
Abstract
Ichthyosis covers a wide spectrum of diseases affecting the cornification of the skin. In recent years, new advances in understanding the pathophysiology of ichthyosis have been made. This knowledge, combined with constant development of pathogenesis-based therapies, such as protein replacement therapy and gene therapy, are rather promising for patients with inherited skin diseases. Several ongoing trials are investigating the potency of these new approaches and various studies have already been published. Furthermore, a lot of case series report that biological therapeutics are effective treatment options, mainly for Netherton syndrome and autosomal recessive congenital ichthyosis. It is expected that some of these new therapies will prove their efficacy and will be incorporated in the treatment of ichthyosis.
Similar content being viewed by others
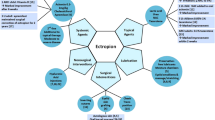


Background
Ichthyosis is a group of heterogeneous disorders affecting the cornification of the skin. Many ichthyosis variants are inherited and a subdivision is made between syndromic and non-syndromic variants (Table 1) [1]. All forms of ichthyosis are characterized by extensive scaling, hyperkeratosis, and often inflammation of the skin, resulting in erythroderma. Many patients report anxiety and depression and most of them experience a quality of life impairment [2,3,4]. Current treatment for ichthyosis is focused on symptom relief and includes emollients, keratolytics, and oral retinoids. The efficacy of these treatments is moderate and is usually not effective on inflammation of the skin [1, 5,6,7]. In the past few years, new advances in understanding the pathophysiology of ichthyosis have been made [8, 9]. Promising developments have been made in pathogenesis-based therapies, such as enzyme replacement therapy and gene therapy, and recent findings concerning the immune profile of ichthyosis patients have given new ground to repurpose biologicals (Fig. 1). The aim of this review is to provide an overview of the current status on pathogenesis-based therapy for ichthyosis. A Pubmed search was performed with the terms ichthyosis, therapeutics, biological products, molecular targeted therapy, enzyme replacement therapy and genetic therapy. Relevant articles were selected based on title and abstract. Selected articles are summarized in Table 2. Information about current ongoing clinical trials was retrieved from www.clinicaltrials.gov and summarized in Table 3.

Overview of the recent developments in molecular treatment of ichthyosis. ABE adenine base editor, ELA2 epidermal elastase 2, HSV-1 herpes-simplex virus-1, IL interleukin, KLK kallikrein-related peptidase, rAAV-2 recombinant adeno-associated virus-2, siRNA small interfering RNA, TALEN transcription activator-like effector nuclease, TG1 transglutaminase-1, TNF-α tumor necrosis factor alpha
Molecular pathology of ichthyosis
Mutations in over 50 genes are known to cause an ichthyosis phenotype (Table 1). [10] Four processes in skin cornification can be involved in the pathophysiology, i.e., the process of desquamation, impairment in the keratin synthesis, impairment in the synthesis of the cornified envelope or impairment in the organization of the stratum corneum extracellular lipid matrix. This leads to an altered epidermal differentiation, a defective epidermal barrier and increased transepidermal water loss (TEWL) [1, 5,6,7]. Below we will focus on the pathophysiology of the ichthyosis subtypes for which pathogenesis-based treatments have emerged in the past years.
X-linked recessive ichthyosis (XLI) is caused by mutations in the steroid sulfatase (STS) gene and is usually present soon after birth. It is characterized by hyperkeratosis and generalized polygonal brown scales. Extracutaneous associations include protracted delivery and cryptorchidism. Steroid sulfatase is responsible for the breakdown of cholesterol sulfate, necessary for corneodesmosome degradation and normal desquamation. A steroid sulfatase deficiency leads to accumulation of cholesterol sulfate in the stratum corneum resulting in impaired desquamation [7, 11].
Autosomal recessive congenital ichthyosis (ARCI) describes a spectrum of ichthyoses including lamellar ichthyosis (LI), congenital ichthyosiform erythroderma (CIE), and Harlequin ichthyosis (HI). The phenotype of each subtype differs. Children are often born with a collodion membrane. LI is characterized by rough, dark brown scaling, palmoplantar keratoderma and scarring alopecia. CIE expresses as erythroderma with fine white scaling. HI presents itself at birth with a very thick and rigid collodion membrane causing ectropion, eclabium, restriction of movements, and high risk of mortality. The genetic defect in ARCI is based on mutations in numerous genes (Table 1), [1, 7, 12, 13]. The most common form of ARCI is caused by mutations in the transglutaminase-1 (TGM1) gene, which encodes for the transglutaminase-1 (TG1) protein [10]. TG1 mediates crosslinking of cytoplasmic proteins onto the plasma membrane to form the cornified envelope. The lipoxygenase-hepoxilin and acylceramide pathways are essential for formation of the corneocyte lipid envelope and extracellular lipid membranes. Mutations in genes involved in these pathways account for most other forms of ARCI. The NIPAL4 and PNPLA1 genes will be elaborated more in detail, as these genes are involved in recent developments regarding replacement therapy [12, 14, 15]. NIPAL4 encodes for ichthyin, a transmembrane protein in the granular layer of the epidermis. It is thought to be a Mg2+ -transporter that contributes to lipid metabolism in the epidermal development. When the ichthyin function is defective, there will be decreased levels of acylceramide and impaired lipid structures of the stratum corneum, that may be related to the skin permeability barrier defect [16, 17]. PNPLA1 plays a role in the synthesis of ω-O-acylceramide. This lipid is a key component for the permeability layer of the epidermis. In the stratum corneum of PNPLA1-deficient humans, ω-O-acylceramide loss is linked to impairment of the corneocyte lipid envelope and the extracellular lipid lamellae [18]. HI is caused by mutations in the ABCA12 gene. ABCA12 is an epidermal keratinocyte lipid transporter, involved in secretion of lipids, and is mainly localized in the granular layer. Mutations in ABCA12 result in a loss of the lipid skin barrier [19, 20].
