
Abstract
Fish health, growth and disease is intricately linked to its associated microbiome. Understanding the influence, source and ultimately managing the microbiome, particularly for vulnerable early life-stages, has been identified as one of the key requirements to improving farmed fish production. One tropical fish species of aquaculture importance farmed throughout the Asia-Pacific region is the giant grouper (Epinephelus lanceolatus). Variability in the health and survival of E. lanceolatus larvae is partially dependent on exposure to and development of its early microbiome. Here, we examined the development in the microbiome of commercially reared giant grouper larvae, its surrounding environment, and that from live food sources to understand the type of bacterial species larvae are exposed to, and where some of the sources of bacteria may originate. We show that species richness and microbial diversity of the larval microbiome significantly increased in the first 4 days after hatching, with the community composition continuing to shift over the initial 10 days in the hatchery facility. The dominant larval bacterial taxa appeared to be predominantly derived from live cultured microalgae and rotifer feeds and included Marixanthomonas, Candidatus Hepatincola, Meridianimaribacter and Vibrio. In contrast, a commercial probiotic added as part of the hatchery’s operating procedure failed to establish in the larvae microbiome. Microbial source tracking indicated that feed was the largest influence on the composition of the giant grouper larvae microbiome (up to 55.9%), supporting attempts to modulate fish microbiomes in commercial hatcheries through improved diets. The marked abundances of Vibrio (up to 21.7% of 16S rRNA gene copies in larvae) highlights a need for rigorous quality control of feed material.
Similar content being viewed by others


Introduction
Groupers are a highly valued and sought after food fish with annual global wild harvest more than doubling over the past 20 years (approximately 184,000 tons in 2001 to 462,000 tons in 2021) [1]. However, with increased fishing pressure there is concern that many grouper species are at risk due to overexploitation [2]. The giant grouper (GG) (Epinephelus lanceolatus) is the largest known ray-finned fish that inhabits coral reefs. Wild populations of GG have been in decline from overfishing [3] owing to their protogynous hermaphroditism (i.e. females develop into males in later life) and late sexual development with an estimated population doubling time of 14 years [4]. As such GG were classified as a vulnerable species (International Union for Conservation of Nature, 1996 and 2006), and they are protected in several countries including Australia [2, 5].
Against the background of diminishing wild stocks, in recent years there has been increased production in farming groupers to supplement production from capture fisheries. In 2019–2020 aquaculture production of groupers accounted for 46.0% of annual global grouper supply compared to just 6.5% in 2001 [1]; however, the grouper aquaculture industry faces several challenges including a lack of reliable methods in housing and conditioning broodstock, difficulties in production of high-quality eggs, poor larval survival rates, requirements for specialized diets, occurrence of spinal deformities and susceptibility to diseases [6, 7]. Furthermore, the survival of grouper larvae in hatcheries is generally lower compared to other finfish [8]. GG larvae are sensitive to their environment including water quality and environmental changes [8, 9], and have small mouth gape requiring specialized diets to ensure adequate nutrition [8]. Although not generally associated with larval rearing, there have been documented cases of groupers in aquaculture systems infected with diverse bacterial pathogens, including well known examples such as Streptococcus [10, 11], Vibrio [12,13,14,15] and Pseudomonas [16].
Recently, the study of animal microbiomes has gained traction and the microbiome is now recognized as an important factor contributing to host health, growth, and nutrition [17]. The fish microbiome has been implicated in various functions ranging from feeding and metabolism, stress, and immune responses, to reproduction and development (reviewed in [18]). Unlike the vertical transmission of microbiomes evident in mammals, aquatic fish predominantly recruit their microbiomes from the surrounding environment, wherein site [19] and diet [20] have been reported to significantly influence composition of the fish’s microbiome beginning from early larval stages [21]. Since site and diet are typically controlled under aquaculture settings, these findings raise the potential of exploiting the fish microbiome as an avenue to enhance aquaculture productivity. In this study, we partnered with The Company One Pty Ltd (TCO), a commercial GG hatchery producer, to profile the microbiome of GG larvae using high throughput 16S rRNA gene amplicon sequencing with the aim of generating microbiome baselines during the first 10 days post hatching. In addition, we generated microbial profiles from tank water, fish feed and other additives to track the source of microorganisms recruited into the GG larvae microbiota. As healthy development of GG, and ultimately aquaculture production success, is heavily dependent on their early life larval stages [8], results from this study will inform aquaculture management decisions by providing insight into the formation of the GG larvae microbiome during early life stages.
Materials and methods
Larval husbandry and hatchery setup
This study was performed in partnership with TCO (Cairns, Australia), the only commercial captive GG hatchery and aquaculture facility in Australia that houses wild broodstock under controlled conditions that allow the broodfish to spawn regularly. As per the company’s operating procedure, fertilized eggs were exposed to ozonated water (75 ppm for 80 s) and stocked into four 10 kL tanks at a volume of 150 mL eggs per tank. The four tanks were within a recirculating aquaculture system (RAS) with a sump connected for mechanical and biological filtration and ultraviolet (UV) treatment (Fig. S1). All tanks were housed in the same room and water temperature maintained at 28 °C. For the first three days, individual tanks were kept static without water exchange. From day 4, water exchange started at 200 L/h/tank, discharging to the central sump for treatment and recirculation. The exchange rate was increased daily to maintain tank water quality. Additionally, new filtered, ozone and UV treated seawater was added to the RAS starting from day 6 and increased daily to maintain overall water quality within the RAS. Measurements including dissolved oxygen, ammonia concentration, pH and temperature were recorded daily.
