
Abstract
Background
Tablet dosage forms containing combination of darunavir a protease inhibitor, cobicistat a cytochrome P450 3A inhibitor, emtricitabine and tenofovir alafenamide which were nucleoside reverse transcriptase inhibitors were approved by USFDA on 1st July 2018 to suppress the viral load in HIV patients. It can be used as a complete regimen for the treatment of HIV-1 infection in adults and paediatric patients weighing at least 40 kg. An UPLC method was developed, and separation was done on SB C8 column of dimensions 50 × 2.1 × 1.8 μ with mobile phase 0.01 N potassium dihydrogen ortho phosphate (pH-4.8) and acetonitrile in 60:40 ratio, at a flow rate of 0.3 mL/min and an injection volume of 2 μL. The column temperature was maintained at 30 °C, and detection wavelength was 267 nm. The method was validated according to ICH guidelines.
Results
The retention times were 1.031, 1.341, 1.630 and 2.153 min, and they were linear in the concentration range of 1.25–7.5 μg/mL, 18.75–112.5 μg/mL, 25–150 μg/mL and 100–600 μg/mL for tenofovir alafenamide, cobicistat, emtricitabine and darunavir, respectively. The intraday and interday precisions were found to be within acceptable limits. LOD was found to be 0.06 μg/mL, 0.51 μg/mL, 1.31 μg/mL and 3.01 μg/mL, and LOQ was 0.19 μg/mL, 1.54 μg/mL, 3.96 μg/mL and 9.13 μg/mL for tenofovir alafenamide, cobicistat, emtricitabine and darunavir. The correlation coefficients were found to be more than 0.999, and recovery was more than 99.52% indicating the method was accurate. Forced degradation studies reveal that the drugs are unstable under acidic conditions. The method was simple, accurate, precise, stable and can be analysed in less runtime of 4 min.
Conclusions
The flexibility, accuracy and precision of the developed method ensure its applicability in routine analysis of tablet dosage forms.
Graphical Abstract

Similar content being viewed by others

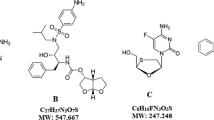

Background
AIDS became the world’s most severe health and development challenge [1]. Treatment for HIV must aim against virus by giving antiretroviral therapy (ART) which was a combination therapy and simultaneously must control opportunistic infections [2]. USFDA approved symtuza on 1st July 2018 the first single-tablet regimen (STR) containing 800 mg of darunavir (DRV) which helps in immune restoration, 150 mg cobicistat (COBI) which inhibits cytochrome P450 3A and acts as pharmacokinetic enhancer of protease inhibitors, 200 mg emtricitabine (FTC) and 10 mg of tenofovir alafenamide (TAF). It reduces the pill burden and combines antiviral efficacy of darunavir with safety of tenofovir [3, 4]. Symtuza can be used for treating of HIV1 infection in adults and adolescents of more than 12 years age [5, 6]. The combination of FTC and TAF with a protease inhibitor shows additive and/or synergistic effect against HIV [7]. The chemical structures of analytes are shown in Fig. 1. Stress investigation details the chemical behaviour of the molecule which helps in the progression of dosage form and packaging [8].

Structures of a tenofovir alafenamide, b cobicistat, c emtricitabine and d darunavir
Literature survey reveals that HPLC analysis was performed to simultaneously determine multiple drug components in STR-based drug products [9,10,11,12,13,14,15,16,17]. To the best of my knowledge, there were no analytical works for the analysis of STR-based tablets containing DRV, COBI, FTC and TAF. Attempts were made to establish a prompt, stable, reliable and accurate along with precise UPLC approach for the simultaneous estimation of DRV, COBI, FTC and TAF. This approach ought to be used in determination of these drugs in bulk and tablet dosage forms.
The drugs were separated and quantitatively determined by using UPLC in which the speed, resolution, sensitivity were enhanced [18]. The four-drug combination can be analysed in a short time which results in less run time and increased sensitivity, and thus reduces the solvent consumption leading to economisation.
Methods
-
1.