Epidermolytic ichthyosis (EI) is a form of ichthyosis that is characterized by congenital erythroderma, hyperkeratosis and blistering. EI is caused by autosomal dominant mutations in the keratin-1 (KRT1) or the keratin-10 (KRT10) gene, encoding for keratin 1 and keratin 10 proteins, respectively [1]. Mutations in the KRT1 and KRT10 genes induce clumping of the keratin intermediate filament (KIF) network in suprabasal keratinocytes, and cellular collapse. The mutations may also interfere with lamellar body secretion and therefore the lipid membrane formation, causing impaired barrier function [12, 21,22,23].
Congenital reticular ichthyosiform erythroderma (CRIE) results in a reticular ichthyosiform phenotype with yellow–brown scaling and erythroderma. It is due to autosomal dominant mutations in the tail regions of the KRT1 and the KRT10 gene, also causing collapse of the KIF network [24, 25]. Affected patients develop multiple confetti-like spots, that are genotypically wild-type and increase in surface and amount with growing age. This phenomenon is called revertant mosaicism and is caused mostly by a mechanism called mitotic recombination that results in somatic homozygosity of the wildtype allele [1, 26, 27].
Netherton syndrome (NS) is characterized by congenital scaly erythroderma, evolving into typical erythematous patches with peripheral scaling (ichthyosis linearis circumflexa), hair shaft abnormalities (trichorrhexis invaginata/bamboo hair) and atopic manifestations. It is an autosomal recessive disorder caused by mutations in the SPINK5 gene [1]. This gene encodes for lympho‐epithelial Kazal‐type‐related inhibitor (LEKTI). LEKTI is a serine protease inhibitor and regulates the degradation of corneodesmosomes by kallikrein-related peptidases (KLKs). KLK5 and matriptase cause a cascade that activates other KLKs, like KLK7 and KLK14, and elastase 2. In NS, there is a LEKTI deficiency resulting in unrestrained KLK activation in the epidermis. This obstructs stratum corneum cohesion and therefore leads to detachment of the stratum corneum, causing a severe permeability barrier defect. Furthermore, KLK5 and KLK14 activation results in the activation of proteinase-activated receptor 2 (PAR-2), which leads to synthesis of pro-inflammatory factors in keratinocytes [1, 28,29,30].
Sjögren-Larsson syndrome (SLS) is an autosomal recessive disorder involving the skin, eyes and central nervous system. The ALDH3A2 gene encodes for fatty aldehyde dehydrogenase (FALDH). This enzyme catalyzes the oxidation of fatty aldehyde from various lipid pathways. Mutations in the ALDH3A2 gene, lead to a FALDH deficiency. The fatty alcohols will accumulate and are diverted to other lipids, which may interfere with normal formation of lamellar body membranes in keratinocytes and lead to abnormal stratum corneum membrane. The accumulated fatty alcohols are thought to interfere with the function of the myelin membranes in the central nervous system, leading to neurological symptoms [1, 31,32,33].
SAM syndrome stands for severe dermatitis, multiple allergies and metabolic wasting syndrome. This congenital form of ichthyosis is due to mutations in the desmoglein-1 (DSG1) or the desmoplakin (DSP) gene, encoding for desmoglein-1 and desmoplakin, respectively. Both proteins are crucial components of desmosomes, necessary to connect the cell surface to the KIF cytoskeleton. Mutations in the DSG1 or DSP gene cause loss of cell-to-cell adhesion and differentiation disturbances [34, 35].
Peeling skin syndrome type 1 (PSS1) has clinically some resemblances with NS. PSS1 is characterized by erythroderma and superficial peeling of the skin. It is caused by mutations in the corneodesmosin gene (CDSN). Lack of corneodesmosin results in subcorneal splitting and detachment of corneocytes [36].
Keratitis-ichthyosis-deafness (KID) syndrome comprises a vascularizing keratitis, erythrokeratoderma skin lesions and a sensorineural hearing loss. KID syndrome is caused by mutations in the connexin-26 (Cx26) gene (GJB2). Gap junctions regulate cellular communication and activities, and are composed of multiple connexins. Cx26 is one of these connexins and is expressed in many epithelial organs, including the inner ear and the skin. Mutations in the GJB2 gene cause a dysfunction of Cx26 and therefore a dysfunction of the gap junctions [37,38,39,40].
CHILD syndrome (congenital hemidysplasia with ichthyosiform erythroderma and limb defects) is a cutaneous mosaicism, caused by mutations in the NAD(P)H steroid dehydrogenase-like (NSDHL) gene, which is involved in the process of cholesterol synthesis. The mutation leads to deficiency of bulk cholesterol, which is thought to adjust the keratinocyte membrane and the skin barrier, resulting in accumulation of upstream potentially toxic metabolic intermediates [41, 42].
Overview of new therapies in development for ichthyosis
pathways
Biological therapy includes a wide range of products, such as monoclonal antibodies that aim on targeting specific marks, e.g., tumor necrosis factor alpha (TNF-α), interleukin-13 (IL-13), interleukin-17 (IL-17) and interleukin-23 (IL-23). There is substantial experience in the usage of biologics in inflammatory skin diseases, such as psoriasis and atopic dermatitis (AD). Since they are already commercially available and dermatologists are experienced in their usage, the path for clinical testing could be rather rapid [9].
In 2011, Fontao et al. [43] provided the first report of a patient with NS who was successfully treated with infliximab, an inhibitor of TNF-α. After 12 weeks of treatment, the skin was clear of inflammatory lesions, however xerosis and ichthyosis did not improve. In 2017, Roda et al. [44] reported another NS patient who experienced a clearance of inflammatory lesions after initiation of infliximab. Nonetheless, TNF-α inhibitors are not recommended in the treatment of NS according to the European guideline of care for ichthyosis, due to paucity of data, risk of non-melanoma skin cancers and recurrent infections [45].
Only recently, the repurposing of other biologics has been proposed. The rationale of repurposing biologics for ichthyosis is based on studies of Paller et al. [46,47,48] In one of their studies (2017) [46], the immune profile of LI, EI, CIE and NS was examined and compared to healthy controls as well as AD and psoriasis patients. An IL-17 dominant profile, similar to psoriasis, was found in all forms of ichthyosis. NS patients showed the highest induction of IL-17 and interleukin-22 (IL-22) pathway genes. The IASI-E (Ichthyosis Area and Severity index—erythema) score showed a significant correlation with IL-17A levels and TEWL. Similar results were shown in the studies that followed, where elevated IL-17 and TNF-α levels in skin and blood samples were found [47, 48]. An important but still unanswered question is whether the cutaneous symptoms in ichthyosis are caused by the overactive Th17 pathway, or if Th17 activation is only a sign of a systemic immune response without a pathogenic relationship to the skin abnormalities [49].