Tank additions including microalgae and copepods
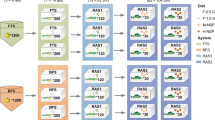
Three types of microalgae were added to the hatchery tanks in batches. Tetraselmis sp. (strain CS317) was added to the tanks on day 1, followed by Tahitian strain of Isochrysis sp. (strain CS177) from day 2 to 9 and Nannochloropsis oceanica (strain CS246) from day 3 to 9 (Fig. 1). The calanoid copepod Parvocalanus crassirostris was added on day 2 at a concentration of 2 adults/mL and 1 nauplii/mL into each hatchery tank. Daily average nauplii counts were recorded (Fig. 1). On days 3, 4, 6, 7, 8 and 9, the rotifer Brachionus rotundiformis was added into each hatchery tank at a concentration of 12.3 ± 0.38 rotifers per tank (Fig. 1). A commercial live Bacillus probiotic, Sanolife® MIC (INVE Aquaculture, USA) was added to tank water (10 g per tank) on days 0, 4 and 7 (Fig. 1).

Timeline of this study. Commercial, microalgae and live feed additions to the tanks are shown on top while samples processed and sent for 16S rRNA gene amplicon sequencing are shown at the bottom. dph indicates number of days after giant grouper (Epinephelus lanceolatus) eggs hatched
Sample collection
On day 0, tank water samples were collected prior to stocking the tanks with GG eggs to obtain starting microbial community profiles (Fig. 1). Tank water was collected by gently submerging a sterile 50 mL Falcon tube just below the surface of the water in each quadrant of the tank (n = 4 per tank, Fig. S1). As eggs were ozonated for stocking, a sample was collected for microbial profiles before and after ozonation. The eggs hatched the next day (day 1), and larvae were collected by gently scooping into sterile 50 mL Falcon tubes. Tank water was removed from the tubes and larvae were washed in autoclaved sterile seawater and immediately frozen at -80 °C. Daily tank inputs including feed (copepods and rotifers, n = 3), microalgae (n = 3), tank water (n = 4 per tank) and larvae samples (n = 3 per tank) were collected from days one to nine (Fig. 1). To ensure reproducibility, tank water samples were collected daily after microalgae addition, but before copepod and rotifers were added to the tanks. Larvae were collected approximately 1–2 h after feeding with copepods or rotifers. Microalgae, copepod and rotifers were sampled by scooping from individual tanks containing homogenous batch preparations of each food source using sterile 50 mL Falcon tubes (n = 3 per batch). All samples were stored at -80 °C immediately.
DNA extraction & quantification
Samples were pre-processed prior to DNA extraction. Tank water and microalgal samples were filtered through sterile Millex® 0.22 μm polyethersulfone syringe filters (Merck KGaA, Germany). The syringe filters were then incubated with 250 µL lysis buffer (20 mM Tris-HCl pH 8.0, 2 mM EDTA pH 8.0, 1.2% Triton X-100, 20 mg/mL lysozyme) for at least 30 min at 37 °C to lyse bacteria captured on the membrane. After this incubation, proteinase K was added to the syringe filters to a final concentration of 2 mg/mL and incubated overnight in a rotating oven at 56 °C. The resulting lysate was eluted with 500 µL of lysis buffer followed by 500 µL Buffer AL (part of the QIAGEN DNeasy Blood and Tissue Kit) and used for downstream DNA extraction. Copepod and rotifer samples were first pelleted at 5000 x g for 5 min at 4 °C. After removing the supernatant, the pellet was resuspended in 1 mL lysis buffer containing 2 mg/mL proteinase K and incubated at 56 °C until completely digested. Larvae were rinsed in sterile sea water, followed by overnight incubation in 500 µL lysis buffer containing 2 mg/mL proteinase K at 56 °C.
After the digestion steps described above, bacterial genomic DNA was extracted from all sample lysates using the DNeasy Blood and Tissue kit (QIAGEN, United States) according to manufacturer’s instructions with DNA eluted in TE buffer. DNA purity was examined by gel electrophoresis (1.2% agarose) and concentration determined using a NanoDrop One UV-Vis Spectrophotometer (Thermo Fisher Scientific, United States). To confirm successful bacterial DNA extraction, DNA from a random subset of samples were tested in a PCR using 16S rRNA primers targeting the prokaryotic V3-V4 region (341F: 5’-CCTACGGGNGGCWGCAG-3’and 806R: 5’-GGACTACNVGGGTWTCTAAT-3’ [22, 23], to check for amplification. PCR was performed using Platinum II Taq Hot-Start DNA Polymerase (Thermo Fisher Scientific, United States) and the presence of PCR amplicons checked on a 1% agarose gel. Concentration of PCR amplicons was measured using a QuantiFluor ONE dsDNA System and Quantus Fluorometer (Promega, United States).
Droplet digital PCR analysis
The QX200 Droplet Digital Polymerase Chain Reaction (ddPCR) System (Bio-Rad Laboratories, United States) was used to determine bacterial load. Universal bacterial 16S rRNA gene primers were used (1406F: 5’-GYACWCACCGCCCGT-3’; 1525R: 5’-AAGGAGGTGWTCCARCC-3’) to quantify total bacterial DNA concentration [24]. Primers specific for the genus Vibrio (567 F: 5’- GGCGTAAAGCGCATGCAGGT-3’; 680R: 5’-GAAATTCTACCCCCCTCTACAG-3’) were used to quantify DNA concentration of Vibrio spp. [25]. Multiplex ddPCR was performed in 25 µL volumes with each reaction consisting of 1X QX200 ddPCR EvaGreen Supermix (Bio-Rad Laboratories, United States), 300 nM of each primer and 5 µL template DNA. For DNA negative controls, 5 µL DNase/RNase free water was added instead of extracted DNA. After preparing the PCR reactions, 20 µL of each reaction mixture was loaded into a sample well of an 8-well cartridge of a droplet generator (Bio-Rad Laboratories, United States) to generate 20,000 droplets according to the manufacturer’s instructions. After generating droplets, PCR amplification was performed using the C1000 Touch Thermal Cycler (Bio-Rad Laboratories, United States) with the following thermocycling conditions: initial enzyme activation at 95 °C for 10 min, 40 cycles of denaturation at 95 °C for 30 s and primer annealing at 56 °C for 1 min; followed by a final inactivation step at 98 °C for 10 min. Droplet fluorescence was measured using a QX200 droplet reader (Bio-Rad Laboratories, United States) and data analysed with the QuantaSoft analysis software (version 1.0.596, Bio-Rad Laboratories, United States). Droplets were considered positive when fluorescence was above the background threshold of the negative droplets of the same sample and of negative controls. The number of molecules of target DNA present in the 20 µL reaction mixture was determined from the ratio of positive/total droplets. Quality controls included the presence of > 10,000 droplets of size and structure checked by the QuantaSoft software, < 10 positive droplets in negative controls and > 5 positive droplets in positive controls. In addition, the lower limit of detection was defined as at least 5 positive droplets, and the upper limit of detection was defined as at least 5 negative droplets [26].