The pharmaceutical-grade standards of TAF (99.7%), COBI (99.8%), FTC (99.9%), DRV (99.9%) are obtained from Spectrum Pharma Lab, Hyderabad.
-
2.
Marketed formulation (symtuza, manufactured by Patheon Inc. for Janssen Pharmaceuticals, New Jersey, and Batch. no. 608) is obtained from Canada market.
-
3.
Chemicals: Acetonitrile (HPLC grade, Merck India Ltd, Mumbai), monopotassium phosphate (AR grade), methanol (HPLC grade) were procured from Merck Chemical division (Mumbai, India). Purified HPLC-grade water was obtained from Milli-Q, USA.
Instrumentation
The separation was done on acquity UPLC system equipped with quaternary pumps, acquity TUV detector along with auto sampler. The data processor was Waters Empower 2 software. The column of SB C8 column of 50 × 2.1, 1.8μ dimensions was used for separation. UV Spectrophotometer (Shimadzu UV 1800, Japan), electronic balance (Afcoset ER-200A, Mumbai, India), ultrasonicator (Enertech) and pH meter (AD 1020, Adwa Hungary ft, Hungary) were used for the study.
UPLC optimised conditions
Analytes can be separated by using combination of 0.01 N potassium dihydrogen ortho phosphate (pH -4.8) buffer and acetonitrile (60:40 v/v) at a flow rate of 0.3 mL/min through column maintained at a temperature of 30 °C at a detection wavelength of 267 nm. The column was equilibrated with mobile phase before injecting the solution. The injection volume was 2 µL.
Composition of analytical solutions
Mobile phase
The buffer and acetonitrile in the ratio of 60:40 v/v were used as mobile phase.
Constitution of 0.01 N potassium dihydrogen ortho phosphate buffer (pH-4.8): 1.36 g of potassium dihydrogen ortho phosphate was weighed accurately and added to 1000 mL of Milli-Q water filtered through 0.45-µ membrane filter sonicated to degas, and made volume up to mark.
Diluent
Water and acetonitrile in the ratio of 50:50 were used as diluent.
Standard solution
1.25 mg, 18.75 mg, 25 mg and 100 mg of tenofovir, cobicistat, emtricitabine and darunavir working standards were accurately weighed and transferred into four different 25-mL clean dry volumetric flask, add 10 mL of diluent, sonicate for 30 min, and make up to the final volume with diluent. The solution contains 50 µg/mL tenofovir, 750 µg/mL cobicistat, 1000 µg/mL emtricitabine, 4000 µg/mL darunavir.
1 mL of above stock solution was pipetted out in to a 10-mL volumetric flask, and then, make up to the final volume with diluent. The solution contains 5 µg/mL tenofovir, 75 µg/mL cobicistat, 100 µg/mL emtricitabine, 400 µg/mL darunavir, and solution was dribbled through 0.45-µm PVDF syringe filter, supplied by Allpure Biotechnology.
Sample solution
Estimate the average weight of 10 symtuza tablets, powder equivalent to one tablet was taken in to 100-mL volumetric flask, and 50 mL of diluent was added, ultrasonicated for 25 min and made volume with water: acetonitrile 50:50v/v (diluent) and filtered. From this solution, pipette out 0.5 mL in to 10 mL of volumetric flask and contrive the volume with diluent. The solution was dribbled through the Allpure brand PVDF syringe filter (0.45 µ). The resultant solution contains 5 µg/mL TAF, 75 µg/mL COBI, 100 µg/mL FTC, 400 µg/mL of DRV.
Validation parameters of the UPLC method
The optimised approach was validated conferring to the ICH guidelines in Q2(R1) [19], and validation results show system suitability of the method developed, sensitivity, accuracy, precision, linearity, detection limit (DL), quantitation limit (QL), robustness and ruggedness. Stress conditions are induced, and forced degradation studies are carried on according to ICH guidelines Q2 (R1). According to ICH guidelines in Q1A (R2) [20], the stability testing of new drug products facilitates the identification of degradation products and the stability of analyte. Guidelines for the photostability studies are outlined in ICH guidelines Q1B [21].
