
A new simple, precise and robust isocratic reverse-phase high performance liquid chromatography (RP-HPLC) method was developed and validated for simultaneous determination of lamivudine, tenofovir disoproxil fumarate (TDF), and doravirine in bulk and pharmaceutical dosage form. The validation included specificity, linearity, system suitability, precision, robustness, LOD and LOQ characteristics. The chromatographic separation was achieved on C18X bridge phenyl column (150 × 4.6 mm, 3 μm particle size) eluted with acetonitrile and hexane-1-sulfonic acid (pH 2.5; 50:50, v/v) at a flow rate of 0.8 mL/min and monitored at 243 nm over a run time of 12 min. The retention times of lamivudine, TDF, and doravirine were found to be 2.45, 7.3, and 8.79 min. respectively. The method was linear in the range of 5 – 100 μg/mL (r2 = 0.999) for lamivudine and TDF and in the range of 1.75 – 35 μg/mL (r2 = 0.999) for doravirine. The percentage recoveries of three drugs were within the acceptable limits (98 – 102%). The method was found to be precise as confirmed by % RSD < 0.6. Forced degradation study was conducted as per ICH guidelines, and the three drugs showed degradation within 21.4 – 33.8% under acidic, basic, oxidative, photolysis, and hydrolysis conditions. The proposed RP-HPLC method can be used for the quantification of lamivudine, TDF, and doravirine in API and tablets without any interference from excipients.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
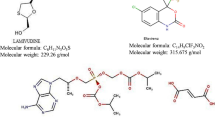
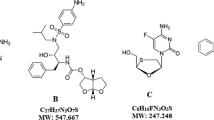
Lamivudine is a synthetic nucleoside analogue used to treat human immunodeficiency virus (HIV-1) infection and hepatitis-B. Chemically, lamivudine is (2R-cis)-4-amino-1-(2-hydroxymethyl-1,3-oxathiolan-5-yl)-(1H)-pyrimidin-2-one (Fig. 1a ). Lamivudine is a negative enantiomer of cytidine and exhibits its activity through an active metabolite triphosphate formed by phosphorylation. Triphosphate acts as a competitive inhibitor of viral DNA polymerase and blocks viral replication [1]. Tenofovir disoproxil fumarate (TDF) is an oral pro-drug of bioavailable tenofovir. This nucleoside analogue reverse transcriptase inhibitor is used to treat HIV infection [2]. Chemically, TDF is 9-[(R)-2[[bis-[[(isopropoxycarbonyl)oxy]methoxy]propyl] adenine fumarate (Fig. 1b ). It exhibits its activity by terminating viral DNA chain elongation, acting an adenosine 5'-monophosphate analogue. TDF inhibits the activity of viral DNA polymerase and terminates DNA chain elongation by competing with natural substance [3]. Doravirine chemically is 3-chloro-5-({1-[(5-hydroxy-4-methyl-4H-1,2,4-triazol-3-yl) methyl]-2-oxo-4-(trifluoromethyl)-1,2-dihydropyridin-3-yl} oxy) benzonitrile (Fig. 1c ). Doravirine is a pyridinone non-nucleoside reverse transcriptase inhibitor used to treat HIV infections in adult patients with no prior antiretroviral treatment history. Doravirine shows its activity by inhibiting viral replication through non-competitive inhibition of HIV-1 reverse transcriptase [4].

