
Abstract
Alzheimer’s disease (AD) is a progressive neurological disorder that primarily impacts cognitive function. Currently there are no disease-modifying treatments to stop or slow its progression. Recent studies have found that several peripheral and systemic abnormalities are associated with AD, and our understanding of how these alterations contribute to AD is becoming more apparent. In this review, we focuse on amyloid‑beta (Aβ), a major hallmark of AD, summarizing recent findings on the source of brain-derived Aβ and discussing where and how the brain-derived Aβ is cleared in vivo. Based on these findings, we propose future strategies for AD prevention and treatment, from a novel perspective on Aβ metabolism.
Similar content being viewed by others

Background
Alzheimer’s disease (AD) is an age-related neurodegenerative disease that can lead to brain atrophy and neuronal death in the brain [1]. AD is the most common type of dementia. In the 2019 Global Status Report on the Public Health Response to Dementia, the World Health Organization (WHO) highlighted that approximately 55 million patients worldwide are suffering from AD. Without significant medical breakthroughs in the prevention, slowing, and treatment of AD, the number would reach 150 million by 2050. However, after nearly a century of research, effective preventive strategies and treatments for AD are still lacking. A deeper understanding of the pathogenesis of AD may facilitate development of therapeutic strategies for AD.
AD pathogenesis is complex, involving extracellular amyloid-beta (Aβ) protein plaques, intracellular tau, neurofibrillary tangles, cholinergic insufficiency, oxidative stress, mitochondrial dysfunction, inflammation, and hormonal imbalances [2]. Brain Aβ plaques are the major pathological hallmark of AD [3]. Hardy et al. [4] found that amyloid precursor protein (APP) gene mutations can lead to massive Aβ deposition in the brain. They further proposed the Aβ cascade hypothesis, supported by the molecular genetic data of early-onset familial AD. The hypothesis states that when the amount of Aβ generated is greater than the amount that is degraded, accumulation occurs, leading to plaque formation, tau tangle formation, neuronal death, and a cascade of neuroinflammatory responses [5]. The anti-Aβ monoclonal antibodies Aducanumab and Lecanemab have been approved by the United States Food and Drug Administration (FDA) for brain Aβ clearance.
Both human and mouse studies have demonstrated that high levels of Aβ can flow from the brain to the periphery, and physiological catabolism of brain-derived Aβ can occur in the peripheral system [6]. This provides a novel perspective for understanding the pathogenesis of AD and developing therapeutics. In this review, we focus on the systemic role of Aβ in AD, discussing the major source of brain Aβ and associations of brain-derived Aβ aggregates with inflammation in the brain. We also discuss the communications between peripheral and central pools of Aβ, the peripheral pathways of brain-derived Aβ clearance, and how systemic diseases might interfere with brain-derived Aβ clearance. Finally, we summarize therapeutic strategies targeting Aβ.
Where does brain-derived Aβ come from?
The generation and spread of Aβ
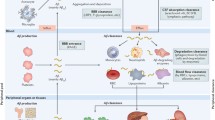
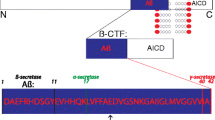
Aβ is a peptide of 38–43 amino acids generated from sequential cleavage of β-APP by β-secretase (BACE1) and γ-secretase [7]. BACE1 is an aspartyl protease that cleaves APP primarily at a single site, β-site1, whereas the γ-secretase complex cleaves the C-terminal fragment (CTF) at multiple sites, with preference for positions 40 and 42, forming the Aβ40 and Aβ42 peptides [8] (Fig. 1a). Previous studies have indicated that Aβ38 and Aβ3–40 are more amyloidogenic than other forms of Aβ, such as Aβ4–40 [8]. APP is a transmembrane protein widely expressed in brain neurons and various peripheral tissues, including blood (platelets) and peripheral organs (skin, intestines, and liver) [9, 10] (Fig. 1a). Alternative splicing of exons 7 and 8 of APP leads to generation of three major isoforms of APP, APP695, APP751, and APP770, comprising 695, 751 and 770 amino acids, respectively [11]. The APP695 isoform is predominantly expressed in neurons and lacks two exons. APP751 and APP770 possess a Kunitz-type serine protease inhibitor domain encoded by exon 7, while APP770 contains an additional immunoregulatory OX-2 antigen domain encoded by exon 8 in its extracellular region. Both APP751 and APP770 are expressed in both the brain and peripheral tissues [12, 13]. Therefore, AD is a disorder of both the central nervous system (CNS) and the peripheral system. Moreover, studies in animal models and cell cultures demonstrate that peripheral Aβ seeds accelerate plaque formation in a prion-like manner via templated seeding and intercellular propagation [14]. One study found that hepatogenic Aβ can enter the bloodstream through triglyceride-rich lipoproteins and further deposit in the brain to result in AD-like pathology, including neurodegeneration and brain atrophy [15]. Similarly, Aβ plaques have also been detected in the gastrointestinal tracts of AD patients and mice [16]. Additionally, when fluorescence-labeled Aβ is injected into the intestines of C57BL/6J mice, the fluorescence-labeled Aβ could spread to the hippocampus, resulting in brain disorder [17]. In another study, AD patient brain lysates were injected into the colons of wild-type (WT) mice, and induced AD-like pathology in the mice [18]. These studies suggest that Aβ in the gastrointestinal tract can also enter the brain parenchyma, resulting in Aβ deposition in the brain. However, it is still unclear how gastrointestinal tract Aβ is transmitted from the intestines to the brain and how Aβ deposition is induced.

The generation and clearing path of Aβ. a The source, synthesis, and clearance of Aβ under normal physiological condition. Aβ is produced by neurons, microglia and astrocytes in the brain, platelets in the blood, and the skin, intestine, and liver in the periphery. Some Aβ is released from the brain to the blood via the blood–brain barrier (BBB), the interstitial fluid (ISF) bulk flow or cerebrospinal fluid (CSF) leakage. Aβ can also be transported to peripheral organs or tissues by carriers, where it is degraded by macrophages or hepatocytes or excreted via the liver or kidney. Under normal physiological conditions, high-density lipoprotein (HDL) prevents Aβ vascular accumulation independently of its primary receptor, scavenger receptor class B type 1 (SR-B1). ApoE promotes Aβ clearance by activating phagocytosis and increasing Aβ migration in microglia, where ApoE4 has a reduced capacity to induce these phenotypes than ApoE3. b Under normal physiological conditions, there is a balance between Aβ production and clearance (healthy brain without Aβ deposits). Under pathophysiological conditions, there is a loss of the balance between Aβ production and clearance, leading to Aβ aggregation in the brain (AD brain with Aβ deposition)
Brain-derived Aβ can enter the peripheral blood through the blood–brain barrier (BBB) or cerebrospinal fluid (CSF) [9]. Given the bidirectional relationship between the brain and the periphery, the question arises concerning whether peripheral Aβ is the primary source of brain Aβ. Bu and colleagues used a parabiosis model between APPswe/PS1dE9 (APP/PS1) transgenic AD mice and their WT littermates, and found that the blood-derived Aβ can enter the brain and induce AD-like pathologies in WT mice, suggesting that the blood-derived Aβ may contribute to AD pathogenesis [19]. Lam and colleagues generated hepatocyte-specific human amyloid transgenic mice that produce pathogenic Aβ specifically in the liver. They found that the liver-derived Aβ travels through the circulatory system and crosses the BBB to enter the brain. The accumulation of Aβ in the brain drives the pathology associated with AD, including capillary dysfunction, inflammation and neurodegeneration [15]. Platelets are the primary source of Aβ in the peripheral circulation, and transplantation of bone marrow cells from APP/PS1 mice into WT mice leads to a continuous increase of human Aβ in the blood and Aβ deposition in the brain [20]. These findings suggest that AD is not simply a result of brain Aβ deposition.
Aβ aggregation
Under normal physiological conditions, brain Aβ production and clearance are maintained at a dynamic balance and confer neuroprotection [9]. However, this balance can be disrupted by excessive production or decreased clearance of Aβ. Microglia dysfunction, low expression of low-density lipoprotein receptor-related protein-1 (LRP-1), apolipoprotein E (ApoE) overexpression, and ApoE binding to LPR-1 in competition with Aβ, can affect the clearance of Aβ [21, 22] (Fig. 1b). In addition, Aβ (especially Aβ42) is a hydrophobic peptide that is prone to aggregation. When Aβ42 exceeds a certain level, its monomers would aggregate into soluble oligomers, fibrils, and even insoluble plaques [23]. The brain Aβ42 patches will continuously recruit Aβ42 monomers and oligomers to form larger plaques [24]. Aβ oligomers are considered as the main neurotoxic components, and there is a negative correlation between the degree of oligomer aggregation and the level of toxicity [25]. The Osaka and Arctic mutations in APP can increase Aβ hydrophobicity, enabling aggregate formation [25, 26]. Studies have shown that the E693Delta APP mutation produces an Aβ variant more resistant to proteolytic degradation and showing enhanced oligomerization but no fibrillization, which is more likely to induce early symptoms of AD, indicating that the level of Aβ aggregates is closely related to the progression of AD [27].
What harmful effects can brain-derived Aβ induce?
Brain-derived Aβ aggregates induce polarization of microglia
Microglia are the primary immune cells in the brain and mainly include M1 and M2 microglia. The M1 type represents the pro-inflammatory phenotype, and M2 represents the anti-inflammatory phenotype [28]. M2 microglia can exhibit both alternative activation and acquired deactivation, which are induced by interleukin (IL)-4 and IL-10, respectively. In the early stage of AD, M2 microglia phagocytose misfolded Aβ and exert a neuroprotective effect [29] (Fig. 2a). However, when the Aβ plaque enrichment exceeds the capacity of microglial phagocytosis, the microglia would shift from the M2 to the M1 type, releasing a large amount of pro-inflammatory factors such as IL-1β and IL-6 [30, 31]. These pro-inflammatory factors can aggravate neuroinflammation, lead to neuronal damage, and further promote the production, aggregation, and deposition of Aβ, thus forming a vicious cycle [31,32,33] (Fig. 2a). The M1 microglia also highly express inducible nitric oxide synthase (iNOS), while reducing iNOS in APP/PS1 mice can reduce Aβ plaques in the brain [34]. In AD pathology, both disease-associated microglia and down-regulation of homeostasis-associated genes are related to the upregulation of AD-related genes, including APOE, triggering receptors expressed on myeloid cells-2 receptor2 (TREM2) and TYROB (tyrosine protein–protein binding protein) [3]. TREM2 is only expressed in microglia in the CNS and is a positive regulator of phagocytosis [35]. TREM2 is a receptor for lipoproteins, and mediates clearance of lipoprotein-Aβ complexes by microglia [36]. TREM2 has a dual influence on AD pathogenesis. It can form a neuroprotective barrier around Aβ aggregates and prevent their spreading outward, resulting in dense Aβ plaques in the early stage of AD. On the other hand, TREM2 also promotes inflammation in the middle and the late stages of AD [31, 37, 38].