System suitability
It is done to check the suitability of method to system as a part of estimating system performance. It was estimated on five replicate injections of freshly prepared standard solutions (5 μg/mL, 75 μg/mL, 100 μg/mL and 400 μg/mL of TAF, COBI, FTC and DRV, respectively), and the parameters like plate count, asymmetry and resolution were estimated. % RSD of area for five replicate injections should be NMT 2%.
Selectivity
Selectivity can be estimated to check for any interference noticed at the retention times of the drugs TAF, COBI, FTC and DRV. Selectivity can be known by comparing the results of standard solution, sample solution, blank sample and placebo sample.
Linearity
By constructing calibration curves of peak area versus concentration range, we can assess linearity of drugs in concentration range of: 1.25–7.5 μg/mL for TAF, 18.75–112.5 μg/mL for COBI, 25–150 μg/mL for FTC and 100–600 μg/mL for DRV. The linearity can be determined by calculating the regression equation from the calibration curve constructed by using six standard concentrations.
Accuracy
It was the closeness of observed value of the approach to the true value. It was estimated as percentage of analyte recovered for assay after adding a known amount of respective drugs. The known amounts of standard solutions of drugs were spiked to pre-analysed sample solutions injected into UPLC system. Accuracy was estimated at 50%, 100% and 150% concentrations in triplicate, and % recovery was estimated.
Precision
System precision or injection repeatability was estimated by injecting six replicate injections of standard solutions containing 5 μg/mL, 75 μg/mL, 100 μg/mL and 400 μg/mL of TAF, COBI, FTC and DRV. The % RSD of peak area response was calculated.
Method precision or repeatability was estimated by injecting sample solutions in six replicate injections of the sample solution containing 5 μg/mL, 75 μg/mL, 100 μg/mL and 400 μg/mL of TAF, COBI, FTC and DRV. Precision of the method was estimated by the RSD of the percentage assay of the analytes.
Intermediate precision was accessed by injecting six replicate samples which contain 5 μg/mL of TAF, 75 μg/mL of COBI, 100 μg/mL of FTC and 400 μg/mL of DRV on same day and consecutive day.
Detection limit and quantification limit
Detection limit also known as limit of detection was the lowest concentrations of TAF, COBI, FTC and DRV which can be reliably detected from background noise, and LOQ was the minimum concentration of the drugs quantified with accuracy and precision. LOD and LOQ can be estimated by the formulas and also from calibration curve.
Robustness
It provides evidence of reliability during usual handling and is determined by analysing different parameters like alternating flow rate of eluent (± 0.01 mL), constitution of mobile phase (± 2%) and changes in column temperature (± 2 °C). The effects of the changes in experimental conditions on retention times and % recoveries were recorded.
Accelerated degradation studies
ICH guidelines recommended stress testing to know the intrinsic stability of drug substance. In accelerated degradation studies, the solution of the standard was subjected to acid hydrolysis (2 N HCl, refluxed at 60o C for 30 min), alkali hydrolysis (2 N NaOH, refluxed at 60 °C for 30 min), peroxide degradation (20% hydrogen peroxide heated at 60 °C for 30 min), thermal degradation (samples arranged in hot air oven at 105 °C for 6 hrs), photolytic degradation (sample in UV chamber for 7 days), neutral degradation (water, refluxed at 60 °C for 6 h). The so stressed samples were observed for the peak areas and compared with peak areas of standard.
Standard solution stability
The results obtained after analysing the standard solution stored after 24 h must be compared with the results obtained for fresh solution (at 0 h). The average %RSD of the area of solution was considered stable if the results do not deviate more than 2% with respect to fresh solution.
Assay procedure
Separately inject 2 µL of standard solution (of pure drugs) and sample solution (extracted from Symtuza tablets) in to the UPLC. The amount of the drugs present was estimated against the peak area of recorded chromatograms.
Results
System suitability
The peaks of TAF, COBI, FTC and DRV were obtained at retention times of 1.031, 1.341, 1.630, 2.153 min, respectively. The system suitability criterions are tabulated in Table 1.
