Martin-Dupont P, et al. Pure red-cell aplasia and antierythropoietin antibodies in patients treated with recombinant erythropoietin. N Engl J Med. 2002;346(7):469–75.
Vultaggio A, Matucci A, Parronchi P, Rossi O, Palandri F, Romagnani S, et al. Safety and tolerability of infliximab therapy: suggestions and criticisms based on wide clinical experience. Int J Immunopathol Pharmacol. 2008;21(2):367–74.
Van Walle I, et al. Immunogenicity screening in protein drug development. Expert Opin Biol Ther. 2007;7(3):405–18.
Davda J, Declerck P, Hu-Lieskovan S, Hickling TP, Jacobs IA, Chou J, et al. Immunogenicity of immunomodulatory, antibody-based, oncology therapeutics. J Immunother Cancer. 2019;7(1):105.
Jurewicz MM, Stern LJ. Class II MHC antigen processing in immune tolerance and inflammation. Immunogenetics. 2019;71(3):171–87.
Attias M, Al-Aubodah T, Piccirillo CA. Mechanisms of human FoxP3(+) Treg cell development and function in health and disease. Clin Exp Immunol. 2019.
Cousens L, Najafian N, Martin WD, de Groot AS. Tregitope: immunomodulation powerhouse. Hum Immunol. 2014;75(12):1139–46.
Garcês S, Demengeot J, Benito-Garcia E. The immunogenicity of anti-TNF therapy in immune-mediated inflammatory diseases: a systematic review of the literature with a meta-analysis. Ann Rheum Dis. 2013;72(12):1947–55.
Salazar-Fontana LI, Desai DD, Khan TA, Pillutla RC, Prior S, Ramakrishnan R, et al. Approaches to mitigate the unwanted immunogenicity of therapeutic proteins during drug development. AAPS J. 2017;19(2):377–85.
Passey C, Mora J, Dodge R, Gibiansky L, Sheng J, Roy A, et al. An integrated assessment of the effects of immunogenicity on the pharmacokinetics, safety, and efficacy of Elotuzumab. AAPS J. 2017;19(2):557–67.
Krieckaert CL, Bartelds GM, Lems WF, Wolbink GJ. The effect of immunomodulators on the immunogenicity of TNF-blocking therapeutic monoclonal antibodies: a review. Arthritis Res Ther. 2010;12(5):217.
Sauna ZE, Lagassé D, Pedras-Vasconcelos J, Golding B, Rosenberg AS. Evaluating and mitigating the immunogenicity of therapeutic proteins. Trends Biotechnol. 2018;36(10):1068–84.
Ross C, Clemmesen KM, Svenson M, Sørensen PS, Koch-Henriksen N, Skovgaard GL, et al. Immunogenicity of interferon-b in multiple sclerosis patients: influence of preparation, dosage, dose frequency, and route of administration. Ann Neurol. 2000;48(5):706–12.
Schellekens, H., Factors influencing the immunogenicity of therapeutic proteins. Nephrol Dial Transplant, 2005. 20 Suppl 6: p. vi3–9.
Rudick RA, Goelz SE. Beta-interferon for multiple sclerosis. Exp Cell Res. 2011;317(9):1301–11.
Barnard JG, Babcock K, Carpenter JF. Characterization and quantitation of aggregates and particles in interferon-beta products: potential links between product quality attributes and immunogenicity. J Pharm Sci. 2013;102(3):915–28.
Ratanji KD, Derrick JP, Dearman RJ, Kimber I. Immunogenicity of therapeutic proteins: influence of aggregation. J Immunotoxicol. 2014;11(2):99–109.
Moussa EM, Panchal JP, Moorthy BS, Blum JS, Joubert MK, Narhi LO, et al. Immunogenicity of therapeutic protein aggregates. J Pharm Sci. 2016;105(2):417–30.
Rubic-Schneider T, Kuwana M, Christen B, Aßenmacher M, Hainzl O, Zimmermann F, et al. T-cell assays confirm immunogenicity of tungsten-induced erythropoietin aggregates associated with pure red cell aplasia. Blood Adv. 2017;1(6):367–79.
Hock MB, Thudium KE, Carrasco-Triguero M, Schwabe NF. Immunogenicity of antibody drug conjugates: bioanalytical methods and monitoring strategy for a novel therapeutic modality. AAPS J. 2015;17(1):35–43.
Xue L, et al. Contribution of enhanced engagement of antigen presentation machinery to the clinical immunogenicity of a human interleukin (IL)-21 receptor-blocking therapeutic antibody. Clin Exp Immunol. 2016;183(1):102–13.