The hazards of brain Aβ aggregates. The diagram shows the dual functions of microglia. a Aβ aggregates promote polarization of microglia from M2 to M1 phenotype in the brain. The M1 microglia promote neuroinflammation through secretion of pro-inflammatory factors, leading to neuronal damage. b Aβ stimulates neuroinflammation, with involvement of nuclear factor-kappa B (NF-κB), endoplasmic reticulum (ER) stress, and the nucleotide-binding oligomerization domain-like receptor protein 3 (NLRP3) inflammasome pathway
In addition, the nuclear factor κB (NF-κB)/C3/C3aR signaling can regulate Aβ aggregation through astrocytes and microglia [35]. Brain Aβ aggregates activate the NF-κB signaling in astrocytes, which release the complement protein C3a [35]. The interaction of C3a with C3a receptors on microglia and neurons promotes Aβ aggregation, and blocking the interaction between C3a and microglia can reduce microglial activation and the Aβ load in the brain [39].
Brain-derived Aβ aggregates stimulate neuroinflammation
Neuroinflammation is mediated mainly by microglia and is one of the main causes of neuronal damage and necrosis [35]. Astrocytes also play an auxiliary role in neuroinflammation. Early misfolding and aggregation of Aβ in the brain can be cleared by binding to receptors on the surface of microglia, such as receptors for advanced glycation end product, Toll-like receptor (TLR), nucleotide-binding oligomerization domain-like receptor protein 3 (NLRP3), and formyl peptide receptor (Fig. 2b) [40]. Loss of TLR4 and NLRP3 inflammasome in mice can reduce Aβ accumulation and production of pro-inflammatory cytokines IL-1β and IL-18 [41]. Additionally, when NLRP3 is activated in response to damage-associated molecular patterns (DAMPs) or pathogen-associated molecular patterns (PAMPs), it recruits ASC (apoptosis-associated speck-like protein containing a caspase recruitment domain [CARD]) through PYD-PYD (Pyrin domain) interactions, leading to the formation of active caspase-1 through CARD-CARD interactions. NLRP3 activation exacerbates the accumulation of Aβ (Fig. 2b) [42], and Aβ aggregates would, in turn, promote the generation of IL, mitogen-activated protein kinase, NF-κB and reactive oxygen species (ROS), thus aggravating neuroinflammation [34, 43, 44]. ROS production has also been identified as a mediator of Aβ-induced endoplasmic reticulum (ER) stress and cytotoxicity [45]. Moreover, ER stress contributes to the progression of AD pathology [46] (Fig. 2b).
Neuroinflammation can also result in the engulfment of misfolded proteins such as Aβ by microglia and astrocytes [47]. Although controversy exists regarding the order of occurrence of Aβ aggregation and neuroinflammation, it is relatively clear that Aβ aggregation accelerates the development of neuroinflammation, which in turn promotes the production and aggregation of Aβ [48]. Neuroinflammation is a chronic response of the innate immune system, resulting in the failure of Aβ clearance.
How can brain-derived Aβ be cleared?
Aβ clearance strategies from the brain to the periphery
Lack of Aβ clearance is considered the main cause of Aβ deposition in patients with sporadic AD [49]. Therefore, accelerating the clearance of Aβ is a promising strategy for treating AD. Studies using the deep cervical lymph node ligation method in AD mice have revealed that brain lymphatic clearance is a physiological mechanism for Aβ transport from the brain to the periphery [50].
Brain-derived Aβ has been detected in the periphery of AD patients and AD mice. Aβ flows from the brain to the periphery mainly through the BBB pathway and the brain lymphatic pathway (Fig. 1a). In the BBB pathway, the capillary lengths in mouse and human brains are approximately 0.6 km and 650 km, respectively, which account for more than 85% of the total length of cerebral blood vessels, providing a large surface area for substance exchange between the blood and the brain [24, 51]. Brain-derived Aβ efflux is usually mediated by brain endothelial cell surface proteins, mainly LRP-1 [52]. Previous studies have suggested that approximately 40%–60% of brain-derived Aβ is cleared in the periphery. Aβ clearance via the BBB is reduced by approximately 30% in AD patients [53, 54]. The brain lymphatic pathways include the perivascular pathway, glymphatic system, meningeal lymphatic vessels, and olfactory/cervical lymphatic drainage, which drains Aβ in the CSF to the deep cervical lymph nodes, allowing Aβ and other macromolecules to drain from the brain to the periphery [27]. The exact contribution of each mechanism to the overall Aβ clearance remains unclear. Nevertheless, these pathways work together to remove pathological proteins from the brain to the periphery. These findings suggest that peripheral tissues and organs are physiologically involved in the clearance of brain-derived Aβ, including the blood, liver, kidney, spleen, and gut (Table 1).
Blood-mediated Aβ clearance
The peripheral sink hypothesis suggests that by increasing Aβ clearance from the blood, a concentration gradient is created, which favors the efflux of Aβ from the brain to the bloodstream, thus facilitating Aβ clearance [27, 55]. As blood components, secreted enzymes are critical for the catabolism of peripheral Aβ. These enzymes include insulin-degrading enzyme [56], neprilysin [57] and angiotensin-converting enzyme [58]. They have an affinity for specific domains of Aβ proteins and can cleave these peptides into shorter, more benign forms. Immune cells in the blood, such as monocytes and macrophages, can recognize Aβ and clear Aβ peptides from circulation. Supplementing blood components by means of bone marrow transplantation [55] or by administering bone marrow-derived cells [44] or albumin [59] can enhance the clearance of peripheral Aβ. In addition, peripheral Aβ can be cleared by blood exchange, including hemodialysis, peritoneal dialysis, and replacement of fresh blood (whole blood therapy) [60].
Liver-mediated Aβ clearance
The liver plays a crucial role in systemic metabolism and detoxification, including the clearance of various substances from the blood. A study found that approximately 13.9% of Aβ42 and 8.9% of Aβ40 are removed by the liver and that LRP-1 mediates the peripheral clearance of Aβ in the liver. LRP-1 overexpression in hepatocytes enhances hepatic Aβ clearance, decreases cerebral Aβ deposition and attenuates cognitive impairment in AD mice [61,62,63]. Moreover, self-protecting biomimetic nanozymes, such as CuxO@EM-K, have been designed to test the efficacy of therapies that can enhance the peripheral sink effect, which could potentially redirect Aβ from the brain to the liver for clearance [64].
Kidney-mediated Aβ clearance
The kidney is thought to be responsible for Aβ clearance by filtering Aβ from the blood to the urine. There is a positive association between chronic kidney disease and a risk of cognitive impairment [6, 65]. Both acute kidney ligation and unilateral nephrectomy aggravate Aβ burdening in the blood and brain, neuroinflammation, and neurodegeneration in APP/PS1 mice. Moreover, chronic furosemide treatment can decrease brain Aβ deposition and ameliorate cognitive deficits in AD mice [66].
Spleen-mediated Aβ clearance
The spleen acts as a blood filter and an immune organ. The mononuclear phagocyte system found primarily in the spleen, has been thought to contribute to the clearance (engulfing and digesting) of Aβ. Splenectomy reduces the monocyte-derived periplaque macrophages and the circulating myeloid cells, and worsens amyloid pathology in APP/PS1 mice [67]. In addition, Aβ levels in the splenic artery are higher than those in the splenic vein, suggesting Aβ clearance when blood flows through the spleen. Splenectomy aggravates AD-related behaviour deficits and pathology in AD mice [68]. Therapies aimed at enhancing Aβ clearance through the spleen and other peripheral organs are a topic of interest. However, further studies are required to comprehensively understand the role of the spleen in Aβ clearance, especially the precise interplay between the peripheral immune system and brain pathology.
Gut-mediated Aβ clearance
Patients with AD often experience an imbalance in gut microbiota, such as an increase in pro-inflammatory gut microbes (e.g., Shigella) and a decrease in anti-inflammatory gut microbes (e.g., Bifidobacterium) [69]. This imbalance leads to abnormal secretion of secondary metabolites of gut microbes, such as lipopolysaccharides (LPS) and short-chain fatty acids (SCFAs). LPS acts on the TLR4-CD14/TLR2 receptors on leukocytes and microglia, leading to NF-κB-mediated increases of cytokines, resulting in elevated Aβ levels, damage to oligodendrocytes, and finally neuronal damage in AD brains [70]. In contrast, SCFAs, which are produced mainly by Clostridium, Lactobacillus, and Bifidobacterium, can cross the BBB via monocarboxylate transporters [71]. They can accelerate the clearance of brain Aβ by regulating the proliferation and differentiation of microglia. They can also directly bind to Aβ to inhibit the formation of Aβ plaques [72, 73]. Therefore, supplementation with probiotics can decrease the brain Aβ burden and ameliorate cognitive impairment in AD mice [66] (Fig. 3). In addition, the gut microflora in 5 × FAD and APP/PS1 mice facilitate entry of peripheral immune cells into the brain, leading to microglial activation and disease progression. Treatment with GV-971 in these mice can restore the gut microbial profile, ameliorate brain immune cell infiltration and inflammation [74], reduce brain Aβ burden and tau hyperphosphorylation, and improve cognitive function [74]. These studies demonstrated that the brain–microbiota axis is involved in the pathogenesis of AD, and it is speculated that the gut has the potential to regulate peripheral Aβ clearance [75, 76].