Selectivity
No interference peaks were recorded at the retention times of drugs in the chromatograms of sample solution. The chromatograms are laid out in Fig. 2.

a Blank, b placebo, c standard and d sample chromatograms
Linearity
Regression lines were linear in concentration range of 1.25–7.5 μg/mL for TAF, 18.75–112.5 μg/mL for COBI, 25–150 μg/mL for FTC and 100–600 μg/mL for DRV. The results are enumerated in Table 2 which confirms the linear relation between concentration and peak area.
Accuracy
According to the ICH guidelines, accuracy must be determined on nine determinations covering minimum of three concentrations. For the determination of accuracy, concentrations at 50%, 100%, 150% levels of labelled claim were determined. The recoveries of known amount of analytes are shown in Table 3. The % mean recovery values were 99.87%, 100.05%, 99.53% and 99.67%, respectively, for TAF, COBI, FTC and DRV which shows the method was accurate.
Precision
The %RSD of percent assay, peak area and intermediate precision were less than 2 which indicates method was repeatable, reproducible and rugged. The outcomes are listed in Table 4.
LOD and LOQ
Sensitivity of the approach can be expressed as LOD and LOQ. The LOD values were observed as 0.06 μg/mL, 0.51 μg/mL, 1.31 μg/mL, 3.01 μg/mL, and LOQ values were found to be 0.19 μg/mL, 1.54 μg/mL, 3.96 μg/mL and 9.13 μg/mL for TAF, COBI, FTC and DRV. The values show the method was sensitive.
Robustness
Robustness was determined by small but deliberate variations in conditions like flow rate, composition of eluent, column temperature. Results due to variance in the conditions were compared with that of optimised chromatographic conditions. Results of robustness are tabulated in Table 5.
Accelerated degradation studies
Stability-indicating performance was estimated by stressing the samples and conducting the forced degradation studies. Peak areas along with % recoveries of TAF, COBI, FTC and DRV after applying the stress conditions are summarised in Table 6.
Stability of standard solution
The stability of UPLC approach was determined by exposing the solution towards sunlight and room temperature for 24 h.
The optimised and valid method was applied for the estimation of TAF, COBI, FTC and DRV in the tablet dosage form, and the results obtained were on par to the labelled amounts. The outcomes are summarised in Table 7.
Discussion
Few HPLC works were done previously on single drugs of the present study, in double combinations and also in combination with other drugs were done [9,10,11,12,13,14,15,16,17], but there were no analytical works developed for STR-based tablets containing DRV, COBI, FTC and TAF till date. In the developed method, the analytes were eluted at retention time of 1.031 min (TAF), 1.341 min (COBI), 1.630 min (FTC) and 2.153 min (DRV) and the run time is 4 min. Analytes exhibit good absorbance at 267 nm; therefore, this wavelength was selected as a detection wavelength in the development of method.
Prior to development of optimised method, several attempts were done with different combinations of mobile phases like methanol and water (50:50 v/v), acetonitrile and 0.1% OPA (40:60 v/v), acetonitrile and 0.01 N potassium dihydrogen phosphate (50:50 v/v) with columns like SB C18 100 × 2.1,1.8μ, hibar C18 100 × 2.1,1.8μ, phenyl C18 50 × 2.1,1.6μ, but the chromatograms obtained were not symmetric and the system suitability parameters like plate count and resolution were not according to the guidelines. Chromatograms with narrow peaks and acceptable symmetry and resolution were observed with SB C8 column of 50 × 2.1, 1.8 μ particle size; eluent used was 0.1% phosphate buffer and acetonitrile in the ratio 60:40 v/v at a flow rate of 0.3 mL/min. The column was maintained at 30 °C, and wave length of detection was 267 nm. The chromatogram of the optimised conditions is shown in Fig. 2.
