
Abstract
Recombinant protein production in Komagataella phaffi (K. phaffi), a widely utilized host organism, can be optimized by enhancing the metabolic flux in the central carbon metabolism pathways. The methanol utilization pathway (MUT) during methanol-based growth plays a crucial role in providing precursors and energy for cell growth and development. This study investigated the impact of boosting the methanol dissimilation pathway, a branch of MUT that plays a vital role in detoxifying formaldehyde and providing energy in the form of NADH, in K. phaffi. This was achieved by integrating two orthologous genes from Hansenula polymorpha into the K. phaffi genome: formaldehyde dehydrogenase (HpFLD) and formate dehydrogenase (HpFMDH). The HpFLD and HpFMDH genes were isolated from the Hansenula polymorpha genome and inserted under the regulation of the pAOX1 promoter in the genome of recombinant K. phaffi that already contained a single copy of model protein genes (eGFP or EGII). The expression levels of these model proteins were assessed through protein activity assays and gene expression analysis. The findings revealed that while both orthologous genes positively influenced model protein production, HpFMDH exhibited a more pronounced upregulation in expression compared to HpFLD. Co-expression of both orthologous genes demonstrated synergistic effects, resulting in approximately a twofold increase in the levels of the model proteins detected. This study provides valuable insights into enhancing the production capacity of recombinant proteins in K. phaffi.
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Komagataella phaffi (K. phaffi), formerly known as Pichia pastoris, is a widely used yeast expression system that has gained significant attention for the production of industrial enzymes. As an efficient host for heterologous protein expression, K. phaffi can achieve high-level production of enzymes with diverse applications in industries such as chemical, food, pharmaceutical, textile, and cosmetics. The advantages of this platform include its capacity for post-translational modifications, high-density growth, efficient secretion, GRAS status, and scalable, cost-effective production compared to more complex systems (Duman-Özdamar and Binay 2021; Offei et al. 2022). Despite its extensive industrial history, there is still a significant need for improvement in this yeast system as a host (Noseda et al. 2013). Recombinant protein production in K. phaffi exerts a metabolic burden on the host cell, affecting energy and redox state (Nocon et al. 2014). For instance, during recombinant protein production, disulfide bond formation and protein folding require ATP and NADPH intensively. Accumulation of misfolded proteins triggers the unfolded protein response and increases chaperone activity, which further consumes energy (Zahrl et al. 2017). Consequently, higher intracellular ATP levels may enhance the cell’s capacity to produce recombinant proteins (Kim et al. 2012). NADH is another crucial cofactor in the redox reactions that occur during protein production, and its availability can affect the efficiency of protein production. Therefore, optimizing the distribution of metabolic flux in central carbon metabolism is crucial for improving recombinant protein production efficiency (Zahrl et al. 2017).
There are various methods to improve or change metabolic flux in a cell to enhance recombinant protein production, such as altering promoters, signal sequences, mRNA stability, and growth conditions (Brown et al. 2019; Xiao et al. 2023; Zahrl et al. 2017). Metabolic engineering is a highly effective approach that involves modifying specific biochemical reactions or introducing new ones using recombinant DNA technology. This technique can improve cell metabolic fluxes and enhance recombinant protein production in different hosts, including K. phaffi (Guo et al. 2021; Zhang et al. 2023). Metabolic engineering is a field of biotechnology that focuses on the design and construction of novel metabolic pathways or the modification of existing pathways to improve the production of specific compounds or to increase the efficiency of specific metabolic processes (Helmy et al. 2020; Mao et al. 2020). Metabolic engineering has been used in K. phaffi to improve recombinant protein production and other metabolic processes (De et al. 2021; Ergün et al. 2022). Some examples of metabolic engineering in K. phaffi include humanizing protein glycosylation, producing high yields of lycopene, producing myoinositol, and manipulating central metabolic fluxes to enhance the production of heterologous proteins (Laukens et al. 2015; Zhang et al. 2022, 2023).
Unlike most yeasts which ferment sugars into ethanol, K. phaffi is a methylotrophic yeast that can utilize methanol as its sole carbon source (Feng et al. 2020; Heistinger et al. 2020). The methanol utilization pathway (MUT) in K. phaffi is different from canonical methylotrophic-like bacteria. In bacteria, methanol is oxidized to formaldehyde by a methanol dehydrogenase enzyme. Formaldehyde is then assimilated into biomass (Müller et al. 2015; Takeya et al. 2020). In contrast, K. phaffi metabolizes methanol to formaldehyde via an alcohol oxidase enzyme that utilizes a flavin adenine dinucleotide (FAD) cofactor. Formaldehyde can enter the metabolic pathway of K. phaffi through two main routes: (I) Assimilation, in which formaldehyde can be directly fixed with dihydroxyacetone (DHA) to form 1,3-bisphosphoglycerate (1,3-BPG), which is then incorporated into the central carbon metabolism, leading to biomass production. (II) Dissimilation, in which formaldehyde is oxidized to CO2 by formaldehyde dehydrogenase (FLD), S-hydroxymethyl glutathione hydrolase (FGH), and formate dehydrogenase (FDH), producing NADH (Fig. 1) (Berrios et al. 2022; Yu et al. 2022b). The production of recombinant proteins or other metabolites in K. phaffi is directly linked to its ability to utilize methanol as the sole carbon source, a process governed by the MUT. Optimizing this pathway through metabolic engineering strategies can lead to improved yields of the desired recombinant products. In methylotrophic yeasts like K. phaffi, the efficient utilization of methanol is crucial for successful recombinant protein production. This is facilitated by the MUT, which encompasses the dissimilation pathway that provides the necessary energy for cellular processes (Meng et al. 2023). By enhancing the MUT through genetic modifications or other approaches, the metabolic flux can be redirected towards the synthesis of recombinant proteins or other valuable metabolites coupled to methanol metabolism (Guo et al. 2021).