The secondary metabolite lipopolysaccharides (LPS) from the gut microbiota, can affect brain Aβ via the gut-brain axis, causing neuronal damage and local inflammation, and promoting local and systemic Aβ deposition. Short-chain fatty acids (SCFAs) from the gut microbiota can enter the bloodstream and maintain BBB homeostasis, inhibiting Aβ deposition.
Representative strategies for targeting Aβ clearance
In the past two decades, the development of AD drugs was particularly challenging. Most AD trials failed to demonstrate efficacy. However, confidence in the potential of AD drug development has not diminished [2]. In the Alzforum database, a total of 388 drugs in development for AD treatment are selected as eligible for scoring. These 388 drugs are categorized into eight therapy types and nine target classes. The largest number of drugs target Aβ (82, 21.19%), followed by those targeting inflammation (50, 12.92%), neurotransmitters and receptors (55, 14.21%), and tau (31, 8.01%) (Fig. 4a). At present, only 2 of the 82 Aβ-targeting drugs have been approved by FDA, with other 39 drugs undergoing clinical trials, 34 drugs having been discontinued, and 7 being inactive (Fig. 4a). Regarding the AD therapy type, most are classified as small-molecule drugs (222, 64.81%) or passive immunotherapy (44, 12.90%) (Fig. 4b). Among the 222 small-molecule drugs, seven have been approved by the FDA: galantamine, memantine, rivastigmine, suvorexant, brexpiprazole, donepezil, and tacrine. A further 110 drugs are undergoing clinical trials, 80 drugs have been discontinued or rejected, and only 24 are inactive. Moreover, among the 44 passive immunotherapy drugs, two have been approved by the FDA, i.e., Aducanumab and Lecanemab, for use as Aβ-clearing drugs, and 24 drugs are undergoing clinical trials (Fig. 4b). Therefore, Aβ-targeting drugs are an important focus of research attention. In the following, we will elaborate on small-molecule drugs and passive immunotherapy.

Therapeutic targets and therapy types for AD (https://www.alzforum.org/therapeutics. Accessed March 28, 2024). a There are 388 drugs in development for AD. They are categorized into nine target classes. Of the 82 drugs targeting Aβ, only 39 are under clinical validation and 2 have obtained FDA approval. b The 388 drugs are categorized into eight therapy types. Among them, 118 small-molecule drugs and 27 passive immunotherapy drugs are in clinical validation or have obtained FDA approval
Small molecules for Aβ clearance
When considering small molecules as an Aβ clearance strategy in AD, the goal is to reduce Aβ peptide accumulation in the brain (Table 2).
Secretory enzyme regulators
The regulatory control of enzymes involved in Aβ secretion, such as BACE and γ-secretase, is an essential area for AD drug development. Drugs that regulate Aβ secretion include the β-secretase inhibitor Lanabecestat [77, 78] and the γ-secretase inhibitor Semagacestat [62], which can inhibit the BACE1 enzyme and the γ-secretase enzyme complex, respectively, further reducing Aβ production [62]. However, these drugs were discontinued in clinical trials because of ineffectiveness or liver toxicity. Several experimental small-molecule inhibitors and modulators targeting secretase enzymes and Aβ aggregation are under clinical trials or preclinical studies (Table 2). While these therapies offer promise, their development is complex, and identifying compounds that can effectively and safely regulate Aβ secretion remains a significant challenge.
Aβ aggregation inhibitors
Aβ aggregation inhibitors encompass many chemical structures, including small molecules and peptides that can bind to Aβ and interfere with its aggregation process. These compounds are designed to target specific regions of Aβ, such as the hydrophobic core involved in aggregation, to inhibit the formation of toxic Aβ oligomers and fibrils. The main Aβ small-molecule inhibitors that have entered clinical trials are 3-APS (III) [79] and 8-HQ (II/III) [80], both of which were discontinued because they did not consistently inhibit Aβ aggregation in the brain to improve cognitive performance in AD patients [81]. In contrast, ALZ-801 has shown good gastrointestinal absorption and stable plasma concentrations, with significant clinical effects in the APOE4/4 high-risk population and dose-dependent preservation of the hippocampal volume [82]. Developing effective Aβ aggregation inhibitors is facing substantial challenges, including BBB penetration, target selectivity, and metabolic stability. Moreover, the potential off-target effects and the safety are crucial issues to be addressed.
Intestinal microbiota regulators
In the process of AD, the imbalance of gut microbiota leads to an abnormal increase in phenylalanine and isoleucine in peripheral blood, which in turn induces the differentiation and proliferation of peripheral pro-inflammatory Th1 cells and promotes their brain invasion [74]. Th1 cells infiltrating the brain and the intrinsic M1 microglia in the brain are activated together, leading to AD-related neuroinflammation [74]. Probiotics, prebiotics, synbiotics, and dietary interventions represent potential regulators of the intestinal microbiota, and their effects on Aβ metabolism and clearance have been investigated [83]. These interventions aim to modulate the composition and activity of the gut microbiota to promote a healthy microbial community and enhance Aβ clearance. However, perhaps because of the widespread use of oral probiotics as supplements, no probiotics are currently in clinical trials. Moreover, the strong acids and bile salts in the gastrointestinal tract may induce leakage of ions and other cellular components that may kill probiotics, resulting in their low efficacy or even ineffectiveness [73]. This poses a challenge to developing methods to effectively transport probiotics around the gastrointestinal barrier to the intestine so that they can exert their biological functions.
Passive immunotherapy for Aβ clearance
Anti-Aβ immunity includes active immunity and passive immunity. Active immunity strategies involve stimulating the immune system to produce anti-Aβ antibodies by administering Aβ or its fragments. This approach has the advantages of long duration and low cost, but adverse immune reactions are difficult to predict [84]. For example, the Aβ vaccines UB-311 and ACC-001 have been discontinued because they led to meningoencephalitis in AD patients [85, 86].
Passive immunotherapy accelerates Aβ clearance by peripheral injection of humanized anti-Aβ monoclonal immunoglobulins. Aβ passive immunotherapy mainly promotes Aβ clearance in the following three ways [87]. (1) The antibody directly binds to Aβ in peripheral blood to reduce the Aβ content in the blood. This leads to an imbalance of Aβ in the periphery and the brain, promotes the outflow of soluble Aβ from the CNS via LRP-1 expressed on the BBB, and indirectly reduces the Aβ load in the brain [88, 89]. (2) The antibody Fc segment binds to the Fc receptor on microglia, which stimulates microglial activation, promoting Aβ clearance [90, 91]. (3) The antibody acts directly on Aβ plaques, fibers, or oligomers in the brain after crossing the BBB, and then degrades and clears Aβ [92].
Targeting soluble Aβ
The aim of targeting soluble Aβ with antibodies is to prevent the aggregation of Aβ peptides into larger, more toxic oligomeric and fibrillar forms. By retaining Aβ in a soluble state, these antibodies may mitigate Aβ-induced neurotoxicity and reduce the formation of Aβ plaques. Several monoclonal antibodies targeting soluble Aβ have been developed and investigated in clinical trials. These antibodies are engineered to specifically recognize and bind to soluble Aβ peptides while avoiding binding to insoluble aggregates. The potential of these antibodies to slow the progression of AD by promoting Aβ clearance has been evaluated in clinical trials (Table 3). Solanezumab is a humanized monoclonal immunoglobulin G (IgG) antibody that recognizes the intermediate domain of Aβ peptides and can specifically recognize soluble Aβ monomers and prevent the formation of Aβ aggregates [93]. A clinical phase III study found that Solanezumab did not improve the cognition of AD patients using the Alzheimer's Disease Assessment Scale (ADAS), which can be used to evaluate the severity of dementia [94]. Lecanemab is a humanized IgG1 form of a mouse monoclonal antibody (mAb), mAb158, that can selectively bind large, soluble Aβ protofibrils and promote their clearance [92]. The results of the latest phase III clinical trial (1795 patients) also showed that compared with the placebo-treated group, the Clinical Dementia Rating Scale (CDR) sum of boxes scores of AD patients treated with Lecanemab for 18 months decreased by 27%, the decline in cognitive level decreased by 26%, and the decline in daily activity function decreased by 36% [95, 96]. Moreover, a significant decrease in Aβ deposition was observed in the brains of AD patients treated with Lecanemab.
Targeting insoluble Aβ
Monoclonal antibodies that target insoluble Aβ have been developed to specifically recognize and bind to insoluble Aβ aggregates. These antibodies are designed to facilitate the clearance of existing plaques through mechanisms such as opsonization and activation of microglia-mediated phagocytosis. These approaches seek to reduce the neurotoxic effects associated with Aβ aggregation and improve cognitive function in individuals with AD. Several monoclonal antibodies targeting insoluble Aβ have been investigated in clinical trials (Table 3). These trials have assessed the safety and efficacy of immunotherapies to clear existing amyloid plaques from the brains of individuals with AD. Gantenerumab is a humanized anti-Aβ monoclonal antibody that recognizes Aβ fibers [97]. Roche currently has a phase I/II trial ongoing with a redesigned version of gantenerumab called trontinemab. Trontinemab is gantenerumab linked to Roche’s “brain shuttle” technology to enhance brain delivery via the TfR. Clinical trial results show that Trontinemab is safe at high doses [98]. At present, Trontinemab is in a Phase I trial that began evaluating multiple doses in 120 people with prodromal or mild-to-moderate AD and a positive amyloid positron emission tomography (PET) scan. Aducanumab is a high-affinity human monoclonal antibody against Aβ that selectively binds to Aβ aggregates and does not bind to Aβ monomers [84]. After intravenous injection, Aducanumab preferentially binds to Aβ in the peripheral blood and brain parenchyma rather than to Aβ on the vascular walls [92]. In a phase III clinical trial, the AD patients treated with Aducanumab showed a 22% reduction in CDR-SB score decline fom baseline compared with the placebo group in the 78th week [99].