Hua F, Comer GM, Stockert L, Jin B, Nowak J, Pleasic-Williams S, et al. Anti-IL21 receptor monoclonal antibody (ATR-107): safety, pharmacokinetics, and pharmacodynamic evaluation in healthy volunteers: a phase I, first-in-human study. J Clin Pharmacol. 2014;54(1):14–22.
de Zafra CL, Quarmby V, Francissen K, Vanderlaan M, Zhu-Shimoni J. Host cell proteins in biotechnology-derived products: a risk assessment framework. Biotechnol Bioeng. 2015;112(11):2284–91.
Bracewell DG, Francis R, Smales CM. The future of host cell protein (HCP) identification during process development and manufacturing linked to a risk-based management for their control. Biotechnol Bioeng. 2015;112(9):1727–37.
Vanderlaan M, Zhu-Shimoni J, Lin S, Gunawan F, Waerner T, van Cott K. Experience with host cell protein impurities in biopharmaceuticals. Biotechnol Prog. 2018;34(4):828–37.
Romer T, Peter F, Saenger P, Starzyk J, Koehler B, Korman E, et al. Efficacy and safety of a new ready-to-use recombinant human growth hormone solution. J Endocrinol Investig. 2007;30(7):578–89.
EMA. Omnitrope: EPAR-Scientific Discussion. 2006 [cited 2019; Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Scientific_Discussion/human/000607/WC500043692.pdf.
Jawa V, Joubert MK, Zhang Q, Deshpande M, Hapuarachchi S, Hall MP, et al. Evaluating immunogenicity risk due to host cell protein impurities in antibody-based biotherapeutics. AAPS J. 2016;18(6):1439–52.
Polumuri SK, Haile LA, Ireland DDC, Verthelyi D. Aggregates of IVIG or Avastin, but not HSA, modify the response to model innate immune response modulating impurities. Sci Rep. 2018;8(1):11477.
Joubert MK, Hokom M, Eakin C, Zhou L, Deshpande M, Baker MP, et al. Highly aggregated antibody therapeutics can enhance the in vitro innate and late-stage T-cell immune responses. J Biol Chem. 2012;287(30):25266–79.
Ahmadi M, Bryson CJ, Cloake EA, Welch K, Filipe V, Romeijn S, et al. Small amounts of sub-visible aggregates enhance the immunogenic potential of monoclonal antibody therapeutics. Pharm Res. 2015;32(4):1383–94.
Verthelyi D, Wang V. Trace levels of innate immune response modulating impurities (IIRMIs) synergize to break tolerance to therapeutic proteins. PLoS One. 2010;5(12):e15252.
Haile LA, Puig M, Polumuri SK, Ascher J, Verthelyi D. In vivo effect of innate immune response modulating impurities on the skin milieu using a macaque model: impact on product immunogenicity. J Pharm Sci. 2017;106(3):751–60.
Haile LA, Puig M, Kelley-Baker L, Verthelyi D. Detection of innate immune response modulating impurities in therapeutic proteins. PLoS One. 2015;10(4):e0125078.
El-Manzalawy, Y. and V. Honavar, Recent advances in B-cell epitope prediction methods. Immunome Res, 2010. 6 Suppl 2: p. S2.
Potocnakova L, Bhide M, Pulzova LB. An Introduction to B-cell epitope mapping and in Silico epitope prediction. J Immunol Res. 2016;2016:6760830.
Vita R, Mahajan S, Overton JA, Dhanda SK, Martini S, Cantrell JR, et al. The immune epitope database (IEDB): 2018 update. Nucleic Acids Res. 2019;47(D1):D339–43.
De Groot AS, McMurry J, Moise L. Prediction of immunogenicity: in silico paradigms, ex vivo and in vivo correlates. Curr Opin Pharmacol. 2008;8(5):620–6.
Moise L, Cousens L, Fueyo J, de Groot AS. Harnessing the power of genomics and immunoinformatics to produce improved vaccines. Expert Opin Drug Discovery. 2011;6(1):9–15.
Wang P, Sidney J, Dow C, Mothé B, Sette A, Peters B. A systematic assessment of MHC class II peptide binding Predictions and evaluation of a consensus approach. PLoS Comput Biol. 2008;4(4):e1000048.
Jensen KK, Andreatta M, Marcatili P, Buus S, Greenbaum JA, Yan Z, et al. Improved methods for predicting peptide binding affinity to MHC class II molecules. Immunology. 2018;154(3):394–406.