As the performance of the system may vary during the normal usage, system suitability tests were performed to check the resolution and reproducibility which were found to adequate. The system suitability results were satisfactory which was determined in terms of injection repeatability, retention time, plate count, tailing and resolution. It ensures the method for the intended use was satisfied and can proceed for the study of validation parameters. Interference peaks from excipients did not co elute on the same retention times of analytes, indicating the optimised condition was selective which is interpreted from Fig. 2. Regression lines were linear in concentration range of 1.25–7.5 μg/mL for TAF, 18.75–112.5 μg/mL for COBI, 25–150 μg/mL for FTC and 100–600 μg/mL for DRV. For TAF, COBI, FTC and DRV the regression coefficients were found to be 0.9993, 0.9996, 0.9998 and 0.9998 indicating the method was linear. The % mean recovery values were 99.87, 100.05, 99.53 and 99.67, respectively, for TAF, COBI, FTC and DRV which shows the method was accurate. The %RSD of percent assay, peak area and intermediate precision were less than 2 which indicates method was repeatable, reproducible and rugged. LOD was found to be 0.06 μg/mL, 0.51 μg/mL, 1.31 μg/mL and 3.01 μg/mL for TAF, COBI, FTC and DRV, and LOQ was 0.19 μg/mL, 1.54 μg/mL, 3.96 μg/mL and 9.13 μg/mL which indicates sensitivity of method. Results due to variance in the conditions are compared with that of optimised chromatographic conditions. The low values of %RSD, which is within limit, indicate the optimised method was robust.
In accelerated stability studies, due to these created stress conditions, the recorded peak areas of TAF, COBI, FTC and DRV were less than the peak area of standard which indicates the degradation of drugs to some extent in all the conditions. In spite of changes in chromatograms, all the responses laid within the limits of assay without any major variations in the analytes retention time and peak area. The conclusions we can draw from the table were that the combination of TAF, COBI, FTC and DRV was more prone to acid hydrolysis. The results suggest that developed method is stability suggesting. The stability samples were analysed, and the %RSD for the peak areas was found to be 0.8, 0.7, 0.5 and 0.67 for TAF, COBI, FTC and DRV, respectively, with no important change in the elution of the peak. It emphasises the stability of solution for 24 h. The label claim of TAF, COBI, FTC and DRV was 10 mg, 150 mg, 200 mg and 800 mg, and the amounts were found to be 9.97 mg, 150 mg, 200.44 mg and 802.4 mg in the marketed formulation.
Conclusions
The chromatographic run time of developed method was 4 min, and the drugs were separated with satisfactory resolution between peaks. Accuracy, precision, linearity, sensitivity, robustness were adequately satisfactory. Satisfactory results for degradation studies show the method was stable and the analytes can be determined in the presence of degradants, interpreting the method was selective. The method allows the determination of TAF, COBI, FTC and DRV in dosage forms and bulk in the shortest possible time and provides economy in cost and time.
To summarise, using single-column and single-phase mobile-phase combination of acetonitrile and phosphate buffer we developed a method to quantify the amount of the four drugs TAF, COBI, FTC and DRV in bulk as well as tablet dosage forms. The method was proved stability indicating and can be used in practice for the dosage forms and to assess the shelf life.
Availability of data and materials
Any data if required will be provided on request.