The binding strength of Lecanemab to 75–300 kDa Aβ fibers was 100 times higher than that of Aducanumab, and the binding strength to 300–500 kDa Aβ fibers was 25 times higher than that of Aducanumab [100]. The clinical cognitive impairment scores of patients taking Lecanemab (27%) were 4% higher than those of individuals taking Aducanumab (23%) [100].
Challenges in clinical trials for passive immunity
BBB penetration
For antibodies that are used to specifically clear Aβ, only 0.1% of them can enter the brain after intravenous injection [101,102,103]. The poor penetration of the BBB by anti-Aβ antibodies results in an antibody level in the brain lower than the concentration needed to continuously inhibit the formation of Aβ aggregates or effectively degrade and clear Aβ. Therefore, improved efficiency of antibody entry in the brain is required to enhance Aβ clearance. Currently, there are three main methods for delivering brain-targeted drugs (Fig. 5a, b). (1) Bypassing the BBB to deliver drugs to the brain, which mainly includes invasive and non-invasive administration. The invasive routes of administration (intraparenchyma administration, convection-enhanced delivery, and intrathecal administration) are performed primarily using specific devices that directly penetrate the skull or lumbar spine [104]. The non-invasive administration is typically performed directly via nasal administration. (2) Enhancing the permeability of the BBB. This method mainly includes high heat [105], high permeability, and focused ultrasound [106]. However, when the BBB is opened, the effect is local, temporary, and non-invasive, and the degree of BBB opening and the amount of medication administered are challenging to evaluate. (3) Crossing the BBB. Receptor-mediated transcytosis is one of the most common strategies for BBB crossing, through TfR, insulin receptors (IR), and LRP-1. The anti-TfR antibody 8D3 is the most commonly used method [107,108,109].

Strategies and shortcomings of passive immunotherapy. a The drugs are mainly enriched in the brain by crossing the BBB through adsorptive-mediated transcytosis (AMT), transporter-mediated transcytosis (TMT), and receptor-mediated transcytosis (RMT). b Methods of drug administration to improve drug entry in the brain. The methods include nasal delivery, convection-enhanced delivery (CED), magnetic resonance-guided focused ultrasound (MRs FUS), intrathecal injection, and intravenous (IV) injection. c Most antibodies induce amyloid-related imaging abnormalities in clinical trials. d Antibody-mediated type III hypersensitivity reactions. e Intravenous injection of antibodies or other drugs via liver metabolism
Low complement response
Although antibodies are well tolerated in vivo, they may also be recognized as a foreign component by the recipient, thus causing an adverse immune response. The adverse reactions caused by peripheral injection of antibodies can be classified into hypersensitivity types I, II, and III according to their pathogenesis [110]. Among them, the type I hypersensitivity is mostly mediated by immunoglobulin E (IgE) antibodies, which react quickly and usually cause physiological function disruption [111]. The type II hypersensitivity is mainly caused by IgG or IgM, which causes hemolysis with the participation of macrophages, natural killer cells, and the complement system [111]. The type III hypersensitivity is mainly caused by the deposition of soluble immune complexes formed by antibodies and antigens in blood vessels, which causes local necrosis, tissue hyperemia, and edema by activating the complement system [111, 112] (Fig. 5d).
Amyloid-related imaging abnormalities (ARIA), including ARIA-edema/effusion (ARIA-E) and ARIA-hemosiderosis/microhemorrhages (ARIA-H), may occur, often during early anti-Aβ monoclonal antibody treatment [113]. In the phase III study of Aducanumab in AD patients, ARIA occurred in 425 (41.3%) of 1029 patients in the 10 mg/kg group [114]. Similarly, in the phase III clinical trial of Lecanemab in AD patients, 12.6% of AD patients developed ARIA, and 17.3% of AD patients had intracranial hemorrhage [114] (Fig. 5c). ARIA is caused by a classical complement reaction activated by antibodies. Therefore, the antibody-mediated adverse immune response is a critical factor that limits the efficacy of passive immunotherapy.
High liver enrichment
Recent studies have shown that AD is associated with abnormal liver function [115]. Anti-Aβ antibodies and antibody-modified nanoparticles are enriched by approximately 33% in the liver after intravenous injection, which will mediate adverse immune reactions and increase the liver burden [116] (Fig. 5e). Moreover, neurotoxic Aβ in the liver also induces and accelerates the pathogenesis of AD [117]. Therefore, in AD immunotherapy, when clearing brain Aβ, reducing the non-specific enrichment of antibodies in the liver is an urgent issue to be addressed [118].
Lower specificity
In AD, Aβ plaques in the brain continuously recruit Aβ monomers and oligomers to form larger plaques [24, 119]. Aβ oligomers are considered to be the main form of neurotoxicity, and Aβ toxicity is negatively correlated with the degree of aggregation [25]. Therefore, an excellent anti-Aβ antibody should have high specificity for soluble Aβ and exhibit strong and continuous inhibition and depolymerization of Aβ aggregates.
Systemic diseases
AD may be a heterogeneous disorder with involvement of biological and psychosocial factors, including immune system dysfunction, hepatic dysfunction, renal insufficiency and diabetes mellitus [120]. First, the immune system dysfunction is considered the most crucial pathological factor in AD. Microglia and macrophages express the class A1 scavenger receptors (Scara1) to bind and phagocytose fibrillar Aβ aggregates. Mononuclear phagocytes also express several Aβ-degrading enzymes, such as insulin-degrading enzyme and neprilysin. In AD, the expression of these phagocytic Aβ receptors and Aβ-degrading enzymes decreases significantly in microglia, resulting in reduced Aβ phagocytosis [121, 122]. In addition, chronic systemic inflammation, such as rheumatoid arthritis and periodontitis, can accelerate the development of AD [123]. Second, the liver is the body’s most crucial organ for protein synthesis and metabolism. Peripheral Aβ is degraded directly in the hepatocytes or indirectly via liver-mediated albumin and Aβ-associated lipoproteins [61]. Studies have shown that the elevated aspartate transaminase/alanine transaminase (ALT) ratio and lower ALT levels are associated with AD pathology (Aβ, Tau) in the brain and poor cognitive performance in participants [124]. Therefore, liver dysfunction is closely related to the pathogenesis of AD. In addition, ApoE is synthesized and secreted in the liver, which regulates Aβ clearance by transport across the BBB, enzymatic degradation, and other pathways [125]. Third, the kidney plays a critical role in the peripheral clearance of Aβ, and soluble Aβ is a normal component of human urine [126]. Clinical studies have shown that patients with chronic kidney disease are more susceptible to cognitive impairment and abnormal deposition of Aβ in the brain [66]. Fourth, diabetes affects the development of AD probably through the disruption of Aβ metabolism in the brain and the periphery [127]. In people with diabetes, excess insulin inhibits IDE-mediated Aβ degradation. In addition, Aβ clearance is also impaired by other mechanisms such as oxidative stress, BBB dysfunction, activation of inflammatory pathways, and hypercholesterolemia [128,129,130].
Conclusion and perspectives
Insoluble Aβ plaques act as a reservoir for soluble Aβ, and the potential removal of insoluble Aβ from the brain could offer several advantages by eliminating all toxic forms of Aβ (oligomers and fibrils) compared to the removal of soluble Aβ. Currently, passive immunotherapy with intravenous monoclonal anti-Αβ antibodies is a promising strategy for AD treatment to remove neurotoxic insoluble Aβ from the brain via the peripheral system. However, this method faces challenges such as antibody-mediated adverse immune responses, limited efficiency in brain penetration, and significant non-specific accumulation in the liver.
The fundamental goal of AD research is to stop and eventually cure the disease. A better understanding of the pathophysiology of AD is critical for AD treatment. Aβ peptides are metabolized (anabolized and catabolized) in both the brain and peripheral tissues. Significantly, abnormal metabolism of Aβ peptides which could communicate bidirectionally between the two regions, could cause both central and systemic abnormalities, which in turn can generate feedback loops. Indeed, this close interaction between the brain and periphery, particularly about Aβ peptide metabolism, offers new insights into the pathogenesis of AD. Therefore, AD can be considered both a disease of the brain and a systemic disease. Understanding the pathophysiology of AD beyond the CNS is essential for the use of different therapeutic approaches or multi-target therapies, learning from unsuccessful clinical trials with Aβ and non-amyloid-based approaches (metabolic and anti-tau therapies). However, clarifying how peripheral processes influence AD pathogenesis, determining the interactions between the brain and the peripheral systems during AD progression, and investigating plasma Aβ as a blood-based biomarker for AD diagnosis, remain challenging. Future studies should explicitly consider systemic therapeutic strategies for prevention, including (1) improving peripheral Aβ peptide clearance (enhancing phagocytosis, proteolytic degradation, and excretion), (2) identifying and managing systemic abnormalities or developing a comprehensive strategy that targets both brain and peripheral abnormalities, and (3) developing rejuvenation factors/Aβ-peptide sequestrants in the blood for systemic rejuvenation therapies. In summary, this review provides new insights into the pathogenesis of AD and potential diagnostic and therapeutic advances.
AD still faces three significant challenges (Fig. 6). First, the etiology of the disease remains unclear and hinders scientific prevention efforts. Second, there is a need for more simple and reliable methods for early diagnosis. Current clinical diagnosis relies heavily on neuropsychological tests and imaging techniques. Due to the low accuracy of cognitive assessments and the high cost of brain imaging, most AD patients receive a diagnosis at late stages. Third, the lack of effective therapeutic drugs complicates treatment and rehabilitation. Early prevention and treatment are crucial for comprehensive improvement of AD.