Methanol utilisation pathway in K. phaffi: The main pathways and the respective enzymes working in the methanol metabolism in methylotrophic yeasts are shown. AOX: alcohol oxidase, CAT: catalase, FLD: formaldehyde dehydrogenase, FGH: S-formylglutathione hydrolase, FDH: formate dehydrogenase, DAS: dihydroxyacetone synthase, DHA: dihydroxyacetone, GAP: glyceraldehyde 3-phosphate, DHAP: dihydroxyacetone phosphate, Xu5P: xylulose 5-phosphate, GSH: glutathione
Hansenula polymorpha, another methylotrophic yeast that can grow on methanol as its sole source of carbon and energy, is another important yeast cell factory for the production of recombinant proteins (Manfrão-Netto et al. 2019). Despite considerable similarities in their MUT, Hansenula polymorpha has some advantages over K. phaffi, such as the derepression of some of its methanol-inducible promoters in the presence of glycerol, which is less pronounced in K. phaffi. Additionally, Hansenula polymorpha has several transcriptional regulators involved in methanol metabolism, such as Mpp1 and Adr1, which are also involved in the regulation of the response to both oleate and methanol in K. phaffi (Mombeni et al. 2020b). Furthermore, Hansenula polymorpha has been found to have a more active methanol oxidase enzyme compared to K. phaffi, contributing to its more efficient dissimilation pathway. Another significant difference in the methanol dissimilation pathway between these two yeasts is the presence of an enzyme in Hansenula polymorpha, called formate dehydrogenase (HpFMDH), which has the activity of both FDH and FGH enzymes, resulting in a shorter dissimilation pathway in this yeast (Bredell et al. 2016; Kurylenko et al. 2018; Van Dijken et al. 1976). However, K. phaffi is more commonly used for recombinant protein production due to specific advantages such as a larger genetic engineering toolkit, better secretion capacity, and more commercial adaptation (Kang et al. 2017; Obst et al. 2017; Staudacher et al. 2022; VijayakumarVenkataraman 2023).
This study examined the effects of increasing the expression of genes involved in the methanol dissimilation pathway from the yeast Hansenula polymorpha on the synthesis of recombinant proteins in K. phaffi. The research involved integrating two important genes, HpFLD and HpFMDH, into the genome of K. phaffi using metabolic engineering techniques. The impact of this integration was assessed using two model proteins, enhanced green fluorescent protein (eGFP) and endoglucanase II (EGII), by measuring the fluorescent intensity of eGFP and the activity of EGII. The results showed that the co-overexpression of these two enzymes significantly influenced the specific methanol uptake and the specific productivity of the recombinant K. phaffi strains, offering valuable insights into enhancing the capacity for producing recombinant proteins in K. phaffi.
Materials and methods
Recombinant K. phaffi co-expressing model proteins and heterologous HpFLD/HpFMDH
The recombinant clones expressing the eGFP and EGII under the control of pAOX1 were obtained from previous work in our lab (the sequences provided in Suppl. 1 and 2). Both genes were codon-optimized according to the preference of K. phaffi, cloned in frame with αMF signal sequence in pPINCα HC vector, transferred to PichiaPink™ strain 1 (ade2), and verified as described previously (Haghighi Poodeh et al. 2022; Mombeni et al. 2020a). To avoid the effect of gene dosage, the single-copy transformants were detected using a simple PCR-based experiment, which has been described in detail in our previous work (Mombeni et al. 2020b).
The genomic DNA of Hansenula polymorpha (strain DL-1) was isolated, qualified, and quantified as described previously (Arjmand et al. 2013). The nucleotide sequences of the HpFLD and HpFMDH genes (Accession No.; AF364077.1 and XM_018356471.1, respectively) were isolated from the genomic DNA through PCR amplification using specific primers (HpFLD-F-EcoRI, HpFLD-R-KpnI) and (HpFMDH-F-EcoRI, HpFMDH-R-KpnI), which included 5’ end restriction enzyme recognition sites as outlined in Table 1.
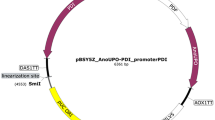
The HpFMDH amplicon, following digestion, was integrated into EcoRI/KpnI double-digested pPICZA plasmids, resulting in the creation of pPICZA-HpFMDH constructs as illustrated in Fig. 2a. Simultaneously, the digested HpFLD amplicon was incorporated into newly engineered pPICZA plasmids that had been altered to include a Kanamycin/Genticin (G418) resistance gene (kmR) in place of zeocin resistance gene (bleoR), as depicted in Fig. 2b, resulting in the formation of pPICKA-HpFLD constructs. To achieve antibiotic-resistant modification, the nucleotide sequence of the kmR gene under the TEF promoter/terminator was isolated from the pNTI647 dCas9-Mxi1 TetR KanMX (pCfB2225) vector through PCR amplification using specific primers (Kan-F-SmaI, Kan-R-SmaI) that contained restriction enzyme recognition sites at their 5′ ends (Table 1).