Abbreviations
- UPLC:
-
Ultra-performance liquid chromatography
- ICH:
-
International Conference on Harmonisation
- FDA:
-
Food and Drug Administration
- STR:
-
Single-tablet regimen
- RSD:
-
Relative standard deviation
- OPA:
-
Ortho phosphoric acid
- DRV:
-
Darunavir
- COBI:
-
Cobicistat
- FTC:
-
Emtricitabine
- TAF:
-
Tenofovir alafenamide
- LOD:
-
Limit of detection
- LOQ:
-
Limit of quantification
References
The Global HIV/AIDS Epidemic/KFF-Kaiser Family Foundation, Global Health Policy Published: Mar 02, 2021. https://www.kff.org/global-health-policy/fact-sheet/the-global-hivaids-epidemic/. Accessed 15 Apr 2021
Valliant AAJ, Naik R (2020) HIV-1 associated opportunistic infections. Stat Pearls [Internet]. https://www.ncbi.nlm.nih.gov/books/NBK539787/. Accessed 15 Apr 2021
Deeks ED (2014) Cobicistat: a review of its use as pharmacokinetic enhancer of atazanavir ad darunavir in patients with HIV1infetion. Drugs 74(2):195–206. https://doi.org/10.1007/s40265-013-0160-x
Lepist EI, Phan TK, Roy A, Tong L, MacLennan K, Murray B, Ray AS (2012) Cobicistat boosts the intestinal absorption of transport substrates, including HIV protease inhibitors and GS-7340, in vitro. Antimicrob Agents Chemother 56(10):5409–5413. https://doi.org/10.1128/AAC.01089-12
Emma D (2018) Darunavir/cobicistat/emtricitabine/tenofovir alafenamide: a review in HIV-1 infection. Drugs 78(10):1013–1024. https://doi.org/10.1007/s40265-018-0934-2
Nicola S, Giorgio B, Elisa C, Andrea G, Alessandra B (2018) Darunavir–cobicistat–emtricitabine–tenofovir alafenamide: safety and efficacy of a protease inhibitor in the modern era. Drug Des 12:3635–3643. https://doi.org/10.2147/DDDT.S147493
Kulakarni R, Hluhanich R, McColl DM, Miller MD, White KL (2014) The combined anti-HIV-1 activities of emtricitabine and tenofovir plus the integrase inhibitor elvitegravir or raltegravir show high levels of synergy in vitro. Antimicrob Agents Chemother 58(10):6145–6150. https://doi.org/10.1128/AAC.03591-14
Blessy M, Patel RD, Prajapati PN, Agrawal PK (2014) Development of forced degradation and stability indicating studies of drugs: a review. J Pharm Anal 4(3):159–165. https://doi.org/10.1016/j.jpha.2013.09.003
Fatima U, Mamatha T, Goud GR (2014) A novel RP-HPLC method development and validation of cobicistat in bulk drug and tablet dosage form. Der Pharm Sin 5(5):99–105
Kalyani K, Anuradha V (2015) A stability-indicating high performance liquid chromatographic method for the determination of cobicistat. Int J Pharm Drug Anal 3(4):117–125
Mane PM, Pranali J, Gaikawad PJ, Patil AV, Mogale AS (2013) A RP-HPLC method for determination of darunavir in bulk and pharmaceutical. Int J Pharm Sci Rev 21(2):20–23
Raveendra Babu G, Lakshmana Rao A, Rao JV (2013) Development and validation of novel HPLC method for estimation of darunavir in pharmaceutical formulations. Int J Res Pharm Chem 3(2):438–443
Aggarwal NN, Bhat KI, Jacob JT (2018) Stability indicating assay method development and validation for tenofovir alafenamide fumarate by RP-HPLC. Pharm Anal Acta. https://doi.org/10.4172/2153-2435.1000601
Madhavi S, Prameela Rani A (2017) Development and validation of RP-UPLC method for simultaneous estimation of cobicistat and darunavir. Res J Pharm Technol 10(12):4343–4349. https://doi.org/10.5958/0974-360X.2017.00796.X
Gummaluri RK, Parthasarathi TVN, Anjanamadhulika G (2016) Simultaneous method for determination of emtricitabine, tenofovir disoproxil fumarate, elvitegravir, and cobicistat in tablets by HPLC. Indian J Pharm Sci 78(4):532–537. https://doi.org/10.4172/pharmaceutical-sciences.1000148
Dhara SB, Bhavini NP, Chhaganbhai NP (2012) RP-HPLC method for simultaneous estimation of tenofovir disoproxil fumarate, lamivudine and efavirenz in combined tablet dosage form. Pharm Methods 3(2):73–78. https://doi.org/10.4103/2229-4708.103876
Chinnalalaiah R, Ravi Kumar P, Srinivasa Rao A (2016) A validated stability indicating RP-HPLC method for the determination of emtricitabine, tenofovir disoproxil fumarate, elvitegravir and cobicistat in pharmaceutical dosage form. J Chromatogr Sci 54(5):759–764. https://doi.org/10.1093/chromsci/bmw004
Ashok K, Gautam S, Anroop N, Rishbha S (2012) UPLC: a preeminent technique in pharmaceutical analysis—a review. Acta Pol Pharm 69(3):371–380
ICH. Harmonized Tripartite Guideline (2005) Validation of Analytical procedures: text and methodology, Q2 (R1). In: International conference on harmonisation, Geneva. https://database.ich.org/sites/default/files/Q2%28R1%29%20Guideline.pdf. Accessed 15 Apr 2021
ICH. Harmonized Tripartite Guideline (2003) Stability testing of new drug substance and products. Q1A (R2). In: International conference on harmonisation, Geneva. https://database.ich.org/sites/default/files/Q1A%28R2%29%20Guideline.pdf. Accessed 15 Apr 2021
International conference on harmonisation of technical requirements for registration of pharmaceuticals for human use, stability testing of new drug substances and products, Q1B; 1996. https://database.ich.org/sites/default/files/Q1B%20Guideline.pdf. Accessed 15 Apr 2021
Acknowledgements
The authors are grateful to Spectrum Pharma Solutions, Hyderabad, for providing the drug samples and providing facilities to carry on the work.