Secukinumab is a recombinant human monoclonal antibody that targets the IL-17A cytokine [50]. Luchsinger et al. [51] reported a case series of four patients with NS that were treated with secukinumab during 3–12 months. Overall, they reported a reduction in IASI from 55 to 88% after 6 months. However, response was less pronounced in two patients with a milder variant of NS. Blanchard et al. [52] reported a case of a patient with NS who had facial erythema and frequent flares of ichthyosis on the trunk and extremities. Treatment with secukinumab resulted in complete clearance of facial erythema, with one mild flare of plaques in three years of follow-up.
Another biological therapeutic that targets IL-17A is ixekizumab, which seems to have higher affinity for IL-17A than secukinumab [53]. Barbieux et al. [54] reported three NS patients, two with the ichthyosis linearis circumflexa (NS-ILC) phenotype and one patient with a scaly erythroderma (NS–SE) phenotype. They all had enhanced levels of IL-17 related cytokines and were treated with ixekizumab for 6 months. IASI-S, Dermatology Life Quality Index (DLQI) and pruritus scores showed a significant reduction of about 50% after 3 months. The IASI-E scores were only reduced for the NS-ILC patients. After 6 months, all scores remained decreased in the NS-ILC patients. Regarding the NS-SE patient, all scores returned back to baseline, except for the IASI-E score.
Ustekinumab indirectly inhibits IL-17 by targeting interleukin-12 (IL-12) and interleukin-23 (IL-23) [55]. Volc et al. [56] reported a 15-year-old girl with NS who was treated with ustekinumab. The skin improved substantially within 4 weeks and no relapse occurred after one year. Ustekinumab has also been used in CARD14 associated papulosquamous eruption (CAPE), an inflammatory dermatosis with palmoplantar hyperkeratosis and erythroderma. The familial variant is caused by mutations in the CARD14 gene, which is known to be an activator of the noncanonical nuclear factor-kappa B (NF-κB) pathway, which leads to inflammation. Affected patients express an increase in CARD14 in the epidermis. Case reports show benificial effects for CAPE patients who have been treated with ustekinumab [57,58,59].
Dupilumab is used in the treatment of AD. It blocks the interleukin-4 receptor and therefore inhibits IL-4 and interleukin-13 (IL-13). IL-4 and IL-13 stimulate the Th2 cells, that regulate the pro-allergic adaptive immune response. Dupilumab has been proposed for NS, since the clinical presentations of NS and eczema show resemblances. A few case reports, that report a total of 5 NS patients, describe that dupilumab is effective in NS [60,61,62,63]. Süssmuth et al. [63] described a 12-year-old girl and an 8-year-old boy with NS that were treated with dupilumab. After 4 months, the Netherton Area Severity Assessment (0–72) dropped from 33 to 11.7 for the girl and from 50.5 to 18 for the boy. For both children, the pruritus dropped from 8 to 3 on the numeric rating scale (NRS; 0–10). These results remained stable after 12 months of treatment. However, Murase et al. [64] reported two cases of NS patients who were treated with dupilumab, where the pruritus gradually reoccurred in the second week after the dupilumab injection. In a case report from Aktas et al. [65] the skin lesions and itch worsened after an initial good response during the first 6 weeks of therapy.
A limited number of case reports describe usage of biological therapy in the other subtypes of ichthyosis. One case reports a 4-year-old boy with ARCI, based on a NIPAL4 mutation, and a large joint inflammatory arthropathy of unknown origin. He was given ustekinumab, which gave a good response at first, but after 12 weeks a relapse occurred. A dosage increase resulted in a clearance of erythema and no further joint complaints [66]. Haiges et al. [67] reported a 20-year-old man with a clinical diagnosis of CIE, but genetic testing did not disclose a mutation in ichthyosis associated genes. Treatment with secukinumab was started based on a biopsy showing an inflammatory infiltrate rich in IL-17. Significant improvements in scaling and erythroderma were reached, however punch biopsies showed that acanthosis, hyper- and parakeratosis were only slightly reduced.
Hernández-Martin et al. [68] presented a case about a 9-month-old girl with a clinical presentation of SAM syndrome, including erythroderma, hyperkeratosis, pruritus, recurrent episodes of sepsis and severe growth retardation. The genetic analysis did not show a mutation in the DSG1 gene. However, the immunofluorescence staining showed abnormalities in the desmosomes. The patients’ IL-17-producing T-cell frequency was 67 times higher compared to healthy controls. She was given 75 mg of secukinumab every week for the first 4 weeks, which was then continued on a monthly basis. At week 35 of treatment, the pruritus had almost disappeared and the weight-for-height z score had increased from − 2.3 SD to + 1.9 SD. Th17 and IL-17 producing T cell levels were more than halved. Another case report described two patients with SAM syndrome due to mutations in the desmoplakin gene. Treatment with ustekinumab resulted in a 58% and 59% reduction in total IASI and a lower TEWL by 16 weeks [69].
Results from a double-blind placebo-controlled study with secukinumab in different subtypes of ichthyosis from Lefferdink et al. [70] have recently been published. Included patients had a diagnosis of EI, NS, CIE or LI and received secukinumab or placebo every 4 weeks for 16 weeks in total, followed by a 16-week open-label phase and a 20-week extension for safety. IASI, the Visual Index for Ichthyosis Severity (VIIS), TEWL, and patient reported outcomes were measured. Furthermore, skin biopsies were taken to study biomarkers of the Th17 pathway. There was no significant difference between secukinumab and placebo group in ichthyosis severity scores, TEWL and patient reported outcome measures at week 16. The group who received secukinumab first, only had a significant reduction in IASI-E and VIIS at week 32 and in VIIS at week 52. A total of five patients (two with NS; two with EI; one with CIE) continued the treatment post-study because of self-perceived improvement. These patients had a median total IASI decrease of 36% (range 29–50%) at the end of the study. Th17 related biomarkers did not show a significant reduction in week 16 and 32 compared to baseline. There was a lack of response in all LI patients, which suggest that only a few ichthyosis subtypes respond to IL-17 treatment. The study was performed on a limited population with different subtypes, resulting in small subgroups. Unfortunately authors did not provide information about genetic mutations of the treated subjects so further analysis per subtype is not possible.