The map of constructed recombinant plasmids, pPICKA- HpFLD and pPICZA-HpFMDH, containing the HpFLD and HpFMDH genes, respectively. Both metabolic genes are expressed under the control of pAOX1, integrated into the K. phaffi genome from AOX1 location on the chromosome
To propagate these constructs, they were introduced into Escherichia coli DH5α via a heat shock method (42 °C for 45 s). Positive clones were then screened for confirmation by employing Zeocin (at 25 µg/ml) or kanamycin (at 50 µg/ml) resistance, in conjunction with PCR validation and subsequent sequencing.
Following linearization with SacI and HindIII, the pPICKA-HpFLD and pPICZA-HpFMDH constructs were electrotransformed into the aforementioned recombinant K. phaffi strains expressing model proteins (EGII and eGFP), as per the established protocol (Arjmand et al. 2013). Clones resistant to zeocin (100 µg/ml) or Genticin (400 µg/ml) were screened for the presence of both the model protein and HpFLD or HpFMDH sequences through PCR using specific primers, followed by sequencing. To distinguish between the native K. phaffi FLD (PpFLD) gene and the newly introduced one, PCR was conducted using the forward primer of pAOX1 in combination with a reverse FLD-specific primer (and vice versa). Five clones from each of the following eight groups were used for expression analysis: (1) Expressing eGFP, (2) Expressing EGII, (3) Expressing eGFP + HpFLD, (4) Expressing EGII + HpFLD, (5) Expressing eGFP + HpFMDH, (6) Expressing EGII + HpFMDH, (7) Expressing eGFP + HpFLD/HpFMDH, (8) Expressing EGII + HpFLD/HpFMDH. The clones were initially grown at 30 °C in YPG medium (1% yeast extract, 2% peptone, and 2% glycerol). After 48 h, equal concentrations of cells from each clone were transferred to fresh inductive YPM medium (1% yeast extract, 2% peptone, and 0.5% methanol). 1% Methanol feeding was continued every 24 h for up to 96 h to keep induction. Densities of the yeast cell cultures were measured at OD600.
Spectrofluorimetery
Spectrofluorimetry was used to determine the intensity of green fluorescence from secreted recombinant eGFP, as well as the effect of HpFLD, HpFMDH, and HpFLD/HpFMDH co-expression on fluorescent emission. The supernatants were isolated on day 4 of induction from clones expressing eGFP alone and those co-expressing eGFP and the candidate genes. These supernatants were tested using a spectrofluorimeter (Shimadzu RF-5000, Japan) with 470 nm excitation and 520 nm emission wavelengths. The supernatant from non-recombinant K. phaffi was used as a negative control.
Endoglucanase assay
Endoglucanase activity was measured using the method of Ghose (Ghose 1987) which is based on the release of reducing sugar from carboxymethyl cellulose (CMC) as the substrate and quantification with dinitrosalicyclic acid (DNS). The endoglucanase reaction involves enzyme-substrate binding, catalysis of glycosidic bond hydrolysis in CMC, and release of reducing sugars. Reaction rate depends on enzyme/substrate concentration, temperature, and pH, which were controlled in this study to ensure consistent, reproducible activity measurements. In this study, 250 µl of supernatant (containing recombinant secreted enzymes) were added to 250 µl of 1% CMC-Na in sodium acetate buffer 50 mM (pH 4.8). After incubating the mixture at 50 °C for 30 min, the reaction was stopped by adding 500 µl of DNS reagent. The resulting mixture was boiled for 5 min to allow color development, and the reducing sugar content was measured by absorbance at 540 nm Citrate buffer replaced the enzyme in negative controls. The absorbance values were directly proportional to the amount of reducing sugars released from the CMC substrate by the endoglucanase activity. One international unit (IU) of CMCase activity was defined as the amount of enzyme releasing 1 mg of reducing sugar per minute per ml under the assay conditions.
Total protein was determined by the Bradford method (Bradford 1976) using bovine serum albumin (BSA) as the standard. (Akbarzadeh et al. 2013).
Real-time PCR
Quantitative real-time PCR was utilized to evaluate the effects of HpFLD and HpFMDH overexpression/co-overexpression on recombinant eGFP and EGII model protein production at the transcriptional level. Yeast cells were harvested on the third day of the methanol induction period from each of the eight aforementioned groups. Total RNA was extracted using QIAzol lysis reagent (Qiagen, 79,306) as described previously (Maceda-López et al. 2021). Extracted RNA was treated with DNase (Sukardi et al. 2019) and reverse transcribed into cDNA using the RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific, K1622) per the manufacturer’s protocol. Real-time PCR was performed with the RealQ Plus 2× Master Mix Green High ROXTM (Ampliqon, 4,309,155) on an ABI Stepone PCR System (Applied Biosystems, US). Cycling conditions were 95 °C for 15 min followed by 40 cycles of 95 °C for 15 s and 60 °C for 30 s with a final extension at 60 °C for 30 min. Relative fold differences were calculated via the 2−ΔΔCT method using actin1 (ACT1) as an endogenous reference. Primers used for real-time PCR are listed in Table 1. Gene sequences and primer locations are provided in Supplementary Fig. 1.