Chemical structures of (a) lamivudine (b) tenofovir disoproxil fumarate, and (c) doravirine.
Varieties of methods are in use for the estimation of TDF as single entity [5, 6] and lamivudine as a single component [7,8,9] in dosage forms and biological samples. Several methods have also been reported for the combination of TDF and lamivudine along with other drugs [10,11,12,13,14] in a variety of matrices. However, no methods have been reported till now for the simultaneous determination of amivudine, TDF, and doravirine in bulk and pharmaceutical dosage form. In this study, efforts were made to develop a new, simple, precise and robust analytical method for the simultaneous determination of lamivudine, TDF, and doravirine in bulk and pharmaceutical dosage form using reverse phase high performance liquid chromatography (RP-HPLC) technique.
Materials And Methods
Chemicals
Pharmaceutical grade reference standards of lamivudine, TDF, and doravirine were obtained as gift samples from Laurus Labs, Hyderabad, India. Fixed dosage combination tablet containing 300mg lamivudine, 300 mg TDF, and 100mg doravirine (DELSTRIGO) was obtained as gift sample from Laurus Labs, Hyderabad, India. All chemicals were HPLC grade purchased from S. D. Fine Chem., Mumbai, India. Milli ‘Q’ water was used throughout the study.
Instrumentation
Chromatographic analysis was carried out on Waters 2695 separation module (Waters Corporation, USA) equipped with auto sampler and waters 2998 PDA detector, and X-bridge Phenyl (150 × 4.6 mm, 5.6 μm particle size) column.
Preparation of Solutions
Buffer solution was prepared from accurately weighed (about 2.5 g) hexane-1-sulfonic acid dissolved in water in 1000 mL volumetric flask. The volume was made up to 1000 mL, the solution pH was adjusted to 2.5 with 0.1% orthophosphoric acid, and the solution was filtered through 0.45 μm membrane filter.
Stock solution. About 5 mg of lamivudine and 5 mg of TDF were accurately weighed in 10 mL volumetric flask. About 5 mg doravirine was accurately weighed in another 10 mL volumetric flask, dissolved and made up with an equal mixture of acetonitrile and buffer as diluent. From this solution, 3.5 mL was pipetted into a 10 mL volumetric flask containing TDF and lamivudine, and finally made up to 10 mL with diluent.
Working standard solutions were prepared from 1 mL stock solution pipetted into a 10 mL volumetric flask and made up to volume with diluent to get 50 μg/mL lamivudine, 50 μg/mL TDF, and 17.5 μg/mL doravirine. This solution was used as standard and further diluted as required.
Preparation of Tablet Samples
Twenty tablets were weighed and powdered. An amount of tablet powder equivalent to 50 mg of lamivudine, 50 mg of TDF, and 17.5 mg of doravirine was accurately weighed and transferred to 100 mL volumetric flask containing 70 mL of diluent. This mixture was subjected to sonication for 30 min to ensure complete extraction of drugs, made up to 100 mL with diluents, and filtered. From this solution, 1mL was taken and diluted to 10 mL with diluent to get final concentration of 50 μg/mL lamivudine, 50 μg/mL TDF, and 17.5 μg/mL doravirine. Optimised chromatographic conditions are listed in Table 1.The chromatogram of lamivudine, TDF, and doravirine standard is presented in Fig. 2.