In conclusion, the use of biological therapies could be beneficial in the treatment of several ichthyosis subtypes. However, the response is mostly seen as reduction of the inflammatory component and pruritus. The effect on scaling seems to be more variable. A larger randomized controlled trial (RCT) and/or large open-label cohort is necessary to rule out a possible publication bias and result in more specific data and correlation with genetic mutations and immunological profile. This could help to predict which subtypes, phenotypically and genetically, show better response to anti IL-17 therapy.
Furthermore, there are case reports that describe a good response at first, but a decline after several weeks [54, 64,65,66]. Perhaps biological therapies in ichthyosis patients require an increased dosage and/or interval compared to the currently used regimen indicated for psoriasis and atopic dermatitis. Most case reports describe effects on young patients. It is possible that also the length of disease affects the therapeutic response. In addition, it would be interesting to further investigate if certain biomarkers, such as increased cytokine levels, could help to choose between the different biological therapies [49]. It is proposed that upstream molecules, such as IL-23 or IL-36 may have a better effect [70]. Several registered clinical trials are ongoing and could provide more information regarding the use of all these different biological therapies. These ongoing clinical trials (Fig. 1) include studies regarding dupilumab (anti IL-4), adalimumab (anti TNF-α), ustekinumab (anti IL-12/IL-23) and imsidolimab (anti IL-36).
Small molecules
Small molecules are used to inhibit certain proteins, such as protein kinases. Because of their size and molecular properties, they are able to interact with specific parts of the targeted protein and therefore inhibit it without disrupting the pathways of other proteins [71]. They are currently used to treat inflammatory skin diseases, such as apremilast in psoriasis treatment and baricitinib for atopic dermatitis. For these indications, small molecules have shown promising results [72, 73]. Regarding ichthyosis, only a few in vitro and mouse-model studies have been conducted in this field.
Preclinical studies
Small molecules could be applicable in NS, where the dysfunctional SPINK5 leads to an unopposed KLK5 activity. Several studies have reported that knockdown or ablation of KLK5 leads to improvements of NS in in vivo mouse-models and in in vitro skin models with human epidermal keratinocytes [74,75,76]. Liddle et al. [30] discovered GSK951, a small molecule inhibitor of KLK5. GSK951 was applied topically for three days to a transgenic murine model expressing human KLK5.After day one, the TEWL was decreased to approximately 50%. However, the mice that were treated topically with the vehicle cream without GSK951 had a two-fold increase of TEWL. Thus, it is not possible to state that the GSK951 causes the TEWL decrease. However, this data does show that topical application of GSK951 is able to inhibit KLK5 activity in the skin. Furthermore, the inflammatory cytokines did show a significant decrease, mainly the IL-17 cytokines.
Chen et al. [77] showed the potency of Sunflower Trypsin Inhibitor (SFTI) to treat NS. SFTI is a small peptide and potent inhibitor of trypsin. Analogues of SFTI can be used to redirect its inhibitory capacities to other proteases. In this study, they showed that the developed SFTI is able to effectively inhibit KLK5, KLK14 and KLK7. The inhibitory potency has not been tested in animal or human skin models so far.
Clinical studies
Mazereeuw-Hautier et al. [78] conducted an in vivo study with NS patients, where they used a protease inhibitor alpha1-antitrypsin, similar to the missing protease inhibitor (LEKTI) in NS. They performed a RCT using recombinant human alpha1-antitrypsin gel and placebo for 21 days. Erythema and scaling were scored, but results did not show statistically significant differences.
A new clinical trial has been initiated in 2021, investigating application of a cream with substance SXR1096 in NS patients during 1 month (NCT05211830). SXR1096 is a potent inhibitor of KLK5, KLK7 and KLK14. Twenty NS patients will participate in this phase I/II trial to assess the safety and effectiveness as compared to placebo [79].
In 2018, Aldeyra Therapeutics Inc [80]. announced their phase III randomized double-blind vehicle-controlled trial with topical application of ADX-102 1% (Reproxalap) in SLS patients (NCT03445650), which has now been completed. ADX-102 is a small molecule, able to degrade aldehydes, and could be helpful to reduce the lipid accumulation as result of the FALDH deficiency in SLS. Results of this trial are not available yet.
Besides the trials described above, two other registered clinical trials are still ongoing (Fig. 1), considering treatments for NS and SLS. These trials include the use of BPR 277, a KLK7 and epidermal elastase 2 (ELA2) small molecule inhibitor in NS, and NS2 cream, an aldehyde binding small molecule for SLS.
Replacement therapy
Enzyme replacement therapy (ERT) entails the replacement of a deficient structural protein or enzyme. It has been used for over 20 years for lysosomal storage disorders like Gaucher and Fabry disease, where it has shown high efficacy [81]. For ichthyosis, it could be applicable for recessive forms, where a deficiency of a specific protein leads to the phenotype. For systemic diseases, as Fabry disease, systemic application is already available, while for ichthyosis only topical treatment has been tested so far [82]. Lipid replacement is also a promising approach for several ichthyoses. Indeed, many of the ichthyoses result from the mutation of genes encoding enzymes or cofactors involved in the metabolism of stratum corneum lipids.
Preclinical studies
TG1 deficient skin in ARCI is a good candidate for ERT [83]. Aufenvenne et al. [82] tested this in vivo with a mouse-model with humanized skin. They prepared liposomes with encapsulated recombinant human TG1. In-situ TG1 activity assays in cryosections showed a restoration of the TG1 activity. TEWL measurements of TG1-treated mice were compared to mice treated with retinoid cream. The TG1 group showed no increase in TEWL compared to normal human skin and indicated a restoration of the epidermal barrier, whereas the retinoid group presented a markedly increased TEWL value.