Results
Construction of recombinant yeasts
The correct construction of the recombinant plasmids pPICKA- HpFLD, pPICZA-HpFMDH, and K. phaffi strains co-expressing the HpFLD orHpFMDH genes along with the target genes eGFP or EGII (Fig. 2) was confirmed by PCR and sequencing. Methanol induction was initiated with equal cell masses for all groups, and OD600 measurements at the end of the induction period indicated that HpFLD or HpFMDH co-expression had no effect on cell growth (Fig. 3).

OD600 at the end of the induction period
Spectrofluorimetery
The spectrofluorimetric analysis demonstrated that co-expression of HpFLD, HpFMDH, or both with eGFP led to enhanced fluorescence intensity compared to eGFP alone (Fig. 4a). Specifically, eGFP- HpFLD, eGFP-HpFMDH, and eGFP-HpFLD/HpFMDH co-expression resulted in 1.4-fold, 1.9-fold, and 2.3-fold increases in measured fluorescence, respectively.

Fluorescence intensity and Normalized endoglucanase activity. (a) Fluorescence intensity in the supernatant of K. phaffi clones expressing eGFP alone (control) or co-expressing eGFP with HpFLD, HpFMDH, or both HpFLD and HpFMDH. Fluorescence was measured by spectrofluorimetry with 470 nm excitation and 520 nm emission wavelengths. Data represent the mean ± standard deviation of five clones with three replicates each. (b) Normalized endoglucanase activity in the supernatant of K. phaffi clones expressing EGII alone or co-expressing EGII with HpFLD, HpFMDH, or both HpFLD and HpFMDH. Enzymatic activity was determined by the CMC-DNS method as described in the text. Data represent the mean ± standard deviation of 5 clones with 3 replicates each
Enzymatic assay
Enzymatic assays demonstrated that co-expression of HpFLD, HpFMDH, or both with EGII resulted in increased endoglucanase activity compared to EGII alone (Fig. 4b). Specifically, EGII-HpFLD, EGII-HpFMDH, and EGII-HpFLD/HpFMDH co-expression led to 1.2-fold, 1.6-fold, and 1.8-fold increases in measured endoglucanase activity, respectively. The activity was determined by quantification of reducing sugars released from the CMC substrate, as described in the methods.
Real-time PCR
Real-time PCR analysis verified overexpression of HpFLD and HpFMDH in recombinant K. phaffi strains containing heterologous genes from Hansenula polymorpha (Fig. 5a and b). Co-expression of HpFLD, HpFMDH, or both under the control of the pAOX1 promoter resulted in enhanced transcription of the reporter genes compared to expression alone. Specifically, eGFP co-expressed with HpFLD, HpFMDH, or HpFLD/HpFMDH showed 1.6-fold, 1.7-fold, and 2.2-fold increased mRNA levels, respectively (Fig. 5c). Similarly, EGII co-expressed with HpFLD, HpFMDH, or HpFLD/HpFMDH exhibited 1.3-fold, 1.5-fold, and 1.8-fold increased transcript levels, respectively (Fig. 5d). The augmentation of eGFP and EGII transcripts with co-expression of the candidate metabolic genes correlated with enhanced fluorescence and enzymatic activity, confirming the positive effects at the transcriptional level.

Real-time PCR analysis of gene expression. (a) HpFLD and (b) HpFMDH overexpression in recombinant K. phaffi strains. (c) eGFP and (d) EGII mRNA levels in clones expressing eGFP or EGII alone (control) versus co-expression with HpFLD, HpFMDH, or HpFLD/HpFMDH. Values were normalized to the endogenous reference gene ACT1. Data represent the mean ± standard deviation of 5 clones with 3 replicates each
Discussion
K. phaffi has become an important host for heterologous protein production, with thousands of proteins expressed at high levels using this expression system (Zhu et al. 2019). A key factor underlying the high recombinant protein yields in K. phaffi is the strongly induced AOX1 promoter. This promoter controls the expression of alcohol oxidase, which is tightly regulated and highly upregulated when K. phaffi is grown on methanol as the sole carbon source (Kielkopf et al. 2021; Türkanoğlu Özçelik et al. 2019). The AOX1 promoter from K. phaffi has been harnessed to drive the high-level production of diverse heterologous proteins by placing this promoter sequence upstream of genes encoding proteins of interest. Upon methanol induction, the engineered genes are strongly transcribed at levels comparable to the native AOX1 gene, allowing recombinant protein overexpression (Cámara et al. 2017; Haghighi Poodeh et al. 2022; Kielkopf et al. 2021; Türkanoğlu Özçelik et al. 2019).
The heightened energy requirements resulting from recombinant protein synthesis in K. phaffi underscore the pivotal role of metabolism as the primary limiting factor influencing recombinant protein production in this organism (Heyland et al. 2011). The essential functions of central metabolic pathways in supplying energy, reducing equivalents, and precursor molecules for cellular growth, maintenance, and recombinant protein synthesis underscore the criticality of employing targeted strain engineering approaches centered on central metabolism to optimize cell growth and protein production requirements (FerrerAlbiol 2014).
In particular, the MUT is directly linked to heterologous protein production in K. phaffi, as methanol induces the AOX1 promoter controlling expression of the recombinant genes. Thus, enhancing MUT pathway flux has the potential to increase energy production and precursor supply, alleviating bottlenecks in recombinant protein synthesis.