Chromatogram of lamivudine, tenofovir disoproxil fumarate, and doravirine (standard).
Method Validation
The developed RP-HPLC method was validated for the linearity, precision, system suitability, robustness, LOD and LOQ characteristics as per ICH guidelines [15].
Linearity
Appropriate aliquots were pipetted from the stock solution to a series of 10 mL volumetric flasks and the volume made up with diluent to get the final concentrations within 5 – 100 μg/mL of lamivudine and TDF and 1.75 – 35 μg/mL of doravirine. Each solution was injected in triplicate. Calibration curves were plotted as the observed peak areas versus corresponding concentration, followed by determination of regression equations and calculation of correlation coefficients.
Precision
The intraday precision was studied using the analysis of six different sample solutions prepared using the same working standard solution. Each solution was injected in triplicate, the peak areas obtained were used to calculate the assay, and the % RSD was computed. Intermediate precision was determined in a similar manner on different days.
System Suitability
System suitability study was carried out with six injections of standard concentration (50 μg/mL lamivudine, 50 μg/mL TDF, and 17.5 μg/mL doravirine) into the HPLC system. Sets of parameters including retention time, number of theoretical plates, tailing factor, and resolution were determined.
Accuracy
Accuracy of the proposed method was estimated from recovery studies. The tablet sample solution was diluted to obtain solutions corresponding to 50, 100, and 150% in triplicate, each solution was injected twice and %recovery of three drugs was calculated.
Robustness
Robustness of the method was analyzed by altering the chromatographic conditions including the mobile phase composition, detection wavelength, etc. Small deliberate changes in the chromatographic conditions were introduced, and the extent to which the method was robust was determined. Deviations of ±0.2 mL/min in the flow rate and ±5mL in the amount of organic solvent in mobile phase composition were tried individually. The sample solution was injected in triplicate for each altered condition and the obtained peak areas were measured to calculate the assay value and %RSD. The system suitability parameters including the tailing factor and number of theoretical plates were monitored during the study.
Limit of Detection (LOD)
LOD is defined as the lowest amount of analyte in the sample which is to be detected and need not be quantified under given experimental conditions. It is determined at a signal/noise ratio of 3:1.
Limit of Quantitation (LOQ)
LOQ is the concentration of analyte in the sample which should be quantified with the precision and accuracy under stated experimental conditions. It is determined at a signal/ noise ratio of 10:1.
Specificity
Specificity of the method is determined by testing standard substances against potential interferences. The method was considered specific when the sample solution was injected and no interferences related to excipients were found. The chromatogram obtained by injecting the tablet solution was compared with the chromatogram obtained by injecting standard solution to study the interferences due to excipients.
Forced Degradation Study
Specificity of the method was also checked in the presence of various degradants through forced degradation studies. To establish the stability indicating nature of the method, the sample solution was subjected to a variety of stress conditions as follows.
Acid degradation. Accurately pipetted out 1mL of sample solution containing 50 μg/mL lamivudine, 50 μg/mL TDF, and 17.5 μg/mL doravirine was transferred to 10 mL volumetric flask, 1mL of 1N HCl was added, refluxed for 30 min, neutralized with 1 mL of 1N NaOH, and made up to 10 mL with diluent. The solution was filtered through 0.45 μm membrane and injected into the chromatographic system in triplicate. Chromatograms were recorded and % degradation was calculated to estimate stability of the sample. Purity of the analyte peak was confirmed based on the peak purity and peak threshold data. The degradation was also carried out with 5N HCl solution.
Base degradation. Accurately pipetted out 1mL of tablet sample was taken in two 10 mL volumetric flasks. To one flask, 1mL of 1N NaOH and to the other 5N NaOH was added. The mixtures were refluxed for 5 hours, neutralized with 1mL 1N HCl/5N HCl and made up to 10 mL with diluent. The solutions were injected into HPLC system, chromatograms were recorded, and statistical data calculated.
Oxidative degradation. Accurately pipetted out 1 mL of tablet sample solution was transferred to 10 mL volumetric flask, 1 mL of 10% H2O2 was added, and the mixture was refluxed for 5hours and made up to volume with diluent. The oxidative degradation was also carried out by taking 1 mL of 30% H2O2. Statistical data were calculated from the peak areas in the recorded chromatograms.
Hydrolytic degradation. Accurately pipetted out 1ml of tablet sample solution was transferred into 10 mL volumetric flask, 1 mL H2O was added, refluxed for 30 min, heated up for 3hours, cooled and made up the solution to 10 mL with diluents. The solution was injected into HPLC column and the degradation was computed from the obtained chromatograms monitoring the system suitability parameters.
Solution Stability
To analyze the stability between analyte, excipients, and diluent, sample solutions were kept for 6, 12, 18 and 24 hours at room temperature (RT) and 2 – 8°C. These solutions were injected into the chromatographic system at regular intervals and the obtained peak areas and retention times of respective drugs were compared to those of the freshly prepared solutions.
Application of the Proposed Method to Dosage Form
The optimized method was applied to determine the purity of marketed formulation (Delstrigo). The tablet sample solution was suitably diluted to obtain various concentrations of the three drugs and injected into the chromatographic system. The corresponding peak areas were determined and assay values were calculated.
Results
In order to develop and validate a chromatographic method for simultaneous quantification of lamivudine, TDF, and doravirine by RP-HPLC, several trials were undertaken. Initially the drug solution was analyzed using a mixture of acetonitrile and 0.1% orthophosphoric acid (pH 3.2; 80:20, v/v) at a flow rate of 1 mL/min, in which case the peak resolution and symmetry were not satisfactory. Several mobile phase compositions including 70:30, 50:50, and 20:80 v/v were tried, but the peak asymmetry and tailing were observed. Then, the buffer composition was changed and a mixture of acetonitrile with hexane-1-sulfonic acid (pH 2.5) at 50:50 v/v provided good peaks at a flow rate of 0.8 mL/min with detection at 243 nm. The retention times of lamivudine, TDF, and doravirine were found to be 2.45, 7.3, and 8.79 min respectively. The proposed method obeyed linearity (Fig. 3) for lamivudine and TDF in the range of 5 – 100 μg/mL (r2 = 0.9996, 0.9995) and for doravirine within 1.75 – 35 μg/mL (r2 = 0.999). The LOD and LOQ value were found to be 0.05 and 0.5 μg/mL for lamivudine and TFD and 0. μ017 μg/mL and 0.17 μg/mL for r2 = doravirine, which indicate proper sensitivity of the proposed method.

Calibration curves of (a) lamivudine (b) tenofovir disoproxil fumarate, and (c) doravirine.
The proposed method was also found to be specific, as the chromatogram (Fig. 4) obtained by injecting tablet solution showed no peaks close to the individual retention times of lamivudine, TFD, and doravirine.

Chromatogram of tablet sample solution
The proposed method gave consistent results indicating its precision as observed from the %RSD data (Table 2) as well as the intermediate precision (Table 3). Accuracy of the method was analyzed using recovery studies for the commercially available formulations. The percentage recovery values (Table 4) were in the range of 99.6 – 100.3% for Lamivudine, 99.3 – 99.7 % for TDF, and 99.4 – 99.7% for doravirine, which indicate that there are no interferences from excipients in formulation.
Small deliberate variations in the chromatographic conditions such as flow rate and mobile phase composition did not produce significant effect on the parameters like tailing factor (<1.2) and number of theoretical plates (>2000). The %RSD calculated for each modified parameter (Table 5) was less than 2 which indicates proper robustness of the method.
The proposed method was also applied to study the forced degradation behaviour of drugs exposed to various stress conditions and to understand the degradation pathway of the drug molecules [16]. Degradation of drug substances between 5 to 20% has been accepted for validation of chromatographic assays [17, 18]. From the obtained results, it was found that all the three drugs showed degradation (within 21.4 – 33.8%) at the stated experimental conditions (Table 6). Three peaks observed in chromatograms measured under acidic and basic conditions (Figs. 5–7) along with two minor degradants in peroxide stress condition (Fig. 8) were well resolved from the drug peaks, indicating specificity of the method. No additional peaks were observed upon hydrolytic degradation (Fig 9).

Chromatogram for blank sample.

Chromatogram for acid degradation study (5N HCl).

Chromatogram for basic degradation study (5N NaOH).

Chromatogram for peroxide degradation study (30% H2O2).

Chromatogram for hydrolytic degradation study.
The results of solution stability studies indicated that the drug solutions were considerably stable over a period of 6 hours and showed only negligible variation in the assay upon exposure up to 24 hours both at room temperature and 2 – 8oC as can be seen from Table 7.
Finally, the proposed method was applied to the quantification of lamivudine, DTF, and doravirine in marketed tablet formulation and assay was found to be 98.7, 98.5 and 99.7% w/w. respectively, in agreement with label claim (Table 8).
Discussion
As there is a developing interest for anti-HIV drugs, it is also necessary to develop a rapid, sensitive and robust analytical method. The statistical investigation of obtained information demonstrated that the proposed RP-HPLC method was accurate, linear, robust and economical. The optimized method is appropriate for determining of pharmaceutical drugs in the marketed formulation with virtually no interferences of excipients. Forced degradation data also confirmed that the degradation of three drugs was within the acceptable range. Hence, the method can be effectively applied to the quality control analysis. In conclusion, this study was aimed at the quantification of lamivudine, TDF, and doravirine in bulk and tablets. The developed method was found to be linear, accurate, robust and reliable. An evident advantage is the simplicity of sample preparation and speed of analysis, since the three compounds were eluted within 10 minutes. The RP-HPLC method also indicated the stability of drugs under various stress conditions, so it can also be employed as stability indicating method. Hence, the developed method can be used for routine analysis in quality control laboratories and for further research.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest. Gollu Gowri (corresponding author) was major contributor in developing and validating the optimised method; Sowjanya Gummadi (co-author) was major contributor in calculating validation parameters and writing manuscript. All authors have read and approved the manuscript.
References
K. Nahiro, J. Antimicrob. Chemother., 51, 1085 – 1089 (2003)
E. G Joel and D. Stanley, Clin. Infect. Dis., 37, 944 – 950 (2003).
B. Maria and H. Maria, Expert Rev. Gastroenterol. Hepatol., 6, 413 – 421 (2012)
C. Nicolas Sluis and T. Gilda, Virus Res., 134, 147 – 156 (2008).
R. Surendra, C. Tejasri, K. Dharmendra, et al., J. Pharm, Biomed. A: Lett., 4, 70 – 73 (2016).
J. Guo, F. Meng, L. Li, et al., Biol. Pharm. Bull., 34, 877 – 882 (2011).
S. O. Choi, N. Rezk, J. S. Kim, and A. D. Kashuba, J. Chromatogr. Sci., 48, 219 – 223 (2010).
M. Alebouyeh and H. Amini, J. Chromatogr. B: Analyt. Technol. Biomed. Life Sci., 975, 40 – 44 (2015).
G. Bahrami, S. Mirzaeei, A. Kiani, and B. Mohammadi, J. Chromatogr. B: Analyt. Technol. Biomed. Life Sci., 823(2), 213 – 7 (2005).
V. S. Akhilesh, K. N. Lila, and R. P. Nihar, J. Pharm. Anal., 1(4), 251–257 (2011).
W. Kromdijk, S. A. Pereira, and H. Rosing, J. Chromatogr. B: Analyt. Technol. Biomed. Life Sci., 919 – 920, 43 – 51 (2013).
C. Waitt, S. Diliiy Penchala, and A. Olagunju, J. Chromatogr. B: Analyt. Technol. Biomed. Life Sci., 1060, 300 – 307 (2017).
D. S. Bhavsar, B. N. Patel, and C. N. Patel, Pharm. Methods, 3(2), 73 – 8 (2012).
R. K. Valluru, B. B. Reddy, S. K. Sumanth, et al., J. Chromatogr. B: Analyt. Technol. Biomed. Life Sci., 931, 117 – 126 (2013).
N. Mallikarjuna Rao and D. Gowri Sankar, Futur. J. Pharm. Sci., 1(2), 73 – 77 (2015).
Dhara S. Bhavsar, B. N. Patel, and C. N. Patel, Pharm. Methods, 3(2), 73 – 78 (2012).
ICH Harmonized Tripartite Guideline, Validation of Analytical Procedures: Text and Methodology, Q2 (R1): International Conference on Harmonization, IFPMA, Geneva, Switzerland (2005).
E. Tamizi and A. Jouyban, Eur. J. Pharm. Biopharm., 98, 26 – 46 (2016).
M. Blessy, D. Ruchi, N. Prajesh, et al., J. Pharm. Anal., 4, 159 – 165 (2014).
Acknowledgements
The authors are thankful to Maharajah’s College of Pharmacy (Vizianagaram, India) and GITAM Institute of Pharmacy (Visakhapatnam, India) for supporting and providing necessary facilities for the research work.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Gollu, G., Gummadi, S. Simultaneous Quantification of Lamivudine, Tenofovir Disoproxil Fumarate and Doravirine in Pharmaceutical Dosage Form by Liquid Chromatography with Diode Array Detection. Pharm Chem J 54, 526–535 (2020). https://doi.org/10.1007/s11094-020-02232-9
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11094-020-02232-9