Author information
M. Satya Venkata Sakuntala is working as Senior Lecturer at Government Polytechnic for women, Kakinada and Research scholar at JNTUK, Kakinada, A.P., India. She has 15 years of teaching experience. She is having membership in various professional societies like IPA, APTI.
Dr. A. Lakshmana Rao is working as Professor and Principal of V.V.Institute of Pharmaceutical Sciences, Gudlavalleru, A.P, India. He is having 20 years of Teaching, Research and Administrative experience. He guided 9 PhD Scholars and 58M.Pharm students. He received many awards and prizes from various organizations. He received Dr.APJ Abdul Kalam Award for Teaching Excellence from Marina Labs, Chennai; Noble Principal of the Year Award from Operant Pharmacy Federation, Rajasthan and Principal of the Year Award from Association of Pharmacy Professionals, Bhopal. He conferred with Meritorious University Best Teacher Award from Jawaharlal Nehru Technological University Kakinada, Kakinada. He is given 10 Guest Lectures in different Universities and colleges. He published 302 papers in various International and National Journals. He published 1 Textbook and 2 eBooks. He is Editor- in- Chief, Reviewer and Editorial Board member for 111 reputed International and National journals. He organised 25 seminars, Conferences and Workshops. He participated in 89 seminars, in various Conferences and Workshops. He presented 32 scientific papers in various Seminars and Conferences and received 12 prizes. He is having 1 Indian Patent in his Credit. He received grants from AICTE and PCI. He is having memberships in various professional societies like IPA, APTI, ABAP, APP, and ISTE etc.
Dr. M. William Carey is working as Head of Pharmacy Section at Government Polytechnic, Visakhapatnam, and A.P, India. He is having 33 years of Teaching out of 10 years of Administrative experience and 15 years of Research experience. He is a highly motivated and dedicated individual supported by a strong academic background and has the ability to work in multidisciplinary environment. He is guiding one PhD student. He published 15 research papers in various International and National journals. He is having membership in various professional societies like APTI, IPA, IPGA, IPS etc and Fellow of Indian Chemist. He is a recipient of Best Teacher award from Govt of Andhra Pradesh in the year 2004 and Best Polytechnic Teacher award by Indian Society for Technical Education, New Delhi in the year 2005 and Principal/Teacher award for the year 2015 by the Association of Pharmaceutical Teachers of India, Bangalore. He served as President of APTI (Association of Pharmaceutical Teachers of India) Andhra Pradesh State Branch from 2003 to 2014.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
MSV ALR and MWC contributed to the research work. Concept was given by ALR. Bench work was done and manuscript prepared by MSV. Analysis of work was done; chromatograms and manuscript were done by MWC and ALR. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
Authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Satya Venkata Sakuntala, M., Lakshmana Rao, A. & William Carey, M. Stability-indicating method development and validation for the concurrent determination of darunavir, cobicistat, emtricitabine and tenofovir alafenamide by UPLC in bulk and tablet dosage forms. Futur J Pharm Sci 7, 236 (2021). https://doi.org/10.1186/s43094-021-00384-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43094-021-00384-3