The MUT pathway in methylotrophic yeasts stands out due to its unique transcriptional regulation. Methanol undergoes oxidation to formaldehyde by AOX within peroxisomes. Subsequently, formaldehyde is detoxified in the dissimilation pathway to CO2 and H2O through a series of three enzymes: FLD, FGH, and FDH. This enzymatic process generates NADH, which is crucial for energy production and the growth on methanol (Geier et al. 2015; Yu et al. 2022a). In Hansenula polymorpha, the activities of FGH and FDH are consolidated into a single enzyme known as HpFMDH. This fusion of FGH and FDH into HpFMDH is highly significant as it optimizes the metabolic pathway, streamlining the conversion of formaldehyde to formate. By combining these enzymes, Hansenula polymorpha enhances the efficiency of the methanol dissimilation pathway while reducing the energy consumption required for enzyme synthesis. This consolidation results in a more efficient process by creating a single multifunctional enzyme unit instead of two separate proteins. This metabolic enhancement not only boosts protein production efficiency but also simplifies the conversion of formaldehyde to formate, leading to improved resource utilization and metabolic flow within the organism (Hartner and Glieder 2006; Van Dijken et al. 1976).
When the methanol-fed batch phase begins, enzymes related to the methanol dissimilation pathway are strongly dominant. On the other hand, enzymes associated with methanol assimilation only increase to a moderate extent during the production phase (Vanz et al. 2012). For instance, in Hansenula polymorpha, the addition of methanol led to a 350-fold upregulation of the HpFMDH gene involved in the dissimilation pathway. In contrast, the dihydroxyacetone synthase (DHAS) gene in the assimilation pathway was only upregulated around 19-fold under the same conditions (van Zutphen et al. 2010). This is because the assimilation of methanol requires more energy and resources than the dissimilation pathway, and the cells need to balance their metabolism to optimize their growth and protein production (Liu et al. 2023). Enhancing the dissimilation pathway in recombinant protein production can indeed improve the efficiency of methanol metabolism, leading to increased ATP production and improved biotransformation capabilities. This enhancement optimizes the energy distributions of methanol metabolism, ensuring more efficient NADH/product coupling and the exploitation of both NADH steps for cofactor regeneration in whole-cell systems (Yu et al. 2022a). By increasing fluxes through the methanol dissimilation pathway, it has been observed that recombinant protein yield can be enhanced, indicating a positive impact on the production of recombinant proteins (Boojari et al. 2023).
In this research endeavor, we conducted an experiment where we overexpressed and co-overexpressed two orthologous genes of HpFLD and HpFMDH from the MUT of Hansenula polymorpha dissimilation pathway, under the regulation of AOX promoter in K. phaffi. This investigation aimed to evaluate the impact of these genetic manipulations on the production of our model proteins in mRNA and protein expression levels.
The sequences of HpFLD and HpFMDH genes were extracted from the Hansenula polymorpha genome and cloned under the control of pAOX1 in the genome of recombinant K. phaffi containing one copy of model protein (eGFP or EGII) genes with the same promoter. The model proteins (generated in our previous works) were integrated into the TRP2 locus of the K. phaffi genome and selected on the selective medium Pichia Adenine Dropout (PAD). While the HpFLD and HpFMDH genes were integrated at the AOX1 locus and selected based on zeocin and genticin resistance.
Both orthologous genes, HpFLD and FMDH, were validated for overexpression through real-time PCR. Despite being expressed under the same AOX1 promoter, HpFMDH demonstrates a more significant expression upregulation compared to HpFLD. Expression studies demonstrated a 1.2, 1.6, and 2-fold increase in EGII enzyme activity and a 1.4, 1.9, and 2.3-fold increase in fluorescence intensity when HpFLD, HpFMDH, or both were co-expressed with the model proteins, respectively. The greater effect of HpFMDH could be due to its higher expression level or increased effectiveness in enhancing the dissimilation pathway. Further molecular studies of methanol metabolic flux will be needed to fully elucidate the underlying mechanism for HpFMDH’s more significant impact. The fluorescence results were confirmed by real-time PCR.
The synergistic effects of overexpressing both HpFLD and HpFMDH, leading to an approximately 2-fold increase, validate the efficacy of enhancing the dissimilation pathway to improve recombinant protein production. This concept underscores the potential benefits of empowering specific pathways to enhance protein expression levels. Leveraging orthologous genes from other methylotrophic yeasts could offer new avenues for optimizing protein expression in K. phaffi and improving its capabilities as a cell factory for producing natural products. This strategy has been recently employed in studies to improve the capabilities of K. phaffi and other yeasts. For example, a heterologous lactate transporter from Bos taurus successfully increased l-lactic acid production from glycerol in recombinant strains of K. phaffi. (de Lima et al. 2016). In addition to orthologous genes, orthologous regulatory sequences have also been effectively utilized for the expression of recombinant proteins in different yeast cells. These regulatory sequences, which include promoters, enhancers, and other cis-acting elements, can significantly impact the expression level and regulation of the target gene (Naseri et al. 2017; Vogl et al. 2020).
The findings of this study highlight the significant potential of optimizing the dissimilation pathway of K. phaffi to enhance recombinant protein production. Overall, in this study, Hansenula polymorpha was utilized as a template for desired modifications to engineer the methanol metabolism pathway in K. phaffi, ultimately boosting recombinant protein yield. While numerous studies have employed orthologous genes or regulatory sequences from other organisms for metabolic engineering, this finding highlights the potential of such cross-species approaches for enhancing industrially strategic recombinant protein production. Evidently, there are many untapped opportunities and novel ideas yet to be explored for further optimizing these pathways in the near future.