More recently, Plank et al. [84] used full-thickness skin equivalents derived from fibroblasts and keratinocytes of TGM1-related ARCI patients. They applied TG1 topically using thermoresponsive nanogels. Improvement of barrier function was tested with Lucifer yellow permeability tests. Before treatment, a 59-fold increase of the amount of Lucifer yellow passing through patient skin equivalents was found, compared to normal keratinocytes. After treatment this was decreased to 1.2. This indicated signs of epidermal barrier function restoration of the TG1-deficient skin in vitro.
Protein replacement therapy has also been studied for PSS1. Valentin et al. [36] conducted an in vitro study with corneodesmosin-deficient human epidermal equivalents. A specific carrier system was developed for delivery of liposome encapsulated recombinant human corneodesmosin. Immunofluorescence showed corneodesmosin staining at the transition of stratum corneum to stratum granulosum. However, the staining of healthy control epidermal equivalents was stronger and more pericellular. Results also showed alterations of the stratum corneum after treatment, comparable to normal equivalents. Furthermore, improved barrier function was achieved, as studied by toluidine blue penetration assay.
Grond et al. [85] investigated in vivo mouse-models with ARCI based on a PNPLA1 deficiency. They found that topical application of epidermal lipids from wild type mice gave a 14-fold increased level of covalent linkage of ω-O-acylceramide to corneocyte proteins in newborn PNPLA1-deficient mice, then in mutant skins treated with epidermal lipids from PNPLA1-deficient mice. Mauldin et al. [86] performed an in vivo study, where they topically applied ω-O-acylceramides in NIPAL4-mutated dogs. As shown by electron microscopy analysis of skin biopsies from affected dogs, the treatment normalized the cornified lipid envelope. However, it failed to correct the clinical phenotype in affected canines. This was probably due to cytotoxicity attributable to accumulation of free fatty acids or other proximal metabolites.
Clinical studies
Lipid replacement could be effective for CHILD syndrome, which is caused by a deficiency in bulk cholesterol. In 2011, Paller et al. [87] proposed topical application of lovastatin and cholesterol. They reported two CHILD affected individuals, who displayed ichthyosiform plaques. They were treated twice daily with a 2% lovastatin/2% cholesterol lotion. After 6 months of treatment, treated areas were almost completely normalized. More recently, Bergqvist et al. [42] presented two cases of CHILD syndrome. One 11-year-old girl with a verruciform xanthoma was treated twice a day with 2% cholesterol and 2% lovastatin cream. After 4 weeks, the lesion had cleared. After 8 months, a recurrence of the lesion was successfully treated and the lesion was still in remission after 7 months being off treatment. However, the cream had no effect on a verruciform xanthoma on the foot of the second patient. Addition of 12% glycolic acid once a day resulted in a substantially decrease of the xanthoma after 12 months. Similar results were reached in another case report about a 2-month-old girl with lesions on the legs, also treated with 2% cholesterol and 2% lovastatin lotion twice a day [88]. Additionally, several case reports have shown promising results of simvastatin monotherapy for CHILD syndrome associated skin lesions. Yu et al. [89] reported four patients who were treated with simvastatin lotion. First, 2.5% simvastatin lotion was used, and after 3 months the individuals received 5% simvastatin. Skin lesions slightly improved after treatment with 2.5% simvastatin, but a marked improvement was seen after switching to 5% simvastatin. Other case reports of simvastatin monotherapy show similar responses [90, 91].
In conclusion, enzyme and lipid replacement are very interesting strategies to improve ichthyosis symptoms, by directly substituting the deficient substance. More studies should be encouraged to look at the clinical effect and consider the possible toxicity arising from accumulation of metabolic intermediates. There are currently no registered ongoing clinical trials.
Gene therapy
Gene therapy is aiming to restore the wild type gene function. In another genodermatosis, epidermolysis bullosa (EB), gene therapy has led to promising results. Junctional EB patients have been successfully transplanted with genetically corrected autologous skin grafts [92, 93]. Gene therapy can be achieved by different approaches. A classical approach is introduction of the wild type gene copy into cells, using a viral vector. However, there are safety concerns regarding insertional mutagenesis that comes with the use of a viral vector [94, 95]. Also, introduction of viruses leads to production of antibodies, which could affect subsequent transduction of the same virus [96].
Another technique is gene editing. With gene editing, sequence specific nucleases are used to induct artificial double strand DNA breaks. This will activate the natural DNA repair system, without the risk of random integration of the transgene as with the use of a viral vector. Examples of nucleases are transcription activator-like effector nuclease (TALEN) and the bacterial CRISPR/Cas9 nuclease system [97]. Base editing is one of the most recent techniques of gene editing. With base editing, single nucleotide variants are introduced into DNA or RNA in living cells. It directly modifies nucleobases by introduction of point mutations, without DNA cleavage. Two forms of DNA base editors are adenine base editors (ABE) and cytosine base editors [98]. Furthermore, one could use small interfering RNA (siRNA) to silence mutated alleles in dominant disorders.
Preclinical studies
In 1997, Freiberg et al. [99] have explored gene therapy for XLI. They transduced the STS gene with a retroviral vector to primary keratinocytes derived from XLI patients. In vitro restoration of STS protein expression and STS enzymatic activity was seen. Transduced XLI keratinocytes were then grafted onto immunodeficient mice. After grafting, the transduced XLI epidermis showed STS expression by immunostaining, and a normalization of TEWL and histologic appearance. No further research regarding gene therapy for XLI has been found after this study.
Akayima et al. [20] have proposed the possibility of gene therapy for HI. They have cultured keratinocytes from HI patients and performed corrective gene transfer of the ABCA12 gene with a pCMV-tag4B vector. Before genetic correction, double immunostaining showed a congested glucosylceramide distribution pattern in HI. Glucosylceramide is a major lipid component of the lamellar granules and is essential for the epidermal permeability barrier. After corrective gene transfer, a significant increase in number of cells that displayed a normal glucosylceramide distribution pattern was seen (from 6.98 to 16.7%), indicating recovery of lamellar granule lipid secretion.