Groot, A.S.D., et al., In silico prediction of HLA-DP and -DQ epitope content is poorly correlated with clinical immunogenicity of therapeutic proteins, manuscript in preparation, in National Biotechnology Conference. 2013, American Association of Pharmaceutical Scientists: San Diego, CA.
Quarmby V, Phung QT, Lill JR. MAPPs for the identification of immunogenic hotspots of biotherapeutics; an overview of the technology and its application to the biopharmaceutical arena. Expert Rev Proteomics. 2018;15(9):733–48.
Purcell AW, Croft NP, Tscharke DC. Immunology by numbers: quantitation of antigen presentation completes the quantitative milieu of systems immunology! Curr Opin Immunol. 2016;40:88–95.
Karle A, Spindeldreher S, Kolbinger F. Secukinumab, a novel anti-IL-17A antibody, shows low immunogenicity potential in human in vitro assays comparable to other marketed biotherapeutics with low clinical immunogenicity. MAbs. 2016;8(3):536–50.
Bartholdy C, Reedtz-Runge SL, Wang J, Hjerrild Zeuthen L, Gruhler A, Gudme CN, et al. In silico and in vitro immunogenicity assessment of B-domain-modified recombinant factor VIII molecules. Haemophilia. 2018;24(5):e354–62.
Caron E, Kowalewski DJ, Chiek Koh C, Sturm T, Schuster H, Aebersold R. Analysis of major histocompatibility complex (MHC) Immunopeptidomes using mass spectrometry. Mol Cell Proteomics. 2015;14(12):3105–17.
Jones TD, Phillips WJ, Smith BJ, Bamford CA, Nayee PD, Baglin TP, et al. Identification and removal of a promiscuous CD4+ T cell epitope from the C1 domain of factor VIII. J Thromb Haemost. 2005;3(5):991–1000.
Nayak S, Sivakumar R, Cao O, Daniell H, Byrne BJ, Herzog RW. Mapping the T helper cell response to acid alpha-glucosidase in Pompe mice. Mol Genet Metab. 2012;106(2):189–95.
Mazor R, Vassall AN, Eberle JA, Beers R, Weldon JE, Venzon DJ, et al. Identification and elimination of an immunodominant T-cell epitope in recombinant immunotoxins based on Pseudomonas exotoxin a. Proc Natl Acad Sci U S A. 2012;109(51):E3597–603.
Jones TD, et al. A deimmunised form of the ribotoxin, alpha-sarcin, lacking CD4+ T cell epitopes and its use as an immunotoxin warhead. Protein Eng Des Sel. 2016.
Alvarez B, Barra C, Nielsen M, Andreatta M. Computational tools for the identification and interpretation of sequence motifs in Immunopeptidomes. Proteomics. 2018;18(12):e1700252.
Tangri S, et al. Rationally engineered therapeutic proteins with reduced immunogenicity. J Immunol. 2005;174(6):3187–96.
McMurry J, et al. Analyzing mycobacterium tuberculosis proteomes for candidate vaccine epitopes. Tuberculosis (Edinb). 2005;85(1–2):95–105.
Weaver JM, et al. Immunodominance of CD4 T cells to foreign antigens is peptide intrinsic and independent of molecular context: implications for vaccine design. J Immunol. 2008;181(5):3039–48.
Blaha DT, Anderson SD, Yoakum DM, Hager MV, Zha Y, Gajewski TF, et al. High-throughput stability screening of Neoantigen/HLA complexes improves immunogenicity Predictions. Cancer Immunol Res. 2019;7(1):50–61.
Castelli FA, Leleu M, Pouvelle-Moratille S, Farci S, Zarour HM, Andrieu M, et al. Differential capacity of T cell priming in naive donors of promiscuous CD4+ T cell epitopes of HCV NS3 and Core proteins. Eur J Immunol. 2007;37(6):1513–23.
Mazor R, Tai CH, Lee B, Pastan I. Poor correlation between T-cell activation assays and HLA-DR binding prediction algorithms in an immunogenic fragment of Pseudomonas exotoxin a. J Immunol Methods. 2015;425:10–20.
Klein L, Robey EA, Hsieh CS. Central CD4(+) T cell tolerance: deletion versus regulatory T cell differentiation. Nat Rev Immunol. 2019;19(1):7–18.
Brinks V, Jiskoot W, Schellekens H. Immunogenicity of therapeutic proteins: the use of animal models. Pharm Res. 2011;28(10):2379–85.
Jiskoot W, et al. Mouse models for assessing protein immunogenicity: lessons and challenges. J Pharm Sci. 2016;105(5):1567–75.