Availability of data and materials
All data supporting the findings of this study are available in the paper and its Supplementary Information.
Abbreviations
- MUT:
-
Methanol utilization pathway
- NADH:
-
Nicotinamide adenine dinucleotide hydrogen
- HpFLD:
-
Hansenula polymorpha formaldehyde dehydrogenase
- HpFMDH:
-
Hansenula polymorpha formate dehydrogenase
- pAOX1:
-
Alcohol oxidase-1 promoter
- eGFP:
-
Enhanced green fluorescent protein
- EGII:
-
Endoglucanase II
- ATP:
-
Adenosine triphosphate
- NADPH:
-
Nicotinamide adenine dinucleotide phosphate
- FAD:
-
Flavin adenine dinucleotide
- DHA:
-
Dihydroxyacetone
- 1,3-BPG:
-
1,3-Bisphosphoglycerate
- FLD:
-
Formaldehyde dehydrogenase
- FGH:
-
S-hydroxymethyl glutathione hydrolase
- FDH:
-
Formate dehydrogenase
- Mpp1:
-
Membrane palmitoylated Protein 1
- αMF:
-
α-mating factor (signal sequence)
- kmR :
-
Kanamycin resistance
- bleoR :
-
Bleomycin resistance
- TEF:
-
Transcription elongation factor 1
- CMC:
-
Carboxymethyl cellulose
- DNS:
-
Dinitrosalicyclic acid
- BSA:
-
Bovine serum albumin
- ACT1:
-
Actin1
- DHAS:
-
Dihydroxyacetone synthase
- PAD:
-
Pichia adenine dropout
References
Akbarzadeh A, Ranaei Siadat SO, Zamani MR, Motallebi M, Barshan Tashnizi M (2013) Comparison of biochemical properties of recombinant endoglucanase II of Trichoderma reesei in methylotrophic yeasts, Pichia pastoris and Hansenula polymorpha. Progress Biol Sci 3(1):108–117
Arjmand S, Lotfi AS, Shamsara M, Mowla SJ (2013) Elevating the expression level of biologically active recombinant human alpha 1-antitrypsin in Pichia pastoris. Electron J Biotechnol 16(1):4–4
Berrios J, Theron CW, Steels S, Ponce B, Velastegui E, Bustos C, Altamirano C, Fickers P (2022) Role of dissimilative pathway of Komagataella phaffii (Pichia pastoris): formaldehyde toxicity and energy metabolism. Microorganisms 10(7):1466
Boojari MA, Rajabi Ghaledari F, Motamedian E, Soleimani M, Shojaosadati SA (2023) Developing a metabolic model-based fed‐batch feeding strategy for Pichia pastoris fermentation through fine‐tuning of the methanol utilization pathway. Microb Biotechnol 16(6):1344–1359
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72(1–2):248–254
Bredell H, Smith JJ, Prins WA, Görgens JF, van Zyl WH (2016) Expression of rotavirus VP6 protein: a comparison amongst Escherichia coli, Pichia pastoris and Hansenula polymorpha. FEMS Yeast Res 16(2):fow001
Brown AJ, Gibson SJ, Hatton D, Arnall CL, James DC (2019) Whole synthetic pathway engineering of recombinant protein production. Biotechnol Bioeng 116(2):375–387
Cámara E, Landes N, Albiol J, Gasser B, Mattanovich D, Ferrer P (2017) Increased dosage of AOX1 promoter-regulated expression cassettes leads to transcription attenuation of the methanol metabolism in Pichia pastoris. Sci Rep 7(1):44302
de Lima PBA, Mulder KCL, Melo NTM, Carvalho LS, Menino GS, Mulinari E, de Castro VH, Dos Reis TF, Goldman GH, Magalhães BS, Parachin NS (2016) Novel homologous lactate transporter improves L-lactic acid production from glycerol in recombinant strains of Pichia pastoris. Microb Cell Fact 15:1–9
De S, Mattanovich D, Ferrer P, Gasser B (2021) Established tools and emerging trends for the production of recombinant proteins and metabolites in Pichia pastoris. Essays Biochem 65(2):293–307
Duman-Özdamar ZE, Binay B (2021) Production of industrial enzymes via Pichia pastoris as a cell factory in bioreactor: current status and future aspects. Protein J 40(3):367–376
Ergün BG, Laçın K, Çaloğlu B, Binay B (2022) Second generation Pichia pastoris strain and bioprocess designs. Biotechnol Biofuels Bioprod 15(1):1–19
Ferrer p, Albiol J (2014) 13 C-based metabolic flux analysis of recombinant Pichia pastoris. Metabolic flux analysis 1191:291–313
Feng D, Stoyanov A, Olliff JC, Wolfe KH, Lahtchev K, Hanson SJ (2020) Carbon source requirements for mating and mating-type switching in the methylotrophic yeasts Ogataea (Hansenula) polymorpha and Komagataella phaffii (Pichia pastoris). Yeast 37(2):237–245
Geier M, Brandner C, Strohmeier GA, Hall M, Hartner FS, Glieder A (2015) Engineering Pichia pastoris for improved NADH regeneration: a novel chassis strain for whole-cell catalysis. Beilstein J Org Chem 11(1):1741–1748
Ghose T (1987) Measurement of cellulase activities. Pure Appl Chem 59(2):257–268
Guo F, Dai Z, Peng W, Zhang S, Zhou J, Ma J, Dong W, Xin F, Zhang W, Jiang M (2021) Metabolic engineering of Pichia pastoris for malic acid production from methanol. Biotechnol Bioeng 118(1):357–371