In 2005 and 2006, Haug et al. [33, 100] have already provided promising results in vitro for gene therapy in SLS. They first conducted a study where they transferred FALDH in keratinocytes using recombinant adeno-associated virus-2 vectors (rAAV-2) in a hamster model. This resulted in a normal FALDH expression. One year later they applied the same technique on keratinocytes from human SLS patients, where they achieved an increase of 60–70% from normal FALDH activity.
March et al. [101] used TALENs for gene editing of the KRT10 gene in EI. In this ex vivo study, they targeted a specific region of the KRT10 gene known to cause a premature stop, leading to a genetic knockout. They tested the TALEN on keratinocytes derived from EI patients with mutant KRT10 alleles, using modified single cell clones and murine xenodraft models. The on-target activity was measured and demonstrated strong gene disruption. Off-target activity was not observed. They suggested that epidermal generation from a single-cell clonal expansion from gene-edited keratinocytes is feasible.
For NS, Krystal Biotech inc. developed a replication-defective herpes simplex virus type 1 (HSV-1) vector encoding human SPINK5 (KB104) for topical administration. HSV-1 can penetrate the skin and does not insert itself into the genome of the host. They presented their results at the annual meeting of the Society for Investigative Dermatology (2019). In the abstract provided of this in vitro study, human keratinocytes that were administered KB104 produced functional SPINK5 and were able to inhibit KLK5. Real time PCR and immunohistochemistry revealed that topical or intradermal administration of KB104 in mice resulted in expression of human SPINK5.
Regarding LI, Choate et al. [102] have investigated potential effects of gene therapy in vitro. They have used a retroviral vector to transfer the TGM1 gene into keratinocytes of LI patients. In > 98% of the cells, expression of functional TG1 protein was achieved, resulting in restoration of epidermal architecture and barrier function.
The potential of gene therapy for TGM1 deficiency has further been explored by Freedman et al. [103] The same strategy as described above for KB104 was used for TG1-deficient ARCI patients, where HSV-1 encoded for human TG1 (KB105). KB105 was analyzed in vitro with patient derived keratinocytes and in vivo, using murine models. This resulted in increased TG1 protein expression, whereas the control group showed no TG1 expression. Assessment of toxicity showed no adverse effects. Investigation of topical gene therapy has also been done by Gurevich et al. [104] for recessive dystrophic epidermolysis bullosa (RDEB), which is caused by mutations in the COL7A1 gene that normally encodes for collagen VII (C7). They investigated beremagene geperpavec (B-VEC), a replication defective HSV-1 vector containing two copies of the COL7A1 coding sequence. First, they evaluated the effect of B-VEC on C7 expression in keratinocytes and fibroblasts cultured of RDEB patients in vitro. Results showed restored C7 expression after 48 h of treatment. They administered intradermal B-VEC injections to C7-deficient mice in vivo, and observed C7 expression at day 3 and 7 by indirect immunofluorescence microscopy, and also measured COL7A1 DNA and C7 transcript expression levels. This was further confirmed in human RDEB skin xenografted onto immunodeficient mice, treated with placebo or B-VEC injections. Control grafts showed dermal-epidermal separation, which was not observed in the xenografts treated with B-VEC. Immunofluorescence showed C7 expression in the xenografts after B-VEC, and was absent in the control grafts.
Dang et al. [105] have investigated the potential of base editing with the ABE system in human embryos. They first generated a homozygous mutant cell model with a c.607C > T mutation in the TGM1 gene. Two different ABEs, called ABEmax-NG and Sc-ABEmax, showed potential in correcting the pathogenic mutation. Subsequently, oocytes and sperm of a couple both carrying a heterozygous c.607C > T TGM1 mutation were used to create heterozygote mutant human embryos by in vitro maturation and intracytoplasmic sperm injection. In the ABEmax-NG group, seven embryos were collected and two of them displayed a completely wild genotype. In the Sc-ABEmax group, five of the eight embryos showed a completely wild genotype. The normalized editing efficiency for the ABE systems was 73.8% and 78.9%, respectively. DNA off-target analysis was performed by whole-genome sequencing and deep sequencing and showed no obvious off-target activity. However, a higher number of RNA single nucleotide variations was seen in embryos injected with the ABE system. Improvement of editors could reduce these RNA off-target effects and thus improve safety.
The technique of siRNA has been explored by Lee et al. [106] for KID syndrome, applied on keratinocytes with mutations in the GJB2 gene. This in vitro study demonstrated inhibition of the mutated GJB2 allele in the keratinocytes, indicated by improvement of its gap junction and hemichannel function. This could potentially improve the skin changes. A human-murine chimeric skin graft model was developed, and a way for a topical approach of siRNA on this in vivo model is currently under investigation.
Clinical studies
To further explore the potential of the above described KB105, Krystal Biotech Inc. presented interim results of a phase 1/2 placebo-controlled trial in TGM1 associated ARCI patients at the annual meeting of the Society for Investigative Dermatology (2021). Three adults were included and received either topical KB105 or placebo. Biopsies were taken from KB105-treated areas to evaluate the effects. Treated areas showed detectable functionally active TG1, confirmed by quantitative real-time PCR, immunofluorescence and in situ analysis. Clinically, the treated areas showed a reduction of two points on the Investigator’s Global Assessment (IGA) scale.
Di et al. [107] generated gene-modified epithelial sheets using keratinocytes of NS patients and transduced them with a lentiviral vector encoding SPINK5. Successful generation of epithelial sheets was achieved in three of the six included patients, and one subject was suited for engraftment. After 1-month, complete healing had occurred with mild hypopigmentation and KLK5 expression similar to that in normal skin. However, KLK5 expression decreased again by month 3, 6 and 12. The treatment was considered safe and feasible.
The results of Gurevich et al. [104] regarding B-VEC for RDEB patients as described above, were further investigated in a phase I/II trial. In this trial, nine RDEB patients received B-VEC treatment. Wound area surface was treated topically with B-VEC or placebo for 12 weeks. B-VEC treated wounds were statistically significant different (P = 0.0026) from placebo treated wounds, based on wound closure responder analysis. However, results showed a numerically favorable trend towards B-VEC regarding time to and duration of wound closure. At weeks 8, 10 and 12, a statistically significant effect (P < 0.025) of reduction in wound surface area was seen. There were no serious or significant adverse events observed. This treatment focusses however on wounds and blisters, and not on the intact epidermis, which is different than in case of ichthyosis patients. One could thus wonder if the penetration and delivery with an HSV vector in ichthyosis would have the same efficacy as it has in EB.