The prospects of AD prevention and therapy. Challenges remain in understanding AD etiology and pathologic mechanisms, finding early biomarkers, and developing early diagnostic techniques and effective drugs. Promising research directions for AD treatment include model establishing, drug discovery and delivery, and physical stimulation. Preventive strategies focus on lifestyle modifications, including improving the living environment, playing puzzle games, promoting communication with others, maintaining a healthy diet, and physical exercise
In terms of prevention, research has shown that specific strategies can reduce the risk or delay the onset of the disease (Fig. 6). (1) Physical activity can improve blood flow to the brain and promote overall well-being [131]. (2) A healthy diet rich in fruits, vegetables, whole grains, lean proteins, and healthy fats is recommended. Some studies suggest that following the Mediterranean diet can positively impact brain health [111]. (3) Mentally stimulating activities such as playing games, reading, learning new skills, or practicing hobbies can keep the brain active and improve cognitive health. A large-scale clinical trial, the Advanced Cognitive Training for Independent and Vital Elderly (ACTIVE) study, has shown that cognitive training improves the cognitive performance of healthy older adults aged 65 and over [132]. (4) Managing chronic conditions like high blood pressure, diabetes, obesity, and high cholesterol can help prevent or delay AD [133]. (5) Exposure to polluted air, especially PM2.5, can lead to inflammation, oxidative stress, and damage to blood vessels, which can affect brain health and potentially contribute to neurodegenerative diseases [134, 135]. Therefore, limiting exposure to polluted air can reduce the risk of AD.
Although currently there is no cure for AD, there are several therapeutic strategies and interventions aimed at alleviating symptoms and improving the general well-being of patients (Fig. 6). Among them, FDA-approved cholinesterase inhibitors and anti-Aβ antibodies are effective in treating emotional and cognitive symptoms in patients, although they may not halt or reverse the progression of AD. There is ongoing research on the use of mRNAs to stimulate specific proteins or molecules in the brain, using lipid nanoparticles to encapsulate mRNA molecules and deliver them into cells to produce target proteins [136]. Drug delivery methods are critical for treatment efficacy. Researchers are investigating nanotechnology to develop nanoparticles that can cross the BBB and deliver drugs directly to the brain. These nanoparticles can improve drug solubility and stability and increase brain uptake [137]. In addition, light [138], sound [139], and gamma wave [140] are increasingly used as non-invasive therapies to improve cognitive function. The efficacy and safety of these therapies for AD need to be thoroughly investigated in well-designed clinical trials. In addition, research is underway to develop various models to understand, diagnose, and treat AD, using biological, animal, cellular, and computational approaches. Each model has its strengths and weaknesses [141] (Fig. 6). Much work is being done to characterize animal models of AD, from rodents to primates, to improve our understanding of the pathophysiology of the disease and to find models suitable for studying potential treatments.
Availability of data and materials
Not applicable.
Abbreviations
- AD:
-
Alzheimer’s disease
- Aβ:
-
Amyloid-beta
- APP:
-
Amyloid precursor protein
- CNS:
-
Central nervous system
- BBB:
-
Blood–brain barrier
- CSF:
-
Cerebrospinal fluid
- LRP-1:
-
Low density lipoprotein receptor related protein-1
- ApoE:
-
Apolipoprotein E
- IL-1β:
-
Interleukin-1β
- IL-6:
-
Interleukin-6
- iNOS:
-
Inducible nitric oxide synthase
- TREM2:
-
Triggering receptor expressed on myeloid cells-2 receptor2
- Tyrob:
-
Tyrosine protein–protein binding
- NF-κB:
-
Nuclear factor κB
- NLRP3:
-
Nucleotide binding oligomerization domain-like receptor protein 3
- ROS:
-
Reactive oxygen species
- TLR4:
-
Toll-like receptor 4
- TfR:
-
Transferrin receptors
- ARIA:
-
Amyloid related imaging abnormalities
References
Yiannopoulou KG, Papageorgiou SG. Current and future treatments in Alzheimer disease: an update. J Cent Nerv Syst Dis. 2020;12:1–12.
Liu N, Liang X, Chen Y, Xie L. Recent trends in treatment strategies for Alzheimer’s disease and the challenges: a topical advancement. Ageing Res Rev. 2024;94:1–12.
Long JM, Holtzman DM. Alzheimer disease: an update on pathobiology and treatment strategies. Cell. 2019;179:312–39.
Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256:184–9.
Gulisano W, Maugeri D, Baltrons MA, Fa M, Amato A, Palmeri A, et al. Role of amyloid-beta and tau proteins in Alzheimer’s disease: Confuting the amyloid cascade. J Alzheimers Dis. 2018;64:611–31.
Xiang Y, Bu XL, Liu YH, Zhu C, Shen LL, Jiao SS, et al. Physiological amyloid-beta clearance in the periphery and its therapeutic potential for Alzheimer’s disease. Acta Neuropathol. 2015;130:487–99.
Kent SA, Spires-Jones TL, Durrant CS. The physiological roles of tau and Abeta: implications for Alzheimer’s disease pathology and therapeutics. Acta Neuropathol. 2020;140:417–47.
Li N, Liu K, Qiu Y, Ren Z, Dai R, Deng Y, et al. Effect of presenilin mutations on APP cleavage; insights into the pathogenesis of FAD. Front Aging Neurosci. 2016;8:51.
Wang J, Gu BJ, Masters CL, Wang YJ. A systemic view of Alzheimer disease—insights from amyloid-beta metabolism beyond the brain. Nat Rev Neurol. 2017;13:612–23.
Puig KL, Swigost AJ, Zhou X, Sens MA, Combs CK. Amyloid precursor protein expression modulates intestine immune phenotype. J Neuroimmune Pharmacol. 2012;7:215–45.
D’uscio LV, He T, Katusic ZS. Expression and processing of amyloid precursor protein in vascular endothelium. Physiology (Bethesda). 2017;32:20–32.
Ponte P, Gonzalez-Dewhitt P, Schilling J, Miller J, Hsu D, Greenberg B, et al. A new A4 amyloid mRNA contains a domain homologous to serine proteinase inhibitors. Nature. 1988;331:525–7.
Zheng H, Koo EH. The amyloid precursor protein: beyond amyloid. Mol Neurodegener. 2006;1:5–16.
Ruiz-Riquelme A, Lau HHC, Stuart E, Goczi AN, Wang Z, Schmitt-Ulms G, et al. Prion-like propagation of beta-amyloid aggregates in the absence of APP overexpression. Acta Neuropathol Commun. 2018;6:26–42.
Kauwe G, Tracy TE. Amyloid beta emerges from below the neck to disable the brain. PLoS Biol. 2021;19:1–4.
Harris JC, Zhang Q, Tondon R, Alipour Z, Stashek K. Characterization of amyloidosis in the gastrointestinal tract with an emphasis on histologically distinct interstitial patterns of deposition and misinterpretations. Am J Surg Pathol. 2024;48:302–8.
Sun Y, Sommerville NR, Liu JYH, Ngan MP, Poon D, Ponomarev ED, et al. Intra-gastrointestinal amyloid-beta1-42 oligomers perturb enteric function and induce Alzheimer’s disease pathology. J Physiol. 2020;598:4209–23.
Chen C, Zhou Y, Wang H, Alam A, Kang SS, Ahn EH, et al. Gut inflammation triggers C/EBPbeta/delta-secretase-dependent gut-to-brain propagation of Abeta and tau fibrils in Alzheimer’s disease. EMBO J. 2021;40:1–21.
Bu XL, Xiang Y, Jin WS, Wang J, Shen LL, Huang ZL, et al. Blood-derived amyloid-beta protein induces Alzheimer’s disease pathologies. Mol Psychiatry. 2018;23:1948–56.
Sun HL, Chen SH, Yu ZY, Cheng Y, Tian DY, Fan DY, et al. Blood cell-produced amyloid-beta induces cerebral Alzheimer-type pathologies and behavioral deficits. Mol Psychiatry. 2021;26:5568–77.
Verghese PB, Castellano JM, Garai K, Wang Y, Jiang H, Shah A, et al. ApoE influences amyloid-beta (Abeta) clearance despite minimal apoE/Abeta association in physiological conditions. Proc Natl Acad Sci U S A. 2013;110:E1807–16.
Ma Q, Zhao Z, Sagare AP, Wu Y, Wang M, Owens NC, et al. Blood-brain barrier-associated pericytes internalize and clear aggregated amyloid-beta42 by LRP1-dependent apolipoprotein E isoform-specific mechanism. Mol Neurodegener. 2018;13:57.
Chen GF, Xu TH, Yan Y, Zhou YR, Jiang Y, Melcher K, et al. Amyloid beta: structure, biology and structure-based therapeutic development. Acta Pharmacol Sin. 2017;38:1205–35.
Montagne A, Zhao Z, Zlokovic BV. Alzheimer’s disease: a matter of blood-brain barrier dysfunction? J Exp Med. 2017;214:3151–69.
Sengupta U, Nilson AN, Kayed R. The Role of Amyloid-beta oligomers in toxicity, propagation, and immunotherapy. EBioMedicine. 2016;6:42–9.
Tomiyama T, Nagata T, Shimada H, Teraoka R, Fukushima A, Kanemitsu H, et al. A new amyloid beta variant favoring oligomerization in Alzheimer’s-type dementia. Ann Neurol. 2008;63:377–87.
Cheng Y, Tian DY, Wang YJ. Peripheral clearance of brain-derived Abeta in Alzheimer’s disease: pathophysiology and therapeutic perspectives. Transl Neurodegener. 2020;9:16–27.
Tang Y, Le W. Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol Neurobiol. 2016;53:1181–94.
Takata K, Kitamura Y, Saeki M, Terada M, Kagitani S, Kitamura R, et al. Galantamine-induced amyloid-beta clearance mediated via stimulation of microglial nicotinic acetylcholine receptors. J Biol Chem. 2010;285:40180–91.
Sim TM, Tarini D, Dheen ST, Bay BH, Srinivasan DK. Nanoparticle-based technology approaches to the management of neurological disorders. Int J Mol Sci. 2020;21:6070–100.
Leng F, Edison P. Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here? Nat Rev Neurol. 2021;17:157–72.
Cai Z, Hussain MD, Yan LJ. Microglia, neuroinflammation, and beta-amyloid protein in Alzheimer’s disease. Int J Neurosci. 2014;124:307–21.
Kinney JW, Bemiller SM, Murtishaw AS, Leisgang AM, Salazar AM, Lamb BT. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement. 2018;4:575–90.
Nathan C, Calingasan N, Nezezon J, Ding A, Lucia MS, La Perle K, et al. Protection from Alzheimer’s-like disease in the mouse by genetic ablation of inducible nitric oxide synthase. J Exp Med. 2005;202:1163–72.