Ogishi M, Yotsuyanagi H. Quantitative prediction of the landscape of T cell epitope immunogenicity in sequence space. Front Immunol. 2019;10:827.
Schultz HS, Reedtz-Runge SL, Bäckström BT, Lamberth K, Pedersen CR, Kvarnhammar AM, et al. Quantitative analysis of the CD4+ T cell response to therapeutic antibodies in healthy donors using a novel T cell:PBMC assay. PLoS One. 2017;12(5):e0178544.
Reich K, Blauvelt A, Armstrong A, Langley RG, Fox T, Huang J, et al. Secukinumab, a fully human anti-interleukin-17A monoclonal antibody, exhibits minimal immunogenicity in patients with moderate-to-severe plaque psoriasis. Br J Dermatol. 2017;176(3):752–8.
Spindeldreher S, Maillère B, Correia E, Tenon M, Karle A, Jarvis P, et al. Secukinumab demonstrates significantly lower immunogenicity potential compared to Ixekizumab. Dermatol Ther (Heidelb). 2018;8(1):57–68.
Wullner D, Zhou L, Bramhall E, Kuck A, Goletz TJ, Swanson S, et al. Considerations for optimization and validation of an in vitro PBMC derived T cell assay for immunogenicity prediction of biotherapeutics. Clin Immunol. 2010;137(1):5–14.
Lawlor DA, Ward FE, Ennis PD, Jackson AP, Parham P. HLA-A and B polymorphisms predate the divergence of humans and chimpanzees. Nature. 1988;335(6187):268–71.
Hughes AL, Hughes MK. Natural selection on the peptide-binding regions of major histocompatibility complex molecules. Immunogenetics. 1995;42(4):233–43.
Hedrick PW. Evolutionary genetics of the major histocompatibility complex. Am Nat. 1994;143(6):945–64.
Tsai S, Santamaria P. MHC class II polymorphisms, autoreactive T-cells, and autoimmunity. Front Immunol. 2013;4:321.
Lenz TL, Deutsch AJ, Han B, Hu X, Okada Y, Eyre S, et al. Widespread non-additive and interaction effects within HLA loci modulate the risk of autoimmune diseases. Nat Genet. 2015;47(9):1085–90.
Dieli-Crimi R, Cenit MC, Nunez C. The genetics of celiac disease: a comprehensive review of clinical implications. J Autoimmun. 2015;64:26–41.
Elfaki ME, Khalil EA, de Groot AS, Musa AM, Gutierrez A, Younis BM, et al. Immunogenicity and immune modulatory effects of in silico predicted L. donovani candidate peptide vaccines. Hum Vaccin Immunother. 2012;8(12):1769–74.
Kagina BM, Mansoor N, Kpamegan EP, Penn-Nicholson A, Nemes E, Smit E, et al. Qualification of a whole blood intracellular cytokine staining assay to measure mycobacteria-specific CD4 and CD8 T cell immunity by flow cytometry. J Immunol Methods. 2015;417:22–33.
Jawa V, Cousens LP, Awwad M, Wakshull E, Kropshofer H, de Groot AS. T-cell dependent immunogenicity of protein therapeutics: preclinical assessment and mitigation. Clin Immunol. 2013;149(3):534–55.
Azam A, et al. Healthy donors exhibit a CD4 T cell repertoire specific to the immunogenic human hormone H2-Relaxin before injection. J Immunol. 2019.
Delluc S, Ravot G, Maillere B. Quantitative analysis of the CD4 T-cell repertoire specific to therapeutic antibodies in healthy donors. FASEB J. 2011;25(6):2040–8.
Delluc S, Ravot G, Maillere B. Quantification of the preexisting CD4 T-cell repertoire specific for human erythropoietin reveals its immunogenicity potential. Blood. 2010;116(22):4542–5.
Higbee RG, et al. An immunologic model for rapid vaccine assessment -- a clinical trial in a test tube. Altern Lab Anim. 2009;37(Suppl 1):19–27.
Dhir V, Fort M, Mahmood A, Higbee R, Warren W, Narayanan P, et al. A predictive biomimetic model of cytokine release induced by TGN1412 and other therapeutic monoclonal antibodies. J Immunotoxicol. 2012;9(1):34–42.
Dauner A, Agrawal P, Salvatico J, Tapia T, Dhir V, Shaik SF, et al. The in vitro MIMIC(R) platform reflects age-associated changes in immunological responses after influenza vaccination. Vaccine. 2017;35(41):5487–94.
Kraus T, et al. Evaluation of a 3D human artificial lymph node as test model for the assessment of immunogenicity of protein aggregates. J Pharm Sci. 2019.