Haghighi Poodeh S, Ranaei Siadat SO, Arjmand S, Khalifeh Soltani M (2022) Improving AOX1 promoter efficiency by overexpression of Mit1 transcription factor. Mol Biol Rep 49(10):9379–9386
Hartner FS, Glieder A (2006) Regulation of methanol utilisation pathway genes in yeasts. Microb Cell Fact 5:1–21
Heistinger L, Gasser B, Mattanovich D (2020) Microbe Profile: Komagataella phaffii: a methanol devouring biotech yeast formerly known as Pichia pastoris. Microbiology 166(7):614–616
Helmy M, Smith D, Selvarajoo K (2020) Systems biology approaches integrated with artificial intelligence for optimized metabolic engineering. Metabolic Eng Commun 11:e00149
Heyland J, Fu J, Blank LM, Schmid A (2011) Carbon metabolism limits recombinant protein production in Pichia pastoris. Biotechnol Bioeng 108(8):1942–1953
Kang Z, Huang H, Zhang Y, Du G, Chen J (2017) Recent advances of molecular toolbox construction expand Pichia pastoris in synthetic biology applications. World J Microbiol Biotechnol 33:1–8
Kielkopf CL, Bauer W, Urbatsch IL (2021) Expression of cloned genes in Pichia pastoris using the methanol-inducible promoter AOX1. Cold Spring Harbor Protocols. https://doi.org/10.1101/pdb.prot102160
Kim H-J, Kwon YD, Lee SY, Kim P (2012) An engineered Escherichia coli having a high intracellular level of ATP and enhanced recombinant protein production. Appl Microbiol Biotechnol 94(4):1079–1086
Kurylenko OO, Ruchala J, Vasylyshyn RV, Stasyk OV, Dmytruk OV, Dmytruk KV, Sibirny AA (2018) Peroxisomes and peroxisomal transketolase and transaldolase enzymes are essential for xylose alcoholic fermentation by the methylotrophic thermotolerant yeast, Ogataea (Hansenula) polymorpha. Biotechnology for biofuels 11:1–16
Laukens B, De Wachter C, Callewaert N (2015) Engineering the Pichia pastoris N-glycosylation pathway using the GlycoSwitch technology. Glyco-Engineering: Methods and Protocols. https://doi.org/10.1007/978-1-4939-2760-9_8
Liu S, Dong H, Hong K, Meng J, Lin L, Wu X (2023) Improving methanol utilization by reducing Alcohol Oxidase Activity and adding co-substrate of Sodium citrate in Pichia pastoris. J Fungi 9(4):422
Maceda-López LF, Villalpando-Aguilar JL, García-Hernández E, de Dios EÁ, Andrade-Canto SB, Morán-Velázquez DC, Rodríguez-López L, Hernández-Díaz D, Chablé-Vega MA, Trejo L, Góngora-Castillo E, López-Rosas I, Simpson J, Alatorre-Cobos F (2021) Improved method for isolation of high-quality total RNA from Agave tequilana Weber roots. 3 Biotech 11(2):1–10
Manfrão-Netto J, Gomes A, Parachin N (2019) Advances in using Hansenula polymorpha as chassis for recombinant protein production. Front Bioeng Biotechnol. https://doi.org/10.3389/fbioe.2019.00094
Mao W, Yuan Q, Qi H, Wang Z, Ma H, Chen T (2020) Recent progress in metabolic engineering of microbial formate assimilation. Appl Microbiol Biotechnol 104:6905–6917
Meng J, Liu S, Gao L, Hong K, Liu S, Wu X (2023) Economical production of Pichia pastoris single cell protein from methanol at industrial pilot scale. Microb Cell Fact 22(1):198
Müller JE, Meyer F, Litsanov B, Kiefer P, Potthoff E, Heux S, Quax WJ, Wendisch VF, Brautaset T, Portais J-C, Vorholt JA (2015) Engineering Escherichia coli for methanol conversion. Metabolic engineering 28:190–201
Mombeni M, Arjmand S, Siadat SOR, Alizadeh H, Abbasi A (2020a) pMOX: a new powerful promoter for recombinant protein production in yeast Pichia pastoris. Enzyme Microb Technol. https://doi.org/10.1016/j.enzmictec.2020.109582
Mombeni M, Arjmand S, Siadat SOR, Alizadeh H, Abbasi A (2020b) pMOX: a new powerful promoter for recombinant protein production in yeast Pichia pastoris. Enzym Microb Technol 139:109582
Naseri G, Balazadeh S, Machens F, Kamranfar I, Messerschmidt K, Mueller-Roeber B (2017) Plant-derived transcription factors for orthologous regulation of gene expression in the yeast Saccharomyces cerevisiae. ACS Synth Biol 6(9):1742–1756
Nocon J, Steiger MG, Pfeffer M, Sohn SB, Kim TY, Maurer M, Rußmayer H, Pflügl S, Ask M, Haberhauer-Troyer C, Ortmayr K, Hann S, Koellensperger G, Gasser B, Lee SY, Mattanovich D (2014) Model based engineering of Pichia pastoris central metabolism enhances recombinant protein production. Metab Eng 24:129–138
Noseda DG, Recúpero MN, Blasco M, Ortiz GE, Galvagno MA (2013) Cloning, expression and optimized production in a bioreactor of bovine chymosin B in Pichia (Komagataella) pastoris under AOX1 promoter. Protein Exp Purif 92(2):235–244
Obst U, Lu T, Sieber V (2017) A modular toolkit for generating Pichia pastoris secretion libraries. ACS Synth Biol 6:1016–1025
Offei B, Braun-Galleani S, Venkatesh A, Casey WT, O’Connor KE, Byrne KP, Wolfe KH (2022) Identification of genetic variants of the industrial yeast Komagataella phaffii (Pichia pastoris) that contribute to increased yields of secreted heterologous proteins. PLoS Biol 20(12):e3001877
Türkanoğlu Özçelik A, Yılmaz S, Inan M (2019) Pichia pastoris promoters. Methods Mol Biol. https://doi.org/10.1007/978-1-4939-9024-5_3
Staudacher J, Rebnegger C, Dohnal T, Landes N, Mattanovich D, Gasser B (2022) Going beyond the limit: increasing global translation activity leads to increased productivity of recombinant secreted proteins in Pichia pastoris. Metab Eng 70:181–195
Sukardi P, Rosita RE, Siregar AS, Januar CS, Harisam MT, Hidayat F, Bessho Y, Prayogo NA (2019) The effect of endosulfan (insecticide) on expression of vitelogenin gene in female silver sharkminnow (Osteochilus hasseltii CV). IOP Conference Series: Earth Environ Sci; IOP Publishing, Indonesia, 20-22 November 2019
Takeya T, Yamakita M, Hayashi D, Fujisawa K, Sakai Y, Yurimoto H (2020) Methanol production by reversed methylotrophy constructed in Escherichia coli. Biosci Biotechnol Biochem 84(5):1062–1068
Van Dijken J, Oostra-Demkes G, Otto R, Harder W (1976) S-Formylglutathione: the substrate for formate dehydrogenase in methanol-utilizing yeasts. Arch Microbiol 111:77–83
Vanz A, Lünsdorf H, Adnan A, Nimtz M, Gurramkonda C, Khanna N, Rinas U (2012) Physiological response of Pichia pastoris GS115 to methanol-induced high level production of the Hepatitis B surface antigen: catabolic adaptation, stress responses, and autophagic processes. Microb Cell Fact 11(1):103
van Zutphen T, Baerends RJ, Susanna KA, De Jong A, Kuipers OP, Veenhuis M, Van der Klei IJ (2010) Adaptation of Hansenula polymorpha to methanol: a transcriptome analysis. BMC Genomics 11:1–12
Vijayakumar VE, Venkataraman K (2023) A systematic review of the potential of Pichia pastoris (Komagataella Phaffii) as an alternative host for biologics production. Molecular Biotechnology. https://doi.org/10.1007/s12033-023-00803-1
Vogl T, Fischer JE, Hyden P, Wasmayer R, Sturmberger L, Glieder A (2020) Orthologous promoters from related methylotrophic yeasts surpass expression of endogenous promoters of Pichia pastoris. AMB Express 10:1–9
Xiao C, Xue S, Pan Y, Liu X, Huang M (2023) Overexpression of genes by stress-responsive promoters increases protein secretion in Saccharomyces cerevisiae. World J Microbiol Biotechnol 39(8):203
Yu Y-f, Yang J, Zhao F, Lin Y, Han S (2022a) Comparative transcriptome and metabolome analyses reveal the methanol dissimilation pathway of Pichia pastoris. BMC Genomics 23(1):366
Yu Y-f, Yang J, Zhao F, Lin Y, Han S (2022b) Comparative transcriptome and metabolome analyses reveal the methanol dissimilation pathway of Pichia pastoris. BMC Genomics 23(1):1–14
Zahrl RJ, Peña DA, Mattanovich D, Gasser B (2017) Systems biotechnology for protein production in Pichia pastoris. FEMS Yeast Res 17(7):fox068
Zhang Q, Wang X, Luo H, Wang Y, Wang Y, Tu T, Qin X, Su X, Huang H, Yao B, Bai Y, Zhang J (2022) Metabolic engineering of Pichia pastoris for myo-inositol production by dynamic regulation of central metabolism. Microb Cell Fact 21(1):112
Zhang X, Chen S, Lin Y, Li W, Wang D, Ruan S, Yang Y, Liang S (2023) Metabolic Engineering of Pichia pastoris for high-level production of Lycopene. ACS Synth Biol 12(10):2961–2972
Zhu T, Sun H, Wang M, Li Y (2019) Pichia pastoris as a versatile cell factory for the production of industrial enzymes and chemicals: current status and future perspectives. Biotechnol J 14(6):1800694
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
M.K.S. conducted the experiments, performed data analysis, wrote the manuscript draft, S.A. conceived and designed the study, performed data analysis, wrote and revised the manuscript, S.O.R.S. supervised the study, provided scientific support, assisted with device preparation, A.B. supervised the study, S.H.M. advised the process. All authors have read and agreed to the final version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it.The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Khalifeh Soltani, M., Arjmand, S., Ranaei Siadat, S. et al. Hansenula polymorpha methanol metabolism genes enhance recombinant protein production in Komagataella phaffi. AMB Expr 14, 88 (2024). https://doi.org/10.1186/s13568-024-01743-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13568-024-01743-y