Singh D. Astrocytic and microglial cells as the modulators of neuroinflammation in Alzheimer’s disease. J Neuroinflamm. 2022;19:206–21.
Song W, Hooli B, Mullin K, Jin SC, Cella M, Ulland TK, et al. Alzheimer’s disease-associated TREM2 variants exhibit either decreased or increased ligand-dependent activation. Alzheimers Dement. 2017;13:381–7.
Jay TR, Miller CM, Cheng PJ, Graham LC, Bemiller S, Broihier ML, et al. TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer’s disease mouse models. J Exp Med. 2015;212:287–95.
Jay TR, Hirsch AM, Broihier ML, Miller CM, Neilson LE, Ransohoff RM, et al. Disease progression-dependent effects of TREM2 deficiency in a mouse model of Alzheimer’s disease. J Neurosci. 2017;37:637–47.
Lian H, Yang L, Cole A, Sun L, Chiang AC, Fowler SW, et al. NFkappaB-activated astroglial release of complement C3 compromises neuronal morphology and function associated with Alzheimer’s disease. Neuron. 2015;85:101–15.
Zhang W, Xiao D, Mao Q, Xia H. Role of neuroinflammation in neurodegeneration development. Signal Transduct Target Ther. 2023;8:267–99.
Fiebich BL, Batista CRA, Saliba SW, Yousif NM, De Oliveira ACP. Role of microglia TLRs in neurodegeneration. Front Cell Neurosci. 2018;12:329–39.
Dominic A, Le NT, Takahashi M. Loop between NLRP3 inflammasome and reactive oxygen species. Antioxid Redox Signal. 2022;36:784–96.
Wang WY, Tan MS, Yu JT, Tan L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann Transl Med. 2015;3:136–51.
Malm TM, Koistinaho M, Parepalo M, Vatanen T, Ooka A, Karlsson S, et al. Bone-marrow-derived cells contribute to the recruitment of microglial cells in response to beta-amyloid deposition in APP/PS1 double transgenic Alzheimer mice. Neurobiol Dis. 2005;18:134–42.
Hashimoto S, Saido TC. Critical review: involvement of endoplasmic reticulum stress in the aetiology of Alzheimer’s disease. Open Biol. 2018;8:1–15.
Barbero-Camps E, Fernandez A, Baulies A, Martinez L, Fernandez-Checa JC, Colell A. Endoplasmic reticulum stress mediates amyloid beta neurotoxicity via mitochondrial cholesterol trafficking. Am J Pathol. 2014;184:2066–81.
Wyss-Coray T, Mucke L. Inflammation in neurodegenerative disease-a double-edged sword. Neuron. 2002;35:419–32.
Forloni G, Balducci C. Alzheimer’s disease, oligomers, and inflammation. J Alzheimers Dis. 2018;62:1261–76.
Soria Lopez JA, Gonzalez HM, Leger GC. Alzheimer’s disease. Handb Clin Neurol. 2019;167:231–55.
Wang L, Zhang Y, Zhao Y, Marshall C, Wu T, Xiao M. Deep cervical lymph node ligation aggravates AD-like pathology of APP/PS1 mice. Brain Pathol. 2019;29:176–92.
Bell RD, Sagare AP, Friedman AE, Bedi GS, Holtzman DM, Deane R, et al. Transport pathways for clearance of human Alzheimer’s amyloid beta-peptide and apolipoproteins E and J in the mouse central nervous system. J Cerebr Blood F Met. 2007;27:909–18.
Deane R, Wu Z, Zlokovic BV. RAGE (yin) versus LRP (yang) balance regulates Alzheimer amyloid beta-peptide clearance through transport across the blood-brain barrier. Stroke. 2004;35:2628–31.
Da Mesquita S, Louveau A, Vaccari A, Smirnov I, Cornelison RC, Kingsmore KM, et al. Functional aspects of meningeal lymphatics in ageing and Alzheimer’s disease. Nature. 2018;560:185–91.
Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, et al. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science. 2010;330:1774–8.
Das MM, Godoy M, Chen S, Moser VA, Avalos P, Roxas KM, et al. Young bone marrow transplantation preserves learning and memory in old mice. Commu Biol. 2019;2:73–83.
Miller BC, Eckman EA, Sambamurti K, Dobbs N, Chow KM, Eckman CB, et al. Amyloid-beta peptide levels in brain are inversely correlated with insulysin activity levels in vivo. Proc Natl Acad Sci U S A. 2003;100:6221–7.
Huang SM, Mouri A, Kokubo H, Nakajima R, Suemoto T, Higuchi M, et al. Neprilysin-sensitive synapse-associated amyloid-beta peptide oligomers impair neuronal plasticity and cognitive function. J Biol Chem. 2006;281:17941–51.
Zou K, Yamaguchi H, Akatsu H, Sakamoto T, Ko M, Mizoguchi K, et al. Angiotensin-converting enzyme converts amyloid beta-protein 1–42 (Abeta(1–42)) to Abeta(1–40), and its inhibition enhances brain Abeta deposition. J Neurosci. 2007;27:8628–35.
Dou Y, Zhao D, Yang F, Tang Y, Chang J. Natural phyto-antioxidant albumin nanoagents to treat advanced Alzheimer’s disease. ACS Appl Mater Interfaces. 2021;13:30373–82.
Urayama A, Moreno-Gonzalez I, Morales-Scheihing D, Kharat V, Pritzkow S, Soto C. Preventive and therapeutic reduction of amyloid deposition and behavioral impairments in a model of Alzheimer’s disease by whole blood exchange. Mol Psychiatry. 2022;27(10):4285–96.
Sehgal N, Gupta A, Valli RK, Joshi SD, Mills JT, Hamel E, et al. Withania somnifera reverses Alzheimer’s disease pathology by enhancing low-density lipoprotein receptor-related protein in liver. Proc Natl Acad Sci U S A. 2012;109:3510–5.
Sagare A, Deane R, Bell RD, Johnson B, Hamm K, Pendu R, et al. Clearance of amyloid-beta by circulating lipoprotein receptors. Nat Med. 2007;13:1029–31.
Cheng Y, He CY, Tian DY, Chen SH, Ren JR, Sun HL, et al. Physiological beta-amyloid clearance by the liver and its therapeutic potential for Alzheimer’s disease. Acta Neuropathol. 2023;145:717–31.
Ma M, Liu Z, Gao N, Pi Z, Du X, Ren J, et al. Self-Protecting biomimetic nanozyme for selective and synergistic clearance of peripheral amyloid-beta in an Alzheimer’s disease model. J Am Chem Soc. 2020;142:21702–11.
Etgen T, Chonchol M, Forstl H, Sander D. Chronic kidney disease and cognitive impairment: a systematic review and meta-analysis. Am J Nephrol. 2012;35:474–82.
Tian DY, Cheng Y, Zhuang ZQ, He CY, Pan QG, Tang MZ, et al. Physiological clearance of amyloid-beta by the kidney and its therapeutic potential for Alzheimer’s disease. Mol Psychiatry. 2021;26:6074–82.
Yan P, Kim KW, Xiao Q, Ma X, Czerniewski LR, Liu H, et al. Peripheral monocyte-derived cells counter amyloid plaque pathogenesis in a mouse model of Alzheimer’s disease. J Clin Invest. 2022;132:1–9.
Yu ZY, Chen DW, Tan CR, Zeng GH, He CY, Wang J, et al. Physiological clearance of Abeta by spleen and splenectomy aggravates Alzheimer-type pathogenesis. Aging Cell. 2022;21:1–12.
Seo DO, Holtzman DM. Gut microbiota: from the forgotten organ to a potential key player in the pathology of Alzheimer’s disease. J Gerontol A Biol Sci Med Sci. 2020;75:1232–41.
Zhan X, Stamova B, Sharp FR. Lipopolysaccharide associates with amyloid plaques, neurons and oligodendrocytes in Alzheimer’s disease brain: a review. Front Aging Neurosci. 2018;10:42–56.
Sittipo P, Choi J, Lee S, Lee YK. The function of gut microbiota in immune-related neurological disorders: a review. J Neuroinflammation. 2022;19:154–71.
Correa-Oliveira R, Fachi JL, Vieira A, Sato FT, Vinolo MA. Regulation of immune cell function by short-chain fatty acids. Clin Transl Immunology. 2016;5:73–81.
Liu N, Yang C, Liang X, Cao K, Xie J, Luo Q, et al. Mesoporous silica nanoparticle-encapsulated Bifidobacterium attenuates brain Abeta burden and improves olfactory dysfunction of APP/PS1 mice by nasal delivery. J Nanobiotechnology. 2022;20:439–46.
Wang X, Sun G, Feng T, Zhang J, Huang X, Wang T, et al. Sodium oligomannate therapeutically remodels gut microbiota and suppresses gut bacterial amino acids-shaped neuroinflammation to inhibit Alzheimer’s disease progression. Cell Res. 2019;29:787–803.
Zhuang ZQ, Shen LL, Li WW, Fu X, Zeng F, Gui L, et al. Gut microbiota is altered in patients with Alzheimer’s disease. J Alzheimers Dis. 2018;63:1337–46.
Freudenthaler S, Hegenbart U, Schonland S, Behrens HM, Kruger S, Rocken C. Amyloid in biopsies of the gastrointestinal tract-a retrospective observational study on 542 patients. Virchows Arch. 2016;468:569–77.
Folch J, Ettcheto M, Petrov D, Abad S, Pedros I, Marin M, et al. Review of the advances in treatment for Alzheimer disease: strategies for combating beta-amyloid protein. Neurologia. 2018;33:47–58.
Imbimbo BP, Watling M. Investigational BACE inhibitors for the treatment of Alzheimer’s disease. Expert Opin on Investig Drugs. 2019;28:967–75.
Aisen PS, Saumier D, Briand R, Laurin J, Gervais F, Tremblay P, et al. A Phase II study targeting amyloid-beta with 3APS in mild-to-moderate Alzheimer disease. Neurology. 2006;67:1757–63.