Natural gene therapy
Revertant mosaicism is a phenomenon in which cells containing mutated copies of the gene undergo a second genetic event that leads to spontaneous correction of the gene [108]. This manifests itself on the skin as areas that seem phenotypically normal. This mechanism has been described in all forms of EB. Attempts to use revertant mosaicism as a therapy have not yet been very successful. However, one case concerning a junctional EB patient reports punch grafting of the revertant spot, with transplantation of the biopsy specimen. This resulted in re-epithelization of the wound with wild type skin [109]. One ichthyosis subtype, CRIE, displays the same phenomenon, but no studies have been published about the usage of reverted keratinocytes as therapy for CRIE [95, 108].
Gene therapy has not yet been explored as widely as biologicals have. The few studies that have been published show that gene therapy could be a safe and effective option, but further investigation is essential. Ongoing, registered clinical trials (Fig. 1) include the above described KB105 for TGM1-related ARCI, and a study about grafting of autologous epidermal sheets generated from genetically modified skin stem cells with a lentiviral vector for NS.
Conclusion and future
In the past few years, relevant research regarding the molecular pathophysiology of ichthyosis has been conducted. This knowledge provides a foundation to target defective genes or the proteins that they encode for. These approaches include small molecules, protein and lipid replacement, and gene replacement and editing therapy. To date, these approaches are still in (pre-)clinical stage, ranging from phase I to III for clinical research. There is a need for more safety data, before these treatment options can be considered for application in humans. Only a few gene therapies are currently considered in the treatment of ichthyosis. Several therapies, such as the use of siRNA and CRISPR/Cas9, are already in the pre-clinical testing for EB, where they show promising results. These therapies have not yet been explored for ichthyosis, but could lead to new perspectives in the search for a cure. Next to the development of new therapies, drugs used for other diseases have been repurposed as well. Due to increasing knowledge about the immune profile, usage of biological therapeutics was considered in the treatment of ichthyosis. These biologics are already widely prescribed for other indications and are considered relatively safe. A growing amount of reports is available on this topic. Nonetheless, the inflammatory dysregulation in ichthyotic skin has only been explored in a few ichthyosis forms and with a relatively small patient number. Further research should investigate biomarkers in larger cohorts including all ichthyosis variants. Studies with long-term follow-up are necessary to show if the effects of biologics in ichthyosis are durable. Different treatment strategies, acting on different levels of the underlying pathophysiology, might be combined in one patient to optimize the clinical results. Furthermore, the applicability of pathogenesis-based therapies should be addressed, since they are likely to be gene-, or even individual-specific.
Availability of data and materials
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
Abbreviations
- ABE:
-
Adenine base editor
- AD:
-
Atopic dermatitis
- ARCI:
-
Autosomal recessive congenital ichthyosis
- B-VEC:
-
Beremagene geperpavec
- CAPE:
-
CARD14 associated papulosquamous eruption
- CIE:
-
Congenital ichthyosiform erythroderma
- CHILD syndrome:
-
Congenital hemidysplasia with ichthyosiform erythroderma and limb defects syndrome
- CRIE:
-
Congenital reticular ichthyosiform erythroderma
- C7:
-
Collagen VII
- DSG1:
-
Desmoglein-1
- DSP:
-
Desmoplakin
- EI:
-
Epidermolytic ichthyosis
- ELA2:
-
Epidermal elastase 2
- ERT:
-
Enzyme replacement therapy
- FALDH:
-
Fatty aldehyde dehydrogenase
- HI:
-
Harlequin ichthyosis
- IASI:
-
Ichthyosis assessment severity index
- IASI-E:
-
Ichthyosis assessment severity index-erythema
- IASI-S:
-
Ichthyosis assessment severity index-scaling
- IGA:
-
Investigator’s Global Assessment
- IL:
-
Interleukin
- JAK:
-
Janus kinase
- Ki :
-
Inhibitory constant
- KID syndrome:
-
Keratitis–ichthyosis–deafness syndrome
- KLK:
-
Kallikrein-related peptidase
- KRT1:
-
Keratin-1
- KRT10:
-
Keratin-10
- LEKTI:
-
Lympho‐epithelial Kazal‐type‐related inhibitor
- LI:
-
Lamellar ichthyosis
- NS:
-
Netherton syndrome
- ILC:
-
Ichthyosis linearis circumflexa
- SE:
-
Scaly erythroderma
- NRS:
-
Numeric rating scale
- PAR-2:
-
Protease-activated receptor 2
- PSS1:
-
Peeling skin syndrome type 1
- Raav-2:
-
Recombinant adeno-associated virus-2
- RDEB:
-
Recessive dystrophic epidermolysis bullosa
- SAM:
-
Severe dermatitis, multiple allergies and metabolic wasting
- siRNA:
-
Small interfering RNA
- SLS:
-
Sjögren–Larsson syndrome
- SFTI:
-
Sunflower trypsin inhibitor
- STS:
-
Steroid sulfatase
- TALEN:
-
Transcription activator-like effector nuclease
- TEWL:
-
Transepidermal water loss
- TGM1:
-
Transglutaminase-1 (gene)
- TG1:
-
Transglutaminase-1 (protein)
- tNG:
-
Thermoresponsive nanogels
- Th:
-
T-helper cell
- TNF-α:
-
Tumor necrosis factor alpha
- VIIS:
-
Visual Index for Ichthyosis Severity
- XLI:
-
X-linked recessive ichthyosis
References
Oji V, Tadini G, Akiyama M, Blanchet Bardon C, Bodemer C, Bourrat E, et al. Revised nomenclature and classification of inherited ichthyoses: results of the first ichthyosis consensus conference in soreze 2009. J Am Acad Dermatol. 2010;63(4):607–41.
Sun Q, Ren I, Zaki T, Maciejewski K, Choate K. Ichthyosis affects mental health in adults and children: a cross-sectional study. J Am Acad Dermatol. 2020;83(3):951–4.
Troiano G, Lazzeri G. A review of quality of life of patients suffering from ichthyosis. J Prev Med Hyg. 2020;61(3):E374–8.
Mazereeuw-Hautier J, Dreyfus I, Barbarot S, Serrentino L, Bourdon-Lanoy E, Ezzedine K, et al. Factors influencing quality of life in patients with inherited ichthyosis: a qualitative study in adults using focus groups. Br J Dermatol. 2012;166(3):646–8.
Mazereeuw-Hautier J, Vahlquist A, Traupe H, Bygum A, Amaro C, Aldwin M, et al. Management of congenital ichthyoses: European guidelines of care, part one. Br J Dermatol. 2019;180(2):272–81.
Oji V, Traupe H. Ichthyosis: clinical manifestations and practical treatment options. Am J Clin Dermatol. 2009;10(6):351–64.
DiGiovanna JJ, Robinson-Bostom L. Ichthyosis: etiology, diagnosis, and management. Am J Clin Dermatol. 2003;4(2):81–95.
Shwayder T. Disorders of keratinization: diagnosis and management. Am J Clin Dermatol. 2004;5(1):17–29.
Paller AS. profiling immune expression to consider repurposing therapeutics for the ichthyoses. J Invest Dermatol. 2019;139(3):535–40.
Fischer J, Bourrat E. Genetics of inherited ichthyoses and related diseases. Acta Derm Venereol. 2020;100(7):adv00096.
Fernandes NF, Janniger CK, Schwartz RA. X-linked ichthyosis: an oculocutaneous genodermatosis. J Am Acad Dermatol. 2010;62(3):480–5.
Vahlquist A, Fischer J, Torma H. Inherited nonsyndromic ichthyoses: an update on pathophysiology, diagnosis and treatment. Am J Clin Dermatol. 2018;19(1):51–66.
Youssefian L, Vahidnezhad H, Saeidian AH, Sotoudeh S, Mahmoudi H, Daneshpazhooh M, et al. Autosomal recessive congenital ichthyosis: CERS3 mutations identified by a next generation sequencing panel targeting ichthyosis genes. Eur J Hum Genet. 2017;25(11):1282–5.
Zhang H, Ericsson M, Westrom S, Vahlquist A, Virtanen M, Torma H. Patients with congenital ichthyosis and TGM1 mutations overexpress other ARCI genes in the skin: part of a barrier repair response? Exp Dermatol. 2019;28(10):1164–71.
Munoz-Garcia A, Thomas CP, Keeney DS, Zheng Y, Brash AR. The importance of the lipoxygenase-hepoxilin pathway in the mammalian epidermal barrier. Biochim Biophys Acta. 2014;1841(3):401–8.
Honda Y, Kitamura T, Naganuma T, Abe T, Ohno Y, Sassa T, et al. Decreased skin barrier lipid acylceramide and differentiation-dependent gene expression in ichthyosis gene Nipal4-knockout mice. J Invest Dermatol. 2018;138(4):741–9.
Casal ML, Wang P, Mauldin EA, Lin G, Henthorn PS. A defect in NIPAL4 Is associated with autosomal recessive congenital ichthyosis in american bulldogs. PLoS ONE. 2017;12(1):e0170708.
Hirabayashi T, Murakami M, Kihara A. The role of PNPLA1 in omega-O-acylceramide synthesis and skin barrier function. Biochim Biophys Acta Mol Cell Biol Lipids. 2019;1864(6):869–79.
Kelsell DP, Norgett EE, Unsworth H, Teh MT, Cullup T, Mein CA, et al. Mutations in ABCA12 underlie the severe congenital skin disease harlequin ichthyosis. Am J Hum Genet. 2005;76(5):794–803.
Akiyama M, Sugiyama-Nakagiri Y, Sakai K, McMillan JR, Goto M, Arita K, et al. Mutations in lipid transporter ABCA12 in harlequin ichthyosis and functional recovery by corrective gene transfer. J Clin Invest. 2005;115(7):1777–84.
Arin MJ. The molecular basis of human keratin disorders. Hum Genet. 2009;125(4):355–73.
Arin MJ, Oji V, Emmert S, Hausser I, Traupe H, Krieg T, et al. Expanding the keratin mutation database: novel and recurrent mutations and genotype-phenotype correlations in 28 patients with epidermolytic ichthyosis. Br J Dermatol. 2011;164(2):442–7.
Vodo D, Sarig O, Peled A, Samuelov L, Malchin N, Grafi-Cohen M, et al. Recessive epidermolytic ichthyosis results from loss of keratin 10 expression, regardless of the mutation location. Clin Exp Dermatol. 2018;43(2):187–90.
Choate KA, Lu Y, Zhou J, Choi M, Elias PM, Farhi A, et al. Mitotic recombination in patients with ichthyosis causes reversion of dominant mutations in KRT10. Science. 2010;330(6000):94–7.
Choate KA, Lu Y, Zhou J, Elias PM, Zaidi S, Paller AS, et al. Frequent somatic reversion of KRT1 mutations in ichthyosis with confetti. J Clin Invest. 2015;125(4):1703–7.
Lim YH, Choate KA. Expanding the mutation spectrum of ichthyosis with confetti. J Invest Dermatol. 2016;136(10):1941–3.
Guerra L, Diociaiuti A, El Hachem M, Castiglia D, Zambruno G. Ichthyosis with confetti: clinics, molecular genetics and management. Orphanet J Rare Dis. 2015;10:115.
Petrova EHA. Advances in understanding of Netherton syndrome and therapeutic implications. Expert Opin Orphan Drugs. 2020;8(11):455–87.
Wiegmann H, Valentin F, Tarinski T, Liebau E, Loser K, Traupe H, et al. LEKTI domains D6, D7 and D8+9 serve as substrates for transglutaminase 1: implications for targeted therapy of Netherton syndrome. Br J Dermatol. 2019;181(5):999–1008.
Liddle J, Beneton V, Benson M, Bingham R, Bouillot A, Boullay AB, et al. A potent and selective kallikrein-5 inhibitor delivers high pharmacological activity in skin from patients with Netherton syndrome. J Invest Dermatol. 2021;141(9):2272–9.
Rizzo WB. Sