
Abstract
Background
Neuroblastoma (NB) is the most common extracranial pediatric solid tumor with a highly variable clinical course, ranging from spontaneous regression to life-threatening disease. Survival rates for high-risk NB patients remain disappointingly low despite multimodal treatment. Thus, there is an urgent clinical need for additional biomarkers to improve risk stratification, treatment management, and survival rates in children with aggressive NB.
Results
Using gene promoter methylation analysis in 48 neuroblastoma tumors with microarray technology, we found a strong association between survival and gene promoter hypermethylation (P = 0.036). Hypermethylation of 70 genes significantly differentiated high-risk survivor patients from those who died during follow-up time. Sixteen genes with relevant roles in cancer biology were further validated in an additional cohort of 83 neuroblastoma tumors by bisulfite pyrosequencing. High promoter methylation rates of these genes were found in patients with metastatic tumors (either stage metastatic (M) or metastatic special (MS)), 18 months or older at first diagnosis, MYCN amplification, relapsed, and dead. Notably, the degree of methylation of retinoblastoma 1 (RB1) and teratocarcinoma-derived growth factor 1 (TDGF1) predicts event-free and overall survival independently of the established risk factors. In addition, low RB1 mRNA expression levels associate with poor prognosis suggesting that promoter methylation could contribute to the transcriptional silencing of this gene in NB.
Conclusions
We found a new epigenetic signature predictive for NB patients’ outcome: the methylation status of RB1 and TDGF1 associate with poorer survival. This information is useful to assess prognosis and improve treatment selection.
Similar content being viewed by others


Background
Neuroblastoma originates from the sympathico-adrenal lineage of the neural crest and is the most common extracranial solid tumor in early childhood. This tumor exhibits contrasting patterns of clinical behavior ranging from spontaneous remission to rapid tumor progression and death. Prognosis classically depends on age at diagnosis [1], tumor stage [2], MYCN oncogene amplification status [3,4], and histology in a lesser extent [5]. Several well-characterized genetic abnormalities associated with neuroblastoma (NB) have been used to predict outcome, for example, DNA content [6], gain of chromosome arm 17q [7], or deletion of chromosome arm 1p [8] and 11q [9,10]. However, current knowledge of the molecular features of NB is not sufficient to explain the observed clinical heterogeneity.
There is an important body of work on NB to find robust biomarkers that could help to improve the standard response criteria and, consequently, patients’ survival. In this regard, several mRNA and miRNA classifiers have been established [11-13]. A recent study from SIOPEN shows that levels of tyrosine hydroxylase (TH) and paired-like homeobox 2B (PHOX2B) or doublecortin (DCX) mRNA in peripheral blood and bone marrow at diagnosis are independent predictors of survival [14]. Notably, high levels of TH and PHOX2B mRNA in peripheral blood identify ultrahigh-risk NB patients. Despite these new discoveries, survival rates in children with high-risk NB remain disappointingly low.
Epigenetic modifications, particularly the methylation of the 5′ position of cytosines, within CpGs dinucleotides at gene promoter regions, are essential regulatory mechanisms for normal cell development and may modulate gene expression without altering the DNA sequence. In the last decade, DNA-methylation studies have focused on identifying epigenetically modified genes to further understand NB pathogenesis and to find prognostic methylation markers. In this regard, global methylation studies have demonstrated that a methylator phenotype, characterized by the methylation of multiple CpG islands, is a hallmark of NB with poor prognosis [15]. Several tumor suppressor genes such as caspase 8 (CASP8), Ras association (RalGDS/AF-6) domain family member 1 (RASSF1A), cycling D2 (CCND2), CD44, O-6-methylguanine-DNA methyltransferase (MGMT), and PYD and CARD domain containing (TMS1), have been shown to be silenced in NB by aberrant hypermethylation of their promoters [16-18]. In general, screening studies in NB showed that frequently methylated genes are related to apoptotic pathways as well as to cell cycle regulation.
Recent studies in oncology research have increased genome coverage allowing the identification of new epigenetic biomarkers. In NB, two recent studies have investigated DNA methylation patterns using genome-wide technologies [19,20]. However, the number of clinically relevant epigenetic biomarkers is still very low. In here, we used methylation microarrays to identify robust and independent epigenetic biomarkers in NB.
Results
Genome-wide promoter methylation screening
With the purpose of identifying DNA methylation biomarkers, we first analyzed 48 primary NB tumors using the Infinium HumanMethylation27 BeadChip microarray. Clinical, biological, and follow-up data are summarized in Table 1. Patients were classified following the International Neuroblastoma Risk Group Staging System (INRGSS) guidelines as 10 L1 (stage 1 localized); 16 L2 (stage 2 localized), 18 M (metastatic), and 4 MS (metastatic special). Twenty patients relapsed: 1 L1 presented a local relapse and is alive and disease free, 7 L2 (5 presented local relapses, 1 metastatic and 1 combined, 4 of them died of disease); 11 M stages (2 presented local relapses, 3 presented metastatic relapses and 6 combined relapses, all of them died of disease). One MS stage presented a metastatic relapse and died due to disease progression.
Data distribution according to the methylation probe intensity from the Illumina array was shown to be bimodal (Figure 1A). Using minAS, a method for feature selection in multivariate data, cutoff values for data discretization were defined as follows: ≤0.3 hypomethylated ‘0’, >0.3 < 0.7 intermediate ‘0.5’, and >0.7 hypermethylated ‘1’.

DNA promoter methylation profiling from 48 NB primary tumors. (A) Data distribution according to the probe intensity. (B) Association between gene promoter hypermethylation and patients’ status. (C) Heat map representation of DNA promoter methylation data of the 70 genes differentially methylated in the cohort of 48 NB patients.
In order to understand to which extent gene promoter methylation is relevant to NB, we analyzed differences in the methylation levels among different patient subgroups. The most striking result of this analysis revealed a significant association between poor survival and global gene promoter hypermethylation (P = 0.036) (Figure 1B). Patients who died during follow-up time had higher promoter hypermethylation rates than survivors (median follow-up time for survivors of 12 years). No association was detected between survival and global hypomethylation of gene promoters. Other types of subgrouping (by age at diagnosis, MYCN status, or relapse) did not reveal any significant changes. However, comparing patients at different NB stages (L1 and MS vs. L2 vs. M) did indicate a near-significant hypermethylation of the runt-related transcription factor 3 (RUNX3) gene (P = 0.05). These results suggest that the overall promoter hypermethylation status at diagnosis could predict survival and that NB stages can be associated with methylation changes at specific genes.
After establishing a significant association between promoter hypermethylation and NB, we then aimed to identify particular methylation events. Comparison of methylation values of high-risk patients who died of disease, and of high-risk patients who survived, revealed 70 genes that significantly differentiate these two groups (Figure 1C and Table 2). Interestingly, only specific CpG sites within promoter regions were methylated (compare total probes vs. significative probes that were methylated on Table 2 and see the scheme on Additional file 1). This result revealed the importance of sufficient CpG probes at promoter regions to capture the dynamics of methylation changes. Sixteen genes with high impact on cancer biology were selected for validation. The methylated probes and P values as well as the methylation level of the selected hypermethylated genes are shown in Table 3. Detailed information of these genes’ promoter methylation sites in each patient is described in Additional file 2. These genes regulate the following: maturation and maintenance of the overall structure of the nervous system (neuronatin (NNAT)); cell cycle progression (cyclin D1 (CCND1), janus kinase 2 (JAK2), retinoblastoma 1 (RB1), leucine-rich repeat C4 protein (LRRC4), and tumor protein P73 (TP73)); cell growth and differentiation (dual specificity phosphatase 2 (DUSP2), paired box 8 (PAX8), and hepsin (HPN)); tumorigenesis (melanoma antigen family A, 2 (MAGEA2), RUNX3, cathepsin Z (CTSZ), teratocarcinoma-derived growth factor 1 (TDGF1), and tetraspanin 32 (TSPAN32)); apoptosis (JAK2 and esophageal cancer-related gene 4 protein (ECRG4)) and DNA repair mechanisms (MGMT).
Validation of the prognostic power of DNA methylation biomarkers by bisulfite pyrosequencing
To validate the specific CpG methylated sites, we performed sequencing analysis of the 16 promoter genes. Primers for bisulfite pyrosequencing were carefully designed flanking the methylated CpG sites detected in the array. Validation of results was carried out with an independent cohort of 83 NB including 12 L1, 21 L2, 42 M, and 8 MS stages. Clinical and biological data from the validation cohort is summarized in Table 4.
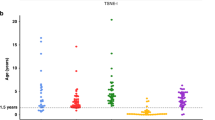
We performed a multivariate analysis of variance (MANOVA) statistical analysis in order to study the methylation variations of selected genes among the established subgroups (see below and “Methods” section) of NB patients. Overall, we found significantly higher gene promoter methylation rates in patients with the following characteristics: metastatic tumors (either stage M or MS) (Figure 2A), aged 18 months or older at first diagnosis (Figure 2B) and MYCN amplification (Figure 2C). The same applies to patients who relapsed or died (Figure 2D). In particular, methylated status of TDGF1 and PAX8 allowed us to differentiate all the clinical subgroups mentioned above (Figure 2). In addition, high methylation rates of RUNX3, ECRG4, CTSZ, and RB1 also distinguished all NB subgroups but not MYCN-amplified status (Figure 2). We also found highest methylation rates of LRRC4 and CCND1 in patients older than 18 months (Figure 2B). Furthermore, DUSP2, TP73, JAK2, MGMT, and HPN methylation rates were significantly higher in MYCN-amplified patients than in non-amplified patients (Figure 2C). These results demonstrate that these set of methylated gene promoters allow the discrimination of specific NB subgroups defined in Figure 2.

Difference in methylation status of indicated genes (% units) among clinical variables in NB patients. Global P value tests the hypothesis of equal methylation status in both groups. (A) Stages M - MS vs. L1-L2. (B) Patients younger than 18 months at diagnosis vs. patients older than 18 months at diagnosis. (C) MYCN-amplified patients vs. patients with no-MYCN amplification. (D) Patients with events (relapse/death) vs. patients without events.
To study the influence of the degree of the 16 validated genes’ promoter methylation on survival, we evaluated them as independent prognostic variables by the elastic net penalized Cox’s regression model. Higher gene promoter methylation rates of RB1, PAX8, and TDGF1 remained as independent predictors of overall survival (OS) after adjusting for known prognostic factors (Table 5A). On the other hand, RB1 and TDGF1 but not PAX8 predicted worse event-free survival (EFS) (Table 5B). Information of TDGF1 and RB1 promoter methylation sites in the validation cohort of patients is described in Additional file 3. These regressions also confirmed the well-established stage, MYCN status, and age as independent predictors, thus further supporting the consistency of this model. The penalized coefficient of each independent variable directly associates with survival. Thus, MYCN amplification and staging have the highest influence on survival followed by the degree of TDGF1 and RB1 promoter methylation. Age at diagnosis had a lower impact in EFS and none in OS.
RB1 expression in NB correlates with survival
Taking into account that RB1 and TDGF1 promoter methylation is one of the mechanisms responsible for the downregulation of these genes in other tumors [21,22], we explored RB1 and TDGF1 expression in NB, the two only independent predictors of EFS and OS that we found in this study. For this purpose, we analyzed a new cohort of 251 NB patients using the R2: microarray analysis and visualization platform (http://r2.arnc.nl). The results of this analysis are displayed in Figure 3. The Kaplan-Meier plots show the significant association between low expression levels of RB1 and poorer outcome (Figure 3A). Based on these findings, promoter methylation could contribute to the transcriptional silencing of RB1 in NB. On the other hand, low expression levels of TDGF1 associates with better outcome in this patient cohort (Figure 3B). Since our results show that high methylation levels of TDGF1 negatively affect patient survival, the TDGF1 expression-Kaplan-Meier plots suggest that this particular epigenetic event on cg10242476 is not involved in TDGF1 downregulation but rather in its expression. In order to investigate the positive correlation between CpG methylation and TDGF1 expression, we explored the correlation between TDGF1 expression and the DNA methyltransferases DNMT1, DNMT3A, and DNMT3B. As shown in Additional file 4, we found a significant correlation between TDGF1 and DNMT1 and DNMT3B but not between TDGF1 and DNMT3A in the cohort of patients with MYCN amplification. These correlations further support that DNA methylation might as well be implicated in the upregulation of TDGF1 expression.

Results of the analysis of a new cohort of 251 NB patients using the R2. Expression graphs (on the right) and Kaplan-Meier plots (on the left) for RB1 (A) and TDGF1 (B) obtained using the R2: microarray analysis and visualization platform in a cohort of 251 NB patients.
Discussion
Most of the DNA methylation studies in NB are experimentally limited because they used previously selected candidate genes based on their implication in cancer development or tumor biology [16,23-25]. A recent report described the signature of hypomethylated regions within non-promoter regulatory sites [20], thus complementing our findings on hypermethylated promoter regions. These characteristic profiles may reflect specific chromatin remodeling events that could contribute to the generation of chromosomal instability. Another recent genome-wide study has proposed new epigenetic biomarkers of interest in NB patients [19,26]. However, authors only used cell lines in the selection phase of potential prognostic DNA methylation biomarkers. Although candidate genes were further validated in 89 primary tumor samples, this approach may have skipped many candidate biomarkers occurring on primary tumors due to the passage-dependent epigenetic changes on cell lines. Lau et al. applied a genome-wide screen of DNA methylation changes using NB primary tumors [26]. In their study, they pre-selected candidate genes which may resulted in the loss of novel epigenetic biomarkers. In here, we adopted a non-targeted approach based on genome-wide screen of DNA-methylation changes. Our results reveal 70 candidate genes that showed epigenetic changes within the high-risk group. We further validated 16 out of 70 candidate genes, leaving for future validation studies the remaining group.
The two patient cohorts used in this study included a representative distribution of all the INRG-based NB subgroups, being among the highest and well-characterized NB cohorts used for genome-wide epigenetic studies so far reported to our knowledge. Interestingly, the degree of methylation of the proposed biomarkers is able to distinguish between different subgroups of NB: patients who were older than 18 months at diagnosis from younger than 18 months at diagnosis, patients with MYCN-amplified tumors from MYCN non-amplified tumors, patients with metastatic tumors from localized tumors and relapsed or dead patients from relapse-free survivors. These findings support the idea that aberrant DNA methylation could be related to NB pathogenesis. Notably, only RB1 and TDGF1 remained as independent prognostic predictors of poorer OS and EFS. Therefore, our predictive epigenetic biomarkers constitute a new set of robust risk predictors of the disease.
TDGF1 promoter is hypomethylated and highly expressed in human-induced pluripotent stem (iPS) and embryonic stem (ES) cells [27]. Using the same Illumina array, our results reveal a different CpG site not only localized downstream but also within the first exon (5′-UTR) of TDGF1 gene (cg10242476) that significantly predicts EFS and OS in NB patients. Importantly, TDGF1 has been shown to be regulated by the ES cell-related transcription factors Oct4/Nanog and to a lesser extent by the DNA methylation status of the promoter region [28]. TDGF1 has been found overexpressed in a variety of human tumors such as breast, colorectal, and gastric cancers, and high expression levels of this gene are associated with poorer prognosis in those tumors [29]. Using the microarray analysis and visualization platform R2 (http://r2.arnc.nl), we confirmed that high TDGF1 expression levels are also associated with poor prognosis in NB (Figure 3B). Following this reasoning, higher methylation of this CpG would correlate with higher TDGF1 expression and, therefore, with poorer prognosis. Interestingly, this CpG site is localized in the first exon of TDGF1 transcript variant 1 (NM_003212) and coincides with the first intron of TDGF1 transcript variant 2 (NM_001174136). One plausible explanation for this correlation is that intronic CpG methylation results in TDGF1 transcript variant 2 expression. In this regard, regulation of imprinted IGF2R expression is mediated by methylation of an intronic CpG island [30]. Taken all together, these results suggest that intronic CpG (cg10242476) methylation in TDGF1 transcript variant 2 positively regulates its expression in NB whereas CpG (cg27371741) hypomethylation in the first exon of TDGF1 transcript variant 1 regulates its expression in embryonic stem cells. The strong association between hypermethylation and poor prognosis reflects the consequences of epigenetic changes occurring in high-risk NB.
Loss of RB1 expression is associated with a higher grade of malignancies and seems to be a prognostic indicator in a variety of human tumors [31,32]. Hypermethylation of RB1 CpG island is a common epigenetic event associated with the development of malignant nervous system tumors [22]. A clear correlation between loss of RB1 expression and promoter hypermethylation was found in glioblastomas [32]. Although hypermethylation of RB1 promoter has previously been reported in NB, no association with patient survival was included in the study [33]. Moreover, the methylation-specific PCR approach used to detect RB1 promoter methylation does not allow to study the broad CpG sites analyzed with a genome-wide approach. We describe for the first time that the degree of RB1 promoter methylation associates with poorer prognosis in NB patients. In agreement with our findings, using the publicly available R2 platform, we found that low expression levels of RB1 associates with poorer outcome. These results suggest that RB1 promoter methylation could contribute to its silencing and enhance NB development and aggressiveness.
Conclusions
Biomarkers are playing an increasing role in the management of NB patients and, together with drug targets, represent the future analytical platforms for personalized clinical intervention. In this work, we demonstrate that high promoter methylation rates of TDGF1 and RB1 genes are independent predictive biomarkers of NB aggressiveness and disease progression. Our findings highlight the use of methylation profiling to identify risk-independent prognostic markers in NB and reinforce the connection between epigenetic events and NB biology. Taking into account that survival rates remain sadly low in high-risk NB patients, our epigenetic biomarkers are valuable tools for future patient stratification and treatment management.
Methods
Patients and samples
Tumor samples were resected from 131 children diagnosed with NB between years 1996 and 2010 in Spanish cooperative hospitals. Patients were included in different national and European studies (LNESG I and II, INES, EUNS, N-AR-99, N-II-92, and HR-NBL1) and carefully selected in order to have all NB subtypes represented (Table 1). Forty-eight NB tumor samples were used for genome-wide promoter methylation analysis, and an additional cohort of 83 tumor samples were used for validation. Staging and risk stratification was established according to International Neuroblastoma Risk Group (INRG) criteria [2]. However, very low- and low-risk patients were joined for the statistical analysis and were considered as low-risk group. Samples were centrally reviewed and classified according to the International Neuroblastoma Pathology Committee (INPC) system [5,34]. Biological studies included status of MYCN and 1p, both studied by FISH according to ENQUA guidelines [35,36]. Parents or guardians signed an informed consent statement for sample and data management. The study was approved by the Hospital La Fe Research Institute Ethical Committee.
Genome-wide promoter methylation profiling
Genomic DNA was extracted from frozen tumor tissues by a standard proteinase K and phenol-chloroform extraction protocol. The quality and quantity of the extracted DNA was measured by A260 spectrophotometric absorbance. Genomic DNA bisulphite modification was carried out following the manufacturer’s instructions (Zymo Research). Promoter methylation analysis was performed using the Infinium HumanMethylation27 BeadChip (Illumina Inc., CA, USA) at the Spanish National Cancer Center (CEGEN-CNIO, Madrid, Spain). The Infinium HumanMethylation27 BeadChip allowed us to interrogate 27,578 highly informative CpG sites per sample, located within the proximal gene promoter regions of transcription. A file containing all CpG sites used in the Illumina array HumanMethylation27 BeadChip can be localized at http://support.illumina.com/array/array_kits/infinium_humanmethylation27_beadchip_kit/downloads.html. BeadStudio software (version 3, Illumina Inc, USA) [37,38] was used to analyze the data. For each CpG site, we calculated the beta-value (b-value), a quantitative measure of DNA methylation levels ranging from 0 for completely unmethylated to 1 for completely methylated cytosines.
Bisulfite pyrosequencing
Promoter methylation data from the array was validated by bisulfite pyrosequencing. Genomic DNA was bisulfite modified using EZ DNA Methylation Gold Kit™ (Zymo Research). A subsequent PCR amplification was performed using biotinylated primers designed with the PyroMark Assay Design 2.0 software, Qiagen (Table 6). The pyrosequencing and data analysis were performed in a PyroMark Q24 System version 2.0.6 (Qiagen) following the manufacturer’s instructions.
Statistical analysis
The methylation status at each array probe was established by analyzing the distribution of b-values for all samples and CpG sites and transforming data into discrete values. The cutoff values for hyper- and hypomethylation were established using the minAS method [39] and considering the bimodal distribution of b-values. Differential methylation analysis was done at the single CpG, CpG island, and gene-centric levels. For the analysis of CpG island, individual probes were considered independent observation, whereas the gene-level analysis simply counted the percentage of CpG and CpG island associated to each gene that were declared significant. Differential methylation across the NB subgroups was determined by either a proportion test when several groups were compared or by Fisher’s exact test when only two subgroups were involved. Subgroups were established and compared based on clinical and biological parameters such as age at first diagnosis (younger vs. older than 18 months), MYCN status (MYCN-amplified vs. non-amplified tumors), stage (L1-L2 vs. M-MS), risk groups (low and intermediate vs. high risk), relapse, and death (patients with events (relapse/death) vs. patients without events). Nominal P values were corrected for multiple tests using the Benjamini and Hochberg FDR procedure [40].
Data distribution from pyrosequencing analysis was not bimodal and varies among genes; therefore, variables were analyzed as continuous. The relationship between the methylation status and NB risk factors was evaluated using a MANOVA test. A Cox elastic net analysis [41] was performed to evaluate the influence on survival of gene promoter methylation. This novel statistical analysis method is especially suited for analyzing data with many variables and few observations by performing variable selection. This is done by penalizing predictors’ coefficients towards zero according to their association with survival. Coefficients from variables with less influence on survival were more penalized, dropped to zero, and excluded from the predictive model.
For all the above-mentioned statistical tests, R software (version 3.0.2) and package glmnet (version 1.9-5) were used. For EFS analysis, time to event was defined as the time from diagnosis until the time of first occurrence of relapse, progression, or death. For OS, time to event was defined as time until death or until last contact if the patient was alive. P values <0.05 were considered statistically significant.
References
London WB, Castleberry RP, Matthay KK, Look AT, Seeger RC, Shimada H, et al. Evidence for an age cutoff greater than 365 days for neuroblastoma risk group stratification in the Children’s Oncology Group. J Clin Oncol. 2005;23:6459–65.
Monclair T, Brodeur GM, Ambros PF, Brisse HJ, Cecchetto G, Holmes K, et al. The International Neuroblastoma Risk Group (INRG) staging system: an INRG Task Force report. J Clin Oncol. 2009;27:298–303.
Brodeur GM, Seeger RC, Schwab M, Varmus HE, Bishop JM. Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science. 1984;224:1121–4.
Seeger RC, Brodeur GM, Sather H, Dalton A, Siegel SE, Wong KY, et al. Association of multiple copies of the N-myc oncogene with rapid progression of neuroblastomas. N Engl J Med. 1985;313:1111–6.
Shimada H, Ambros IM, Dehner LP, Hata J, Joshi VV, Roald B, et al. The International Neuroblastoma Pathology Classification (the Shimada system). Cancer. 1999;86:364–72.
Look AT, Hayes FA, Shuster JJ, Douglass EC, Castleberry RP, Bowman LC, et al. Clinical relevance of tumor cell ploidy and N-myc gene amplification in childhood neuroblastoma: a Pediatric Oncology Group study. J Clin Oncol. 1991;9:581–91.
Caron H. Allelic loss of chromosome 1 and additional chromosome 17 material are both unfavourable prognostic markers in neuroblastoma. Med Pediatr Oncol. 1995;24:215–21.
Lampert F, Rudolph B, Christiansen H, Franke F. Identical chromosome 1p breakpoint abnormality in both the tumor and the constitutional karyotype of a patient with neuroblastoma. Cancer Genet Cytogenet. 1988;34:235–9.
Cohn SL, Pearson AD, London WB, Monclair T, Ambros PF, Brodeur GM, et al. The International Neuroblastoma Risk Group (INRG) classification system: an INRG Task Force report. J Clin Oncol. 2009;27:289–97.
Spitz R, Hero B, Simon T, Berthold F. Loss in chromosome 11q identifies tumors with increased risk for metastatic relapses in localized and 4S neuroblastoma. Clin Cancer Res. 2006;12:3368–73.
De Preter K, Mestdagh P, Vermeulen J, Zeka F, Naranjo A, Bray I, et al. Vandesompele J: miRNA expression profiling enables risk stratification in archived and fresh neuroblastoma tumor samples. Clin Cancer Res. 2011;17:7684–92.
Schulte JH, Schowe B, Mestdagh P, Kaderali L, Kalaghatgi P, Schlierf S, et al. Accurate prediction of neuroblastoma outcome based on miRNA expression profiles. Int J Cancer. 2010;127:2374–85.
Vermeulen J, De Preter K, Naranjo A, Vercruysse L, Van Roy N, Hellemans J, et al. Predicting outcomes for children with neuroblastoma using a multigene-expression signature: a retrospective SIOPEN/COG/GPOH study. Lancet Oncol. 2009;10:663–71.
Viprey VF, Gregory WM, Corrias MV, Tchirkov A, Swerts K, Vicha A, et al. Neuroblastoma mRNAs predict outcome in children with stage 4 neuroblastoma: a European HR-NBL1/SIOPEN study. J Clin Oncol. 2014;32:1074–83.
Abe M, Watanabe N, McDonell N, Takato T, Ohira M, Nakagawara A, et al. Identification of genes targeted by CpG island methylator phenotype in neuroblastomas, and their possible integrative involvement in poor prognosis. Oncology. 2008;74:50–60.
Alaminos M, Davalos V, Cheung NK, Gerald WL, Esteller M. Clustering of gene hypermethylation associated with clinical risk groups in neuroblastoma. J Natl Cancer Inst. 2004;96:1208–19.
Grau E, Martinez F, Orellana C, Canete A, Yanez Y, Oltra S, et al. Hypermethylation of apoptotic genes as independent prognostic factor in neuroblastoma disease. Mol Carcinog. 2011;50:153–62.
Yang Q, Kiernan CM, Tian Y, Salwen HR, Chlenski A, Brumback BA, et al. Methylation of CASP8, DCR2, and HIN-1 in neuroblastoma is associated with poor outcome. Clin Cancer Res. 2007;13:3191–7.
Decock A, Ongenaert M, Hoebeeck J, De Preter K, Van Peer G, Van Criekinge W, et al. Genome-wide promoter methylation analysis in neuroblastoma identifies prognostic methylation biomarkers. Genome Biol. 2012;13:R95.
Mayol G, Martin-Subero JI, Rios J, Queiros A, Kulis M, Sunol M, et al. DNA hypomethylation affects cancer-related biological functions and genes relevant in neuroblastoma pathogenesis. PLoS One. 2012;7:e48401.
Bianco C, Salomon DS. Targeting the embryonic gene Cripto-1 in cancer and beyond. Expert Opin Ther Pat. 2010;20:1739–49.
Gonzalez-Gomez P, Bello MJ, Alonso ME, Arjona D, Lomas J, de Campos JM, et al. CpG island methylation status and mutation analysis of the RB1 gene essential promoter region and protein-binding pocket domain in nervous system tumours. Br J Cancer. 2003;88:109–14.
Grau E, Martinez F, Orellana C, Canete A, Yanez Y, Oltra S, et al. Epigenetic alterations in disseminated neuroblastoma tumour cells: influence of TMS1 gene hypermethylation in relapse risk in NB patients. J Cancer Res Clin Oncol. 2010;136:1415–21.
Hoebeeck J, Michels E, Pattyn F, Combaret V, Vermeulen J, Yigit N, et al. Aberrant methylation of candidate tumor suppressor genes in neuroblastoma. Cancer Lett. 2009;273:336–46.
Michalowski MB, de Fraipont F, Plantaz D, Michelland S, Combaret V, Favrot MC. Methylation of tumor-suppressor genes in neuroblastoma: The RASSF1A gene is almost always methylated in primary tumors. Pediatr Blood Cancer. 2008;50:29–32.
Lau DT, Hesson LB, Norris MD, Marshall GM, Haber M, Ashton LJ. Prognostic significance of promoter DNA methylation in patients with childhood neuroblastoma. Clin Cancer Res. 2012;18:5690–700.
Nishino K, Toyoda M, Yamazaki-Inoue M, Makino H, Fukawatase Y, Chikazawa E, et al. Defining hypo-methylated regions of stem cell-specific promoters in human iPS cells derived from extra-embryonic amnions and lung fibroblasts. PLoS One. 2010;5:e13017.
Watanabe K, Meyer MJ, Strizzi L, Lee JM, Gonzales M, Bianco C, et al. Cripto-1 is a cell surface marker for a tumorigenic, undifferentiated subpopulation in human embryonal carcinoma cells. Stem Cells. 2010;28:1303–14.
Rangel MC, Karasawa H, Castro NP, Nagaoka T, Salomon DS, Bianco C. Role of Cripto-1 during epithelial-to-mesenchymal transition in development and cancer. Am J Pathol. 2012;180:2188–200.
Wutz A, Smrzka OW, Schweifer N, Schellander K, Wagner EF, Barlow DP. Imprinted expression of the Igf2r gene depends on an intronic CpG island. Nature. 1997;389:745–9.
Cryns VL, Thor A, Xu HJ, Hu SX, Wierman ME, Vickery Jr AL, et al. Loss of the retinoblastoma tumor-suppressor gene in parathyroid carcinoma. N Engl J Med. 1994;330:757–61.
Nakamura M, Yonekawa Y, Kleihues P, Ohgaki H. Promoter hypermethylation of the RB1 gene in glioblastomas. Lab Invest. 2001;81:77–82.
Gonzalez-Gomez P, Bello MJ, Lomas J, Arjona D, Alonso ME, Aminoso C, et al. Aberrant methylation of multiple genes in neuroblastic tumours. Relationship with MYCN amplification and allelic status at 1p. Eur J Cancer. 2003;39:1478–85.
Burgues O, Navarro S, Noguera R, Pellin A, Ruiz A, Castel V, et al. Prognostic value of the International Neuroblastoma Pathology Classification in Neuroblastoma (Schwannian stroma-poor) and comparison with other prognostic factors: a study of 182 cases from the Spanish Neuroblastoma Registry. Virchows Arch. 2006;449:410–20.
Ambros IM, Benard J, Boavida M, Bown N, Caron H, Combaret V, et al. Quality assessment of genetic markers used for therapy stratification. J Clin Oncol. 2003;21:2077–84.
Noguera R, Canete A, Pellin A, Ruiz A, Tasso M, Navarro S, et al. MYCN gain and MYCN amplification in a stage 4S neuroblastoma. Cancer Genet Cytogenet. 2003;140:157–61.
Bibikova M, Le J, Barnes B, Saedinia-Melnyk S, Zhou L, Shen R, et al. Genome-wide DNA methylation profiling using Infinium(R) assay. Epigenomics. 2009;1:177–200.
Kanduri M, Cahill N, Goransson H, Enstrom C, Ryan F, Isaksson A, et al. Differential genome-wide array-based methylation profiles in prognostic subsets of chronic lymphocytic leukemia. Blood. 2010;115:296–305.
Tarazona S, Garcia-Alcalde F, Dopazo J, Ferrer A, Conesa A. Differential expression in RNA-seq: a matter of depth. Genome Res. 2011;21:2213–23.
Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B. 1995;57:289–300.
Simon N, Friedman J, Hastie T, Tibshirani R. Regularization paths for Cox’s proportional hazards model via coordinate descent. J Stat Software. 2011;39:1–13.
Acknowledgements
This work was supported in part by FIS PS09/02323 and Prometeo 2013. Conselleria de Educació Cultura I Esports.
We thank Désirée Ramal for her help in the data management and all the Spanish collaborating hospitals who register their patients in neuroblastoma studies. We thank Ana Gonzalez-Neira (head of the Human Genotyping Unit at CEGEN-CNIO, Madrid, Spain) for her technical support.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
Conception and design were performed by YY, EG, AC, and VC. The methodology was developed by YY, EG, VR, RN, and MH. Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.) was performed by YY1, EG, AC, MH, VS, JFdM, and VC. Analysis and interpretation of data (for example, statistical analysis, biostatistics, computational analysis) were accomplished by DH, EV and AC. Writing, review, and/or revision of the manuscript were done by YY, EG, VR, DH, RN, MH, VS, AC, AC, JFdM, and VC. Administrative, technical, or material support (that is, reporting or organizing data, constructing databases) was provided by YY, EG, and AC. JFdM and VC supervised the study. All authors read and approved the final manuscript.
Additional files
Additional file 1:
Schematic mapping of CpGs sites within the promoter regions of the sixteen genes selected for pyrosequencing validation.
Additional file 2:
Illumina methylation microarray data of the sixteen genes selected for bisulfite pyrosequencing validation.
Additional file 3:
TDGF1 and RB1 methylation data from bisulfite pyrosequencing analysis in the validation cohort of patients.
Additional file 4:
Correlation graphs between TDGF1 expression and DNMT1 (A), DNMT3B (B) and DNMT3A (C) obtained at R2: microarray analysis and visualization platform ( http://r2.amc.nl ) using a cohort of 31 NB patients with MYCN amplification.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
Cite this article
Yáñez, Y., Grau, E., Rodríguez-Cortez, V.C. et al. Two independent epigenetic biomarkers predict survival in neuroblastoma. Clin Epigenet 7, 16 (2015). https://doi.org/10.1186/s13148-015-0054-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-015-0054-8