Yang X, Cai P, Liu Q, Wu J, Yin Y, Wang X, et al. Novel 8-hydroxyquinoline derivatives targeting beta-amyloid aggregation, metal chelation and oxidative stress against Alzheimer’s disease. Bioorg Med Chem. 2018;26:3191–201.
Matlack KE, Tardiff DF, Narayan P, Hamamichi S, Caldwell KA, Caldwell GA, et al. Clioquinol promotes the degradation of metal-dependent amyloid-beta (Abeta) oligomers to restore endocytosis and ameliorate Abeta toxicity. Proc Natl Acad Sci U S A. 2014;111:4013–8.
Tolar M, Abushakra S, Hey JA, Porsteinsson A, Sabbagh M. Aducanumab, gantenerumab, BAN2401, and ALZ-801-the first wave of amyloid-targeting drugs for Alzheimer’s disease with potential for near term approval. Alzheimers Res Ther. 2020;12:95–105.
Zhu G, Zhao J, Zhang H, Wang G, Chen W. Gut microbiota and its metabolites: bridge of dietary nutrients and Alzheimer’s disease. Adv Nutr. 2023;14:819–39.
Song C, Shi J, Zhang P, Zhang Y, Xu J, Zhao L, et al. Immunotherapy for Alzheimer’s disease: targeting beta-amyloid and beyond. Transl Neurodegener. 2022;11:18–35.
Holmes C, Boche D, Wilkinson D, Yadegarfar G, Hopkins V, Bayer A, et al. Long-term effects of Abeta42 immunisation in Alzheimer’s disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet. 2008;372:216–23.
Mantile F, Prisco A. Vaccination against beta-amyloid as a strategy for the prevention of Alzheimer’s disease. Biology (Basel). 2020;9:425–34.
Panza F, Lozupone M, Seripa D, Imbimbo BP. Amyloid-beta immunotherapy for Alzheimer disease: is it now a long shot? Ann Neurol. 2019;85:303–15.
Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B, et al. Clearance of Alzheimer’s amyloid-ss(1–40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J Clin Invest. 2000;106:1489–99.
Deane R, Wu Z, Sagare A, Davis J, Du Yan S, Hamm K, et al. LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron. 2004;43:333–44.
Wilcock DM, Munireddy SK, Rosenthal A, Ugen KE, Gordon MN, Morgan D. Microglial activation facilitates Abeta plaque removal following intracranial anti-Abeta antibody administration. Neurobiol Dis. 2004;15:11–20.
Fuller JP, Stavenhagen JB, Teeling JL. New roles for Fc receptors in neurodegeneration-the impact on immunotherapy for Alzheimer’s Disease. Front Neurosci. 2014;8:235–45.
Sevigny J, Chiao P, Bussiere T, Weinreb PH, Williams L, Maier M, et al. The antibody aducanumab reduces Abeta plaques in Alzheimer’s disease. Nature. 2016;537:50–6.
Samadi H, Sultzer D. Solanezumab for Alzheimer’s disease. Expert Opin Biol Ther. 2011;11:787–98.
Doody RS, Thomas RG, Farlow M, Iwatsubo T, Vellas B, Joffe S, et al. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N Engl J Med. 2014;370:311–21.
Nelson SE, Lopez OL. Lecanemab for Alzheimer disease: is it worth it? Neurology. 2024;102:1–2.
Knopman DS. Lecanemab reduces brain amyloid-beta and delays cognitive worsening. Cell Rep Med. 2023;4:100982–4.
Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6:916–9.
Bohrmann B, Baumann K, Benz J, Gerber F, Huber W, Knoflach F, et al. Gantenerumab: a novel human anti-Abeta antibody demonstrates sustained cerebral amyloid-beta binding and elicits cell-mediated removal of human amyloid-beta. J Alzheimers Dis. 2012;28:49–69.
Budd Haeberlein S, Aisen PS, Barkhof F, Chalkias S, Chen T, Cohen S, et al. Two randomized phase 3 studies of aducanumab in early Alzheimer’s disease. J Prev Alzheimers Dis. 2022;9:197–210.
Boston. Lecanemab sweeps up toxic aβ protofibrils, catches eyes of trialists. ALZFORUM. 2021; 1: 5–9.
Huang Z, Wong LW, Su Y, Huang X, Wang N, Chen H, et al. Blood-brain barrier integrity in the pathogenesis of Alzheimer’s disease. Front Neuroendocrin. 2020;59:100857–60.
Al Ojaimi Y, Blin T, Lamamy J, Gracia M, Pitiot A, Denevault-Sabourin C, et al. Therapeutic antibodies—natural and pathological barriers and strategies to overcome them. Pharmacol Ther. 2022;233:1–23.
Lee CS, Leong KW. Advances in microphysiological blood-brain barrier (BBB) models towards drug delivery. Curr Opin in Biotech. 2020;66:78–87.
Groothuis DR, Benalcazar H, Allen CV, Wise RM, Dills C, Dobrescu C, et al. Comparison of cytosine arabinoside delivery to rat brain by intravenous, intrathecal, intraventricular and intraparenchymal routes of administration. Brain Res. 2000;856:281–90.
Zhang C, Feng W, Vodovozova E, Tretiakova D, Boldyrevd I, Li Y, et al. Photodynamic opening of the blood-brain barrier to high weight molecules and liposomes through an optical clearing skull window. Biomed Opt Express. 2018;9:4850–62.
Anastasiadis P, Gandhi D, Guo Y, Ahmed AK, Bentzen SM, Arvanitis C, et al. Localized blood-brain barrier opening in infiltrating gliomas with MRI-guided acoustic emissions-controlled focused ultrasound. Proc Natl Acad Sci U S A. 2021. https://doi.org/10.1073/pnas.2103280118.
Bien-Ly N, Yu YJ, Bumbaca D, Elstrott J, Boswell CA, Zhang Y, et al. Transferrin receptor (TfR) trafficking determines brain uptake of TfR antibody affinity variants. J Exp Med. 2014;211:233–44.
Xiao G, Gan LS. Receptor-mediated endocytosis and brain delivery of therapeutic biologics. Int J Cell Biol. 2013;2013:1–14.
Zhao Y, Li D, Zhao J, Song J, Zhao Y. The role of the low-density lipoprotein receptor-related protein 1 (LRP-1) in regulating blood-brain barrier integrity. Rev Neurosci. 2016;27:623–34.
Thong BY, Vultaggio A, Rerkpattanapipat T, Schrijvers R. Prevention of drug hypersensitivity reactions: prescreening and premedication. J Allergy Clin Immunol. 2021;9:2958–66.
Baranowski BJ, Marko DM, Fenech RK, Yang AJT, Macpherson REK. Healthy brain, healthy life: a review of diet and exercise interventions to promote brain health and reduce Alzheimer’s disease risk. Appl Physiol Nutr Metab. 2020;45:1055–65.
Hansel TT, Kropshofer H, Singer T, Mitchell JA, George AJ. The safety and side effects of monoclonal antibodies. Nat Rev Drug Discov. 2010;9:325–38.
Hampel H, Elhage A, Cho M, Apostolova LG, Nicoll JAR, Atri A. Amyloid-related imaging abnormalities (ARIA): radiological, biological and clinical characteristics. Brain. 2023;146:4414–24.
Salloway S, Chalkias S, Barkhof F, Burkett P, Barakos J, Purcell D, et al. Amyloid-related imaging abnormalities in 2 phase 3 studies evaluating aducanumab in patients with early Alzheimer disease. JAMA Neurol. 2022;79:13–21.
Bassendine MF, Taylor-Robinson SD, Fertleman M, Khan M, Neely D. Is Alzheimer’s disease a liver disease of the brain? J Alzheimers Dis. 2020;75:1–14.
Matera MG, Calzetta L, Rogliani P, Cazzola M. Monoclonal antibodies for severe asthma: pharmacokinetic profiles. Resp Med. 2019;153:3–13.
Maarouf CL, Walker JE, Sue LI, Dugger BN, Beach TG, Serrano GE. Impaired hepatic amyloid-beta degradation in Alzheimer’s disease. PLoS ONE. 2018;13:1–6.
Liu N, Liang X, Yang C, Hu S, Luo Q, Luo H. Dual-targeted magnetic mesoporous silica nanoparticles reduce brain amyloid-beta burden via depolymerization and intestinal metabolism. Theranostics. 2022;12:6646–64.
Fotuhi SN, Khalaj-Kondori M. Imbalanced clearance of Abeta peptide cause presynaptic plaque formation. Int J Neurosci. 2022;5:1–5.
Avelar-Pereira B, Belloy ME. O’hara R, Hosseini SMH, Alzheimer’s disease neuroimaging I. Decoding the heterogeneity of Alzheimer’s disease diagnosis and progression using multilayer networks. Mol Psychiatry. 2023;28:2423–32.
Zaghi J, Goldenson B, Inayathullah M, Lossinsky AS, Masoumi A, Avagyan H, et al. Alzheimer disease macrophages shuttle amyloid-beta from neurons to vessels, contributing to amyloid angiopathy. Acta Neuropathol. 2009;117:111–24.
Frenkel D, Wilkinson K, Zhao L, Hickman SE, Means TK, Puckett L, et al. Scara1 deficiency impairs clearance of soluble amyloid-beta by mononuclear phagocytes and accelerates Alzheimer’s-like disease progression. Nat Commun. 2013;4:2030–49.
Gao HM, Hong JS. Why neurodegenerative diseases are progressive: uncontrolled inflammation drives disease progression. Trends Immunol. 2008;29:357–65.
Nho K, Kueider-Paisley A, Ahmad S, Mahmoudiandehkordi S, Arnold M, Risacher SL, et al. Association of altered liver enzymes with Alzheimer disease diagnosis, cognition, neuroimaging measures, and cerebrospinal fluid biomarkers. JAMA Neurol. 2019;2:1–19.
Yamazaki Y, Zhao N, Caulfield TR, Liu CC, Bu G. Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat Rev Neurosci. 2019;15:501–18.
Ghiso J, Calero M, Matsubara E, Governale S, Chuba J, Beavis R, et al. Alzheimer’s soluble amyloid beta is a normal component of human urine. FEBS Lett. 1997;408:105–8.
Nguyen TT, Ta QTH, Nguyen TKO, Nguyen TTD, Giau VV. Type 3 diabetes and its role implications in Alzheimer’s disease. Int J Mol Sci. 2020;21:1–16.
Tamaki C, Ohtsuki S, Terasaki T. Insulin facilitates the hepatic clearance of plasma amyloid beta-peptide (1–40) by intracellular translocation of low-density lipoprotein receptor-related protein 1 (LRP-1) to the plasma membrane in hepatocytes. Mol Pharmacol. 2007;72:850–5.
Farris W, Mansourian S, Chang Y, Lindsley L, Eckman EA, Frosch MP, et al. Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc Natl Acad Sci U S A. 2003;100:4162–7.
Sims-Robinson C, Kim B, Rosko A, Feldman EL. How does diabetes accelerate Alzheimer disease pathology? Nat Rev Neurol. 2010;6:551–60.
Mandolesi L, Polverino A, Montuori S, Foti F, Ferraioli G, Sorrentino P, et al. Effects of physical exercise on cognitive functioning and wellbeing: biological and psychological benefits. Front Psychol. 2018;9:509–20.
Rebok GW, Ball K, Guey LT, Jones RN, Kim HY, King JW, et al. Ten-year effects of the advanced cognitive training for independent and vital elderly cognitive training trial on cognition and everyday functioning in older adults. J Am Geriatr Soc. 2014;62:16–24.
White CA. Cognitive behavioral principles in managing chronic disease. West J Med. 2001;175:338–42.
Thiankhaw K, Chattipakorn N, Chattipakorn SC. PM2.5 exposure in association with AD-related neuropathology and cognitive outcomes. Environ Pollut. 2022;292:1–3.
Zhang M, Liang C, Chen X, Cai Y, Cui L. Interplay between microglia and environmental risk factors in Alzheimer’s disease. Neural Regen Res. 2024;19:1718–27.
Khare P, Edgecomb SX, Hamadani CM, Tanner EE, Manickam DS. Lipid nanoparticle-mediated drug delivery to the brain. Adv Drug Deliv Rev. 2023;197:114861–71.
Wen MM, El-Salamouni NS, El-Refaie WM, Hazzah HA, Ali MM, Tosi G, et al. Nanotechnology-based drug delivery systems for Alzheimer’s disease management: technical, industrial, and clinical challenges. J Control Release. 2017;245:95–107.
Stepanov YV, Golovynska I, Zhang R, Golovynskyi S, Stepanova LI, Gorbach O, et al. Near-infrared light reduces beta-amyloid-stimulated microglial toxicity and enhances survival of neurons: mechanisms of light therapy for Alzheimer’s disease. Alzheimers Res Ther. 2022;14:84–91.
Hampton T. For Alzheimer pathology, light and sound stimulation may hold promise. JAMA. 2019;322:17–8.
Murdock MH, Yang CY, Sun N, Pao PC, Blanco-Duque C, Kahn MC, et al. Multisensory gamma stimulation promotes glymphatic clearance of amyloid. Nature. 2024;627:149–56.
Blaikie L, Graeme K, Patricia M, Paul KTL. Experimental modelling of Alzheimer’s disease for therapeutic screening. Eur J Med Chem. 2022;5:1–13.
Sahoo BR, Panda PK, Liang W, Tang WJ, Ahuja R, Ramamoorthy A. Degradation of Alzheimer’s amyloid-beta by a catalytically inactive insulin-degrading enzyme. J Mol Biol. 2021;433:166993–7025.
Son SM, Cha MY, Choi H, Kang S, Choi H, Lee MS, et al. Insulin-degrading enzyme secretion from astrocytes is mediated by an autophagy-based unconventional secretory pathway in Alzheimer disease. Autophagy. 2016;12:784–800.
Walker JR, Pacoma R, Watson J, Ou W, Alves J, Mason DE, et al. Enhanced proteolytic clearance of plasma Abeta by peripherally administered neprilysin does not result in reduced levels of brain Abeta in mice. J Neurosci. 2013;33:2457–64.
Rofo F, Metzendorf NG, Saubi C, Suominen L, Godec A, Sehlin D, et al. Blood-brain barrier penetrating neprilysin degrades monomeric amyloid-beta in a mouse model of Alzheimer’s disease. Alzheimers Res Ther. 2022;14:180–99.
Torika N, Asraf K, Roasso E, Danon A, Fleisher-Berkovich S. Angiotensin converting enzyme inhibitors ameliorate brain inflammation associated with microglial activation: possible implications for Alzheimer’s disease. J Neuroimmune Pharmacol. 2016;11:774–85.
Bernstein KE, Koronyo Y, Salumbides BC, Sheyn J, Pelissier L, Lopes DH, et al. Angiotensin-converting enzyme overexpression in myelomonocytes prevents Alzheimer’s-like cognitive decline. J Clin Invest. 2014;124:1000–12.
Liu S, Fan M, Xu JX, Yang LJ, Qi CC, Xia QR, et al. Exosomes derived from bone-marrow mesenchymal stem cells alleviate cognitive decline in AD-like mice by improving BDNF-related neuropathology. J Neuroinflammation. 2022;19:35–55.
Cone AS, Yuan X, Sun L, Duke LC, Vreones MP, Carrier AN, et al. Mesenchymal stem cell-derived extracellular vesicles ameliorate Alzheimer’s disease-like phenotypes in a preclinical mouse model. Theranostics. 2021;11:8129–42.
Li C, Chen YH, Zhang K. Neuroprotective Properties and therapeutic potential of bone marrow-derived microglia in Alzheimer’s disease. Am J Alzheimers Dis Other Demen. 2020;35:1–7.
Chen SH, Tian DY, Shen YY, Cheng Y, Fan DY, Sun HL, et al. Amyloid-beta uptake by blood monocytes is reduced with ageing and Alzheimer’s disease. Transl Psychiatry. 2020;10:423–34.
Krishnan D, Menon RN, Mathuranath PS, Gopala S. A novel role for SHARPIN in amyloid-beta phagocytosis and inflammation by peripheral blood-derived macrophages in Alzheimer’s disease. Neurobiol Aging. 2020;93:131–41.
Jairani PS, Aswathy PM, Krishnan D, Menon RN, Verghese J, Mathuranath PS, et al. Apolipoprotein E polymorphism and oxidative stress in peripheral blood-derived macrophage-mediated amyloid-beta phagocytosis in Alzheimer’s disease patients. Cell Mol Neurobiol. 2019;39:355–69.
Tholen S, Schmaderer C, Chmielewski S, Forstl H, Heemann U, Baumann M, et al. Reduction of amyloid-beta plasma levels by hemodialysis: an anti-amyloid treatment strategy? J Alzheimers Dis. 2016;50:791–7.
Sakai K, Senda T, Hata R, Kuroda M, Hasegawa M, Kato M, et al. Patients that have undergone hemodialysis exhibit lower amyloid deposition in the brain: evidence supporting a therapeutic strategy for Alzheimer’s disease by removal of blood amyloid. J Alzheimers Dis. 2016;51:997–1002.
Kitaguchi N, Tatebe H, Sakai K, Kawaguchi K, Matsunaga S, Kitajima T, et al. Influx of tau and amyloid-beta proteins into the blood during hemodialysis as a therapeutic extracorporeal blood amyloid-beta removal system for Alzheimer’s disease. J Alzheimers Dis. 2019;69:687–707.
Jin WS, Shen LL, Bu XL, Zhang WW, Chen SH, Huang ZL, et al. Peritoneal dialysis reduces amyloid-beta plasma levels in humans and attenuates Alzheimer-associated phenotypes in an APP/PS1 mouse model. Acta Neuropathol. 2017;134:207–20.
Milanesi E, Dobre M, Cucos CA, Rojo AI, Jimenez-Villegas J, Capetillo-Zarate E, et al. Whole blood expression pattern of inflammation and redox genes in mild Alzheimer’s disease. J Inflamm Res. 2021;14:6085–102.
Ramirez S, Koerich S, Astudillo N, De Gregorio N, Al-Lahham R, Allison T, et al. Plasma exchange reduces abeta levels in plasma and decreases amyloid plaques in the brain in a mouse model of Alzheimer’s disease. Int J Mol Sci. 2023;24:1–13.
Navas Guimaraes ME, Lopez-Blanco R, Correa J, Fernandez-Villamarin M, Bistue MB, Martino-Adami P, et al. Liver X receptor activation with an intranasal polymer therapeutic prevents cognitive decline without altering lipid levels. ACS Nano. 2021;15:4678–87.
Wu Y, Dong JH, Dai YF, Zhu MZ, Wang MY, Zhang Y, et al. Hepatic soluble epoxide hydrolase activity regulates cerebral Abeta metabolism and the pathogenesis of Alzheimer’s disease in mice. Neuron. 2023;111:2847–62.
Pritchard AB, Crean S, Olsen I, Singhrao SK. Periodontitis, microbiomes and their role in Alzheimer’s disease. Front Aging Neurosci. 2017;9:336–46.
Acknowledgements
We thank the members of the Toxicology and Brain Science team at the Collage of Public Health, Zhengzhou University for their valuable feedback and suggestions.
Funding
This review was supported by grants from the Startup Fund of Zhengzhou University (32213157), the Key Scientific Research Projects of Colleges and Universities in Henan Province (K20A180031), the Postdoctoral Startup Fund Zhengzhou University (997002), and the National Natural Science Foundation of China (52208131). All the funders have no specific role in the conceptualization, design, data collection, analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
LN: Conceptualization, methodology, data curation, visualization, investigation, writing-original draft preparation, and funding; HA and ZW: software, data curation, visualization and validation. CY: methodology, software, visualization, investigation, supervision, and funding. CH: conceptualization, validation, writing-review & editing, and funding.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liu, N., Haziyihan, A., Zhao, W. et al. Trajectory of brain-derived amyloid beta in Alzheimer’s disease: where is it coming from and where is it going?. Transl Neurodegener 13, 42 (2024). https://doi.org/10.1186/s40035-024-00434-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40035-024-00434-9