16S rRNA gene amplicon sequencing
PCR amplification of the 16S rRNA gene for microbial community profiling and the subsequent sequencing library preparation for amplicon sequencing was performed by the Australian Genome Research Facility (AGRF, Melbourne, Australia). Briefly, extracted genomic DNA were first PCR amplified using 16S rRNA primers 341F (5’-CCTAYGGGRBGCASCAG-3’) and 806R (5’-GGACTACNNGGGTATCTAAT-3’), targeting the V3-V4 regions of the 16S rRNA gene. Sequencing libraries were then constructed using the Illumina Nextera XT Index Kit, pooled, and sequenced on an Illumina MiSeq platform (300 bp paired-end sequencing). Raw data are available at the Sequence Read Archive under accession PRJNA1120584.
16S rRNA amplicon sequence data processing and statistical analyses
Demultiplexed sequence reads were provided by the AGRF in fastq format. All reads were imported into QIIME™ 2 v2022.2 [27] for quality filtering and processing using the DADA2 workflow [28]. Adapter, primer, and low-quality sequences (median quality score < 20) were trimmed, and remaining reads subsequently denoised, merged and checked for chimeras using default settings in DADA2. Alpha diversity metrics (species richness and Shannon’s Index) were calculated based on subsampling sequence counts at a depth of 2930 reads 100 times (Fig. S2). To infer taxonomy of the 16S rRNA gene reads, a classifier was first trained on V3-V4 regions of reference 16S sequences downloaded from the SILVA 16S database v138.1 [29]. The trained classifier was then used to infer taxonomies of the representative 16S sequences output by DADA2. The final 16S counts table based on amplicon sequence variants (ASVs) was exported from QIIME2 and used as input into R v4.2.1 for statistical analyses [30]. All QIIME2 scripts are included in supplementary file 1. A centered log ratio (CLR) transformation was first applied to the counts data. Permutational multivariate analysis of variance (PERMANOVA) and linear models were then used to assess associations between community composition and experiment variables using the CLR transformed counts as Euclidean distances. Principal component analysis (PCA) was used to visualize the clustering of samples based on compositional similarities of their microbial communities. Relative abundances were calculated by dividing each ASV count by total library size (total sum scaling). Microbial source tracking was performed using fast expectation-maximization microbial source tracking (FEAST) [31] on the 16S counts to estimate the proportion of the microbial communities attributable to respective input sources. PERMANOVA and PCA are implemented in the vegan R package v2.4–6 [32]. Figures were edited in Inkscape v0.94 for clarity.
Construction of Vibrio spp. phylogenetic tree
Representative 16S rRNA gene sequences from ASVs classified as Vibrio were used to search for closely related sequences within the SILVA 16S reference database using the online search tool available at https://www.arb-silva.de/aligner/. Sequences reported by the search tool were then aligned with the Vibrio ASVs using MAFFT v7.508 [33], and the resulting sequence alignment was trimmed in silico according to the 341 F and 806R 16S primers using SeqKit v2.6.0 [34]. The trimmed sequence alignment was used to infer a phylogenetic tree using IQTREE2 v2.2.0.3 [35] with 2000 bootstraps. The output tree was midpoint rooted using GenomeTreeTk v0.1.8 (https://github.com/donovan-h-parks/GenomeTreeTk) and visualized in ITOL v6 [36]. All scripts are included in supplementary file 1.
Results
Stable water quality metrics and conditions during study duration
Fluctuations in water temperature, pH, dissolved oxygen, and ammonia concentration were largely consistent across all four tanks over the nine days of sampling. Average water temperature in the four tanks ranged between 27.5 °C and 27.6 °C. Tanks T3 and T4 experienced a drop of about 1.5 °C over a 12 h duration on day 4 due to a water heater malfunction (Fig. S3A), although this did not affect dissolved oxygen, pH or ammonia concentration (Fig. S3B-D). Average dissolved oxygen ranged between 6.75 mg/L and 7.77 mg/L, while pH ranged between 8.11 and 8.46 over the course of the study. Average ammonia concentration over the nine days was slightly higher in tank T1 compared with tanks T2, T3 and T4 (0.29 mg/L vs. 0.21–0.22 mg/L), with all values within the acceptable range of the hatchery SOP.
Development of the larvae-associated microbiota
Amplicon sequencing of DNA generated an average of 6866 sequences per sample. Identical sequences were clustered resulting in 2738 ASVs overall. Each sample was rarefied to 2930 sequences to calculate alpha diversity metrics including microbial community richness, Shannon diversity and Faith’s phylogenetic diversity. Eggs washed in ozonated water prior to stocking, as part of hatchery operational procedures, showed that microbial community richness, but not diversity, was significantly reduced in post-washed eggs compared to before washing (p < 0.05, Kruskal-Wallis test) (Fig. 2A-C). This implies that the population load of some bacterial species is unequally impacted by ozone treatment compared to other species, although treatment does not eliminate them completely. For instance, the most notable change in community composition was a reduced overall proportion of Pseudoalteromonas from an average 69.6% relative abundance to 57.3%, and an increase in Tenacibaculum from 9.6 to 18.4% after washing (Fig. S4A) (Table S1). After stocking washed eggs in the four tanks, larvae were collected daily for nine days to monitor development of the larvae microbiota. Using PCA to visualize compositional shifts in the larvae microbiota (Fig. 2D), we observed a developmental trajectory characterized by rapid increases in microbial species richness and diversity over the first four days post hatching (p < 0.05, linear mixed model). While community composition continued to develop beyond four days post hatching as indicated by the continued shift of samples in the PCA (p < 0.05, pairwise PERMANOVA; p < 0.05, PERMDISP2 for beta dispersion) (Table S2), richness and Shannon diversity largely plateaued around 150 unique ASVs and ∼ 6.5, respectively. Absolute bacterial load in larvae measured using ddPCR indicated that it remained largely stable throughout the study (Fig. S5B).

Microbial community alpha diversity and composition in giant grouper eggs (before and after ozone wash) and larvae. Box and whisker plots of alpha diversity metrics including (A) number of observed species, (B) Shannon diversity, and (C) Faith’s phylogenetic diversity calculated from rarefying samples to a depth of 2930 sequences 100 times. The thick centre within each box represents median values, and the upper and lower boundaries of the boxes represent the upper and lower quartiles, respectively. Whiskers extend to 1.5x interquartile range. (D) Principal component analysis (PCA) visualization of larvae-associated community composition over nine days. The PCA was performed on centered log ratio-transformed 16S counts as Euclidean distances. Each ring represents one replicate coloured by day of sampling. Coloured ellipses represent the 95% confidence limits of the group (sampling day) centroids
Larvae microbiota is primarily comprised of microbes from feed
Although sequences affiliated with Pseudoalteromonas and Tenacibaculum were detected at high relative abundance in washed eggs (57.3% and 18.4%, respectively), they were detected at markedly lower relative abundance in larvae one day post hatching (average 4.1% and 2.4%, respectively) and continued to decline in proportion throughout the study duration (p < 0.01, generalized linear model [GLM]) (Fig. 3) (Table S1). Instead, Alteromonas was the dominant genus in the larvae microbiota on day 1 (84.3% average relative abundance), and it declined in relative abundance (p < 0.01, GLM) and was replaced by Marixanthomonas on day 4 as the most dominant genus (26.3% vs. 8.9% average relative abundance). Alteromonas was also present at high relative abundances in tank water (average 57.9% relative abundance) on day 1, although it failed to establish as its relative abundance declined to < 1% in both tank water and larvae by day 6 (p < 0.01, GLM). By day 9, several other genera in the larvae microbiota had increased in relative abundance, the top five including Marixanthomonas (16.7%), Marivivens (6.6%), Algicola (5.5%) (p < 0.01, GLM), Vibrio (4.6%), and Pseudofulvibacter (4.4%) accounting for 37.8% of the larval microbiota affiliated reads (Fig. 3) (Table S1). Although a Bacillus probiotic product was added to the tanks on days 0, 4 and 7, Bacillus ASVs were detectable in the larvae for only one day after addition at 0.03%, 0.51% and 0.29% on days 1, 5 and 8, respectively (Table S1).

(A) Average relative abundances of microbial phyla detected in the larvae over the study duration. (B) Heat map of giant grouper larvae microbial community composition over the study duration. Labels to the left of the heat map indicate phyla (class for Alphaproteobacteria and Gammaproteobacteria) while labels on the right indicate genera. Only genera whose relative abundances significantly vary over the study duration are included in the heat map (generalized linear model, p < 0.05 and false discovery rate < 0.2). Genera labels are listed for genera with average relative abundance > 0.5% for clarity. See Table S1 for all relative abundance values
To estimate the contribution of the surrounding environment (tank water, feed) to composition of the larvae microbiota, we generated microbial community profiles from tank water, feed and additives, including live cultures of copepods, rotifers, and the alga Tetraselmis, Nannochloropsis and Isochrysis raised at the hatchery (Fig. S4B-E). The resulting tank water and feed microbial community profiles were then used as input sources in a microbial source tracking analysis. The analysis indicated that the larvae microbiota was primarily derived from microorganisms present in feed, with a maximum of 55.9% of the larvae community contributed from feed on day 5 (Fig. 4) (Table S3). The main taxa can be traced, for example, to increasing relative abundances of (i) Marixanthomonas in larvae beginning day 2 (p < 0.01, GLM) coinciding with the addition of Isochrysis feed in which Marixanthomonas was detected at 42.5% relative abundance, (ii) Candidatus Hepatincola and Meridianimaribacter from day 4 and 7, respectively (p < 0.01, GLM), coinciding with addition of rotifer feed in which the two genera were detected at 41.5% and 22.8% relative abundance, respectively, and (iii) Vibrio from day 3 (p < 0.01, GLM) coinciding with addition of copepod and rotifer feed in which Vibrio was detected at 1.1% and 1.6%, respectively (Fig. 3, S3) (Table S1). In contrast, additives consisting of commercial probiotic products did not substantially contribute to development of the larvae microbiome. The contribution from tank water was estimated at an average 9.8%, which was comparatively lower than the contribution from feed likely due to UV treatment and filtration implemented in the RAS to control microbial load in tank water.

Proportion of larvae microbial communities contributed from potential sources inferred using fast expectation-maximization microbial source tracking (FEAST) [31]. See Table S3 for numerical output from FEAST. Commercial product includes all commercial additions including probiotic, oil, nannopaste and emerald while microalgae include Tetraselmis sp., Nannochlropsis oceanica and Isochrysis sp
Vibrio detected in GG larvae possibly related to putative pathogens
Since Vibrio are often implicated in zoonoses in fish [37] and were detected in larvae at appreciable abundances through 16S rRNA gene amplicon sequencing (up to 12.9% of average bacterial load across tanks) and ddPCR (up to 21.7%) (Fig. 3, S5, S6), we thus examined whether the GG larvae Vibrio ASVs were related to known Vibrio pathogens. By comparing the Vibrio ASVs with closely related sequences obtained from the SILVA database sequence search tool, a phylogenetic tree inferred from these sequences indicated several ASVs formed monophyletic clades with putatively pathogenic Vibrio species such as V. coralliilyticus [38], V. tubiashii [39], V. fortis [40], V. proteolyticus [41] and Vibrio sp. 5-BBD-M4 (coral black band disease isolate) (Fig. 5). However, the phylogenetic tree lacked strong bootstrap support (< 95% UFBoot) [42], likely due to the short 16S V3-V4 ASVs used. These results indicate that while the Vibrio ASVs in GG larvae are potentially closely related to putative Vibrio pathogens as reported by the SILVA sequence search tool, their exact species identity requires longer 16S rRNA sequences or genome sequence data to be resolved.

Phylogenetic tree of Vibrio amplicon sequence variants (ASVs) detected in this study and their closest neighbours retrieved from the SILVA 16S database v138.1 (https://www.arb-silva.de/). Reference Vibrio sequences from SILVA are indicated by a grey background, while ASVs from this study are indicated by their respective ASV IDs (string of lowercase alphanumeric characters) generated in QIIME2. Sequences were aligned using MAFFT and phylogenetic tree was inferred using IQTREE2
Discussion
The microbiome of fish bred in captivity is distinct compared with their wild counterparts [43,44,45]; however, the implications on host health and productivity, if any, are still unknown. Research of captive bred fish predominantly focuses on diseases and pathogenic microbes, although in recent years there is growing interest in exploiting the microbiome to guide breeding practices and increase production yields [46,47,48]. In the present study, we examined the microbiome of GG larvae raised in a commercial hatchery over the first 10 days of larval culture and show that the GG’s microbiome composition is primarily influenced by microorganisms from feed sources. While our larvae microbiome community profiles represent that in the entire larvae (as they were too small to subsample individual body sites like the gut), our observations were consistent with microbiome surveys conducted in aquaculture of Atlantic salmon (Salmo salar) [20], yellowtail kingfish (Seriola lalandi) [49] and Nile tilapia (Oreochromis niloticus) [50] in which gut microbiome composition of the fishes reflected microorganisms found in their respective diets, indicating that the fish gut microbiome is markedly influenced by microorganisms in feed. It has been suggested that the influence of diet on fish gut microbiomes is a carry-over effect of bacterial DNA from feed present in the gut [20], and our sampling strategy designed around hatchery operations does not preclude this proposed carry-over as we sampled larvae 2 h after feeding. Nevertheless, in the microbial source tracking analysis we identified a consistent source proportion of the larvae microbiome attributed to larvae (i.e. ASVs shared with larvae-associated microbial communities from the previous day), which suggests that these feed-derived microbes were becoming established in the larvae. In this batch of GG larvae, several bacterial taxa including Marixanthomonas, Candidatus Hepatincola, Meridianimaribacter and Vibrio were inferred to originate from the algae and rotifer feed, but other than Vibrio not much is known about their relevance to fish health and physiology. Marixanthomonas, first isolated from a deep sea brittle star [51], have been reported to be enriched in the gut of giant prawns fed a diet of Thalassiosira weissflogii microalga [52]. Candidatus Hepatincola is likely a parasitic microorganism associated with isopods, insects, marine invertebrates [53] and possibly rotifers as our data suggests, while Meridianimaribacter is likely a lignocellulose degrader largely described in marine sediment [54] and soils [55], but have also been found in healthy shrimp larvae in aquaculture and were thus suggested to be a beneficial microbe [56]. In contrast, the Vibrio genus consists of widely known marine pathogens, many of which are linked to mortality in aquaculture of fishes, shrimp, oysters and other marine invertebrates [57,58,59]. Although it is tempting to ascribe the Vibrio ASVs found here to pathogenic species based on their close sequence similarities, the 16S rRNA gene amplicon data used in this study does not have sufficient length to confidently resolve species identities, nor does it differentiate between live and dead bacterial cells. Other potentially pathogenic taxa such as Pseudomonas and Aeromonas [60] were also detected at low abundances in the larvae. As such, follow up surveys including isolation of pure cultures, phenotyping and animal inoculation assays are needed to investigate the roles and effects of these Vibrio and other microorganisms of interest in GG aquaculture.
As our findings in GG larvae indicate that their microbiome is largely influenced by feed, a universal microbiome-guided aquaculture management strategy is therefore unlikely due to differences in feed and environment between hatcheries. However, it raises the possibility of modulating the GG microbiome through diet. A commercial probiotic consisting of Bacillus was added to tank water on days 1, 4 and 7 as part of the hatchery’s operating procedure, however, Bacillus were only detectable in larvae for one day following application indicating that it likely failed to establish in the larvae microbiome. Although certain probiotics are comprised of inactivated bacteria [61] and are not expected to result in colonization of animal hosts, the Bacillus probiotic used in this study was marketed as viable and tested to germinate within two hours of application (see methods section for product details). It is possible that the nine-day study duration was insufficient for colonization, however, the non-detection in both larvae and tank water samples two days after application indicates that the probiotics product was likely to have been removed from the RAS either by UV and/or filtration. These findings suggest that either: (i) more regular applications are necessary to introduce and maintain microbes of interest into the larvae microbiome (as seen by the transfer of microbes from feed), (ii) microbes have to be ingested to become part of the larvae microbiome, and/or (iii) only specific microbes are able to establish and persist in these fish [62]. Conversely, another implication of the larvae microbiome being largely derived from microorganisms in feed is a need for rigorous quality control of feed material to avoid introducing pathogens into aquaculture systems. For example, the close sequence similarity of Vibrio ASVs in rotifers fed to the GG larvae to pathogenic species warrants further examination to establish whether these specific Vibrio species/strains pose risks to the fish as the presence of opportunistic pathogens does not necessarily result in disease [63]. Rotifers as carriers of putative pathogens could be a widespread issue in aquaculture. A survey of microbiomes of farmed European seabass (Dicentrarchus labrax) and gilthead seabream (Sparus aurata) similarly reported high relative abundances of Vibrio in fish larvae and their rotifer/artemia feed (> 20% relative abundance) and postulated that the Vibrio was transferred from feed to larvae [64], although there was no supporting microbial source tracking data. The increased load of Vibrio and other pathogens in aquaculture systems could pose problems as they can lead to mortality and production losses, and more crucially could result in disease if transmitted to consumers [65]. A previous aquaculture experiment demonstrated that feeding gilthead seabream larvae with live rotifer pretreated with algae-derived antibacterials resulted in reduced relative abundances of known pathogens including Pseudomonas, Klebsiella and Stenotrophomonas in the larvae microbiome [66]. Correspondingly, there was an increase in potentially beneficial alphaproteobacterial taxa such as Paracoccus and Polymorphum, thus highlighting potential benefits of optimizing feeding strategy and quality control over feed material in aquaculture production.
In conclusion, to successfully exploit the microbiome as an avenue to increase agriculture productivity, breeding programs need to be tailored accordingly to accommodate variation across hatcheries, feed and likely also host genetics [67]. As such, longer duration microbiome surveys encompassing independent aquaculture batches are needed to address which microbes establish and persist in GG through to maturity. This includes assessing whether the microbiome from distinct batches of larvae converges as the GG host and its genetics could select for a conserved set of taxa in later life (e.g. akin to a core microbiome), how diet, feeding strategies and probiotics influence the microbiomes through to maturity, and how features of the microbiome (e.g. abundance of known beneficial microorganisms, putative pathogens) translate to host traits relevant to aquaculture production such as animal health, size, flesh colour and texture. Future experiments should include the necessary comparisons (e.g. controlled diets and probiotic treatments in isolated tanks, tank water vs. tank biofilm microbial community composition) to identify the exact microbes and host-microbe interactions that contribute to production. In addition, stringent quality control over feed material will be necessary to control bacterial loads [68]. This information will be crucial for the incorporation of microbiome data into aquaculture management strategies.
Data availability
The dataset supporting the conclusions of this article are available at the Sequence Read Archive under accession PRJNA1120584.
References
FAO Fisheries and Aquaculture - Global Statistical Collections. [https://www.fao.org/fishery/en/statistics]
Sadovy de Mitcheson Y, Craig MT, Bertoncini AA, Carpenter KE, Cheung WWL, Choat JH, Cornish AS, Fennessy ST, Ferreira BP, Heemstra PC, et al. Fishing groupers towards extinction: a global assessment of threats and extinction risks in a billion dollar fishery. Fish Fish. 2013;14:119–36.
Chong-Montenegro C, Thurstan, Ruth H, Pandolfi JM. Diving into archival data: the hidden decline of the giant grouper (Epinephelus lanceolatus) in Queensland, Australia. Aquat Conserv: Mar Freshw Ecosyst. 2024;34:e4094.
Palma P, Takemura A, Libunao GX, Superio J, de Jesus-Ayson EG, Ayson F, Nocillado J, Dennis L, Chan J, Thai TQ, et al. Reproductive development of the threatened giant grouper Epinephelus lanceolatus. Aquaculture. 2019;509:1–7.
Epinephelus lanceolatus. The IUCN Red List of Threatened Species [https://doi.org/10.2305/IUCN.UK.2018-2.RLTS.T7858A100465809.en.].
Canepa M, Berding A, Rossi R, Reason M, Knuckey R. Swim bladder inflation failure and associated spinal deformities in giant grouper Epinephelus lanceolatus. Aquaculture. 2023;577:739963.
Ybanez C Jr, Gonzales R. Challenges and progress of grouper aquaculture in asia: a review. Davao Res J. 2023;14:6–29.
Rimmer MA, Glamuzina B. A review of grouper (Family Serranidae: Subfamily Epinephelinae) aquaculture from a sustainability science perspective. Reviews Aquaculture. 2019;11:58–87.
Caberoy NB, Quinitio GF. Sensitivity of grouper Epinephelus coioides eggs to handling stress at different stages of embryonic development. ISR J Aquacult-Bamid. 1998;50:167–73.
Agnew W, Barnes AC. Streptococcus iniae: an aquatic pathogen of global veterinary significance and a challenging candidate for reliable vaccination. Vet Microbiol. 2007;122:1–15.
Wang P-C, Maekawa S, Chen S-C. Chap. 35 - Streptococcosis. In Aquaculture Pathophysiology. Edited by Kibenge FSB, Baldisserotto B, Chong RS-M: Academic Press; 2022: 439–445.
Amalina NZ, Santha S, Zulperi D, Amal MNA, Yusof MT, Zamri-Saad M, Ina-Salwany MY. Prevalence, antimicrobial susceptibility and plasmid profiling of Vibrio spp. isolated from cultured groupers in Peninsular Malaysia. BMC Microbiol. 2019;19:251.
Chong R. Pathology of Cultured Fish in Hong Kong 1st Edition. Agriculture, Fisheries and Conservation Department: Hong Kong SAR, China; 2015.
Chong RS-M. Chap. 36 - Vibriosis. In Aquaculture Pathophysiology. Edited by Kibenge FSB, Baldisserotto B, Chong RS-M: Academic Press; 2022: 447–464.
Lee KK. Pathogenesis studies on Vibrio alginolyticus in the grouper, Epinephelus malabaricus, Bloch Et Schneider. Microb Pathog. 1995;19:39–48.
Sun Y, Weng S, Xu B, Dong C, He J. Iron loss and hypoxia are involved in lethal Pseudomonas plecoglossicida infections in the orange-spotted grouper Epinephelus coioides. Aquaculture. 2023;563:739015.
Legrand TPRA, Wynne JW, Weyrich LS, Oxley APA. A microbial sea of possibilities: current knowledge and prospects for an improved understanding of the fish microbiome. Rev Aquacult. 2020;12:1101–34.
Butt RL, Volkoff H. Gut microbiota and Energy Homeostasis in Fish. Front Endocrinol 2019, 10.
Kim PS, Shin N-R, Lee J-B, Kim M-S, Whon TW, Hyun D-W, Yun J-H, Jung M-J, Kim JY, Bae J-W. Host habitat is the major determinant of the gut microbiome of fish. Microbiome. 2021;9:166.
Karlsen C, Tzimorotas D, Robertsen EM, Kirste KH, Bogevik AS, Rud I. Feed microbiome: confounding factor affecting fish gut microbiome studies. ISME Commun. 2022;2:14.
Borges N, Keller-Costa T, Sanches-Fernandes GMM, Louvado A, Gomes NCM, Costa R. Bacteriome structure, function, and Probiotics in Fish Larviculture: the Good, the bad, and the gaps. Annu Rev Anim Biosci. 2021;9:423–52.
Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, Fierer N, Knight R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci U S A. 2011;108(Suppl 1):4516–22.
Klindworth A, Pruesse E, Schweer T, Peplies J, Quast C, Horn M, Glöckner FO. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013;41:e1.
Woodcroft BJ, Singleton CM, Boyd JA, Evans PN, Emerson JB, Zayed AAF, Hoelzle RD, Lamberton TO, McCalley CK, Hodgkins SB, et al. Genome-centric view of carbon processing in thawing permafrost. Nature. 2018;560:49–54.
Thompson JR, Randa MA, Marcelino LA, Tomita-Mitchell A, Lim E, Polz MF. Diversity and dynamics of a north atlantic coastal Vibrio community. Appl Environ Microbiol. 2004;70:4103–10.
Dreo T, Pirc M, Ramšak Ž, Pavšič J, Milavec M, Žel J, Gruden K. Optimising droplet digital PCR analysis approaches for detection and quantification of bacteria: a case study of fire blight and potato brown rot. Anal Bioanal Chem. 2014;406:6513–28.
Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. 2019;37:852–7.
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, Holmes SP. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581–3.
Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, Glöckner FO. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007;35:7188–96.
R Core Team. R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2022.
Shenhav L, Thompson M, Joseph TA, Briscoe L, Furman O, Bogumil D, Mizrahi I. Pe’er I, Halperin E: FEAST: fast expectation-maximization for microbial source tracking. Nat Methods. 2019;16:627–32.
Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin P, O’Hara B, Simpson G, Solymos P, Stevens H, Wagner H. Vegan: Community Ecology Package. R Package Version 22 – 1 2022, 2:1–2.
Katoh K, Standley DM. MAFFT multiple sequence alignment Software Version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–80.
Shen W, Le S, Li Y, Hu F. SeqKit: a cross-platform and Ultrafast Toolkit for FASTA/Q file manipulation. PLoS ONE. 2016;11:e0163962.
Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, von Haeseler A, Lanfear R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol. 2020;37:1530–4.
Letunic I, Bork P. Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021;49:W293–6.
Novoslavskij A, Terentjeva M, Eizenberga I, Valciņa O, Bartkevičs V, Bērziņš A. Major foodborne pathogens in fish and fish products: a review. Ann Microbiol. 2016;66:1–15.
Richards GP, Watson MA, Needleman DS, Church KM, Häse CC. Mortalities of Eastern and Pacific Oyster Larvae caused by the pathogens Vibrio coralliilyticus and Vibrio tubiashii. Appl Environ Microbiol. 2015;81:292–7.
Elston RA, Hasegawa H, Humphrey KL, Polyak IK, Häse CC. Re-emergence of Vibrio tubiashii in bivalve shellfish aquaculture: severity, environmental drivers, geographic extent and management. Dis Aquat Organ. 2008;82:119–34.
Green TJ, Siboni N, King WL, Labbate M, Seymour JR, Raftos D. Simulated Marine Heat Wave alters abundance and structure of Vibrio Populations Associated with the Pacific Oyster resulting in a Mass Mortality Event. Microb Ecol. 2019;77:736–47.
Ray A, Kinch LN, Santos MS, Grishin NV, Orth K, Salomon D. Proteomics Analysis reveals previously uncharacterized virulence factors in Vibrio proteolyticus. mBio. 2016;7. https://doi.org/10.1128/mbio.01077-01016
Minh BQ, Nguyen MAT, von Haeseler A. Ultrafast approximation for Phylogenetic Bootstrap. Mol Biol Evol. 2013;30:1188–95.
Alberdi A, Martin Bideguren G, Aizpurua O. Diversity and compositional changes in the gut microbiota of wild and captive vertebrates: a meta-analysis. Sci Rep. 2021;11:22660.
Eichmiller JJ, Hamilton MJ, Staley C, Sadowsky MJ, Sorensen PW. Environment shapes the fecal microbiome of invasive carp species. Microbiome. 2016;4:44.
Liu X, Fan Y, Mo T, Chen Q, Chen W. Comparative study of the gut Microbiota Community between the Farmed and Wild Mastacembelus armatus (zig-Zag eel). Metabolites 2022, 12.
Lorgen-Ritchie M, Uren Webster T, McMurtrie J, Bass D, Tyler CR, Rowley A, Martin SAM. Microbiomes in the context of developing sustainable intensified aquaculture. Front Microbiol. 2023;14:1200997.
Perry WB, Lindsay E, Payne CJ, Brodie C, Kazlauskaite R. The role of the gut microbiome in sustainable teleost aquaculture. Proc R Soc B Biol Sci. 2020;287:20200184.
Vargas-Albores F, Martínez-Córdova LR, Hernández-Mendoza A, Cicala F, Lago-Lestón A, Martínez-Porchas M. Therapeutic modulation of fish gut microbiota, a feasible strategy for aquaculture? Aquaculture. 2021;544:737050.
Wilkes Walburn J, Wemheuer B, Thomas T, Copeland E, O’Connor W, Booth M, Fielder S, Egan S. Diet and diet-associated bacteria shape early microbiome development in Yellowtail Kingfish (Seriola lalandi). Microb Biotechnol. 2019;12:275–88.
Wang M, Fan Z, Zhang Z, Yi M, Liu Z, Ke X, Gao F, Cao J, Lu M. Effects of Diet on the Gut Microbial communities of Nile Tilapia (Oreochromis niloticus) Across their different life stages. Front Mar Sci 2022, 9.
Romanenko LA, Uchino M, Frolova GM, Mikhailov VV. Marixanthomonas ophiurae gen. nov., sp. nov., a marine bacterium of the family Flavobacteriaceae isolated from a deep-sea brittle star. Int J Syst Evol Microbiol. 2007;57:457–62.
Tang Y, Wang R, Tan L, Guo L, Duan Y, Yang L, Jiang S, Zhou F, Jiang S, Huang J. Effects of live microalgae and algae powder on microbial community, survival, metamorphosis and digestive enzyme activity of Penaeus monodon larvae at different growth stages. Aquaculture. 2020;526:735344.
Dittmer J, Bredon M, Moumen B, Raimond M, Grève P, Bouchon D. The terrestrial isopod symbiont ‘Candidatus Hepatincola Porcellionum’ is a potential nutrient scavenger related to Holosporales symbionts of protists. ISME Commun. 2023;3:18.
Wang B, Sun F, Du Y, Liu X, Li G, Lai Q, Luo J, Shao Z. Meridianimaribacter flavus gen. nov., sp. nov., a member of the family Flavobacteriaceae isolated from marine sediment of the South China Sea. Int J Syst Evol Microbiol. 2010;60:121–7.
Lam MQ, Oates NC, Thevarajoo S, Tokiman L, Goh KM, McQueen-Mason SJ, Bruce NC, Chong CS. Genomic analysis of a lignocellulose degrading strain from the underexplored genus Meridianimaribacter. Genomics. 2020;112:952–60.
Zheng Y, Yu M, Liu J, Qiao Y, Wang L, Li Z, Zhang X-H, Yu M. Bacterial Community Associated with Healthy and Diseased Pacific White shrimp (Litopenaeus vannamei) Larvae and Rearing Water across different growth stages. Front Microbiol 2017, 8.
Rønneseth A, Castillo D, D’Alvise P, Tønnesen Ø, Haugland G, Grotkjær T, Engell-Sørensen K, Nørremark L, Bergh Ø, Wergeland HI, Gram L. Comparative assessment of Vibrio virulence in marine fish larvae. J Fish Dis. 2017;40:1373–85.
Zhang X-H, He X, Austin B. Vibrio harveyi: a serious pathogen of fish and invertebrates in mariculture. Mar Life Sci Tech. 2020;2:231–45.
Sanches-Fernandes GMM, Sá-Correia I, Costa R. Vibriosis outbreaks in aquaculture: Addressing Environmental and Public Health Concerns and Preventive Therapies Using Gilthead Seabream Farming as a Model System. Front Microbiol 2022, 13.
Tendencia EA, Lavilla-Pitogo CR. Bacterial diseases. In Diseases of cultured groupers. Edited by Cruz-Lacierda KNER. Tigbauan, Iloilo, Phillipines: Aquaculture Department, Southeast Asian Fisheries Development Center; 2004: 19–28.
Tran NT, Yang W, Nguyen XT, Zhang M, Ma H, Zheng H, Zhang Y, Chan K-G, Li S. Application of heat-killed probiotics in aquaculture. Aquaculture. 2022;548:737700.
Albright MBN, Louca S, Winkler DE, Feeser KL, Haig S-J, Whiteson KL, Emerson JB, Dunbar J. Solutions in microbiome engineering: prioritizing barriers to organism establishment. ISME J. 2022;16:331–8.
Mohamad N, Mustafa M, Amal MNA, Saad MZ, Md Yasin IS, Al-Saari N. Environmental Factors Associated with the Presence of Vibrionaceae in Tropical Cage-cultured Marine fishes. J Aquat Anim Health. 2019;31:154–67.
Najafpour B, Pinto PIS, Sanz EC, Martinez-Blanch JF, Canario AVM, Moutou KA, Power DM. Core microbiome profiles and their modification by environmental, biological, and rearing factors in aquaculture hatcheries. Mar Pollut Bull. 2023;193:115218.
Baker-Austin C, Oliver JD, Alam M, Ali A, Waldor MK, Qadri F, Martinez-Urtaza J. Vibrio spp. infections. Nat Rev Dis Primers. 2018;4:1–19.
Sanches-Fernandes GMM, Califano G, Castanho S, Soares F, Ribeiro L, Pousão-Ferreira P, Mata L, Costa R. Effects of live feed manipulation with algal-derived antimicrobial metabolites on fish larvae microbiome assembly: a molecular-based assessment. Aquac Res. 2022;53:1062–83.
Wilkins LGE, Fumagalli L, Wedekind C. Effects of host genetics and environment on egg-associated microbiotas in brown trout (Salmo trutta). Mol Ecol. 2016;25:4930–45.
Giménez G, Padrós F, Roque A, Estévez A, Furones D. Bacterial load reduction of live prey for fish larval feeding using Ox-Aquaculture©. Aquac Res. 2006;37:1130–9.
Acknowledgements
We thank TCO staff for sampling support, fish and facilities - specifically Anjanette Berding, Ryan Rossi, Lewis Caunce, and Harry Robilliard. Matthew Reason, James Ransom, Tycarra Bianchin.
Funding
This research was funded by the Australian Research Council through its Industrial Transformation Linkage Program (grant IH210100014).
Author information
Authors and Affiliations
Contributions
JYL: conceptualization, sample collection & processing, experimental design, methodology development & execution, data & statistical analyses, manuscript – original draft & edits. YKY: data analysis, manuscript review. MC: sample collection, manuscript review. RK: acquired funding, provided hatchery resources and fish for study, manuscript review. DRJ: acquired funding & resources, manuscript review. DGB: conceptualization, acquired funding & resources, supervision, manuscript review.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lim, J.Y., Yeoh, Y.K., Canepa, M. et al. The early life microbiome of giant grouper (Epinephelus lanceolatus) larvae in a commercial hatchery is influenced by microorganisms in feed. anim microbiome 6, 51 (2024). https://doi.org/10.1186/s42523-024-00339-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s42523-024-00339-y