Dominguez-Villar M, Hafler DA. Regulatory T cells in autoimmune disease. Nat Immunol. 2018;19(7):665–73.
Giese C, Marx U. Human immunity in vitro - solving immunogenicity and more. Adv Drug Deliv Rev. 2014;69-70:103–22.
Shanti, A., J. Teo, and C. Stefanini, In Vitro Immune Organs-on-Chip for Drug Development: A Review. Pharmaceutics, 2018. 10(4).
Groell F, Jordan O, Borchard G. In vitro models for immunogenicity prediction of therapeutic proteins. Eur J Pharm Biopharm. 2018;130:128–42.
Mitra B, Jindal R, Lee S, Xu Dong D, Li L, Sharma N, et al. Microdevice integrating innate and adaptive immune responses associated with antigen presentation by dendritic cells. RSC Adv. 2013;3(36):16002–10.
Baldwin HM, Ito-Ihara T, Isaacs JD, Hilkens CM. Tumour necrosis factor alpha blockade impairs dendritic cell survival and function in rheumatoid arthritis. Ann Rheum Dis. 2010;69(6):1200–7.
de Andres C, et al. Long-term decrease in VLA-4 expression and functional impairment of dendritic cells during natalizumab therapy in patients with multiple sclerosis. PLoS One. 2012;7(4):e34103.
Dahlén R, Strid H, Lundgren A, Isaksson S, Raghavan S, Magnusson MK, et al. Infliximab inhibits activation and effector functions of peripheral blood T cells in vitro from patients with clinically active ulcerative colitis. Scand J Immunol. 2013;78(3):275–84.
Kaech SM, Wherry EJ, Ahmed R. Effector and memory T-cell differentiation: implications for vaccine development. Nat Rev Immunol. 2002;2(4):251–62.
Jenkins MK, Chu HH, McLachlan J, Moon JJ. On the composition of the preimmune repertoire of T cells specific for peptide-major histocompatibility complex ligands. Annu Rev Immunol. 2010;28:275–94.
Arstila TP, Casrouge A, Baron V, Even J, Kanellopoulos J, Kourilsky P. A direct estimate of the human αβ T cell receptor diversity. Science. 1999;286(5441):958–61.
Warren RL, Freeman JD, Zeng T, Choe G, Munro S, Moore R, et al. Exhaustive T-cell repertoire sequencing of human peripheral blood samples reveals signatures of antigen selection and a directly measured repertoire size of at least 1 million clonotypes. Genome Res. 2011;21(5):790–7.
Petrova G, Ferrante A, Gorski J. Cross-reactivity of T cells and its role in the immune system. Crit Rev Immunol. 2012;32(4):349–72.
Zarnitsyna VI, et al. Estimating the diversity, completeness, and cross-reactivity of the T cell repertoire. Front Immunol. 2013;4:485.
Stickler M, et al. An in vitro human cell-based assay to rank the relative immunogenicity of proteins. Toxicol Sci. 2004;77(2):280–9.
Jaber A, Baker M. Assessment of the immunogenicity of different interferon beta-1a formulations using ex vivo T-cell assays. J Pharm Biomed Anal. 2007;43(4):1256–61.
Holgate RG, Weldon R, Jones TD, Baker MP. Characterisation of a novel anti-CD52 antibody with improved efficacy and reduced immunogenicity. PLoS One. 2015;10(9):e0138123.
Rombach-Riegraf V, Karle AC, Wolf B, Sordé L, Koepke S, Gottlieb S, et al. Aggregation of human recombinant monoclonal antibodies influences the capacity of dendritic cells to stimulate adaptive T-cell responses in vitro. PLoS One. 2014;9(1):e86322.
Telikepalli S, Shinogle HE, Thapa PS, Kim JH, Deshpande M, Jawa V, et al. Physical characterization and in vitro biological impact of highly aggregated antibodies separated into size-enriched populations by fluorescence-activated cell sorting. J Pharm Sci. 2015;104(5):1575–91.
Joubert MK, Deshpande M, Yang J, Reynolds H, Bryson C, Fogg M, et al. Use of in vitro assays to assess immunogenicity risk of antibody-based biotherapeutics. PLoS One. 2016;11(8):e0159328.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed Consent
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Duke, B.R., Mitra-Kaushik, S. Current In Vitro Assays for Prediction of T Cell Mediated Immunogenicity of Biotherapeutics and Manufacturing Impurities. J Pharm Innov 15, 202–218 (2020). https://doi.org/10.1007/s12247-019-09412-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12247-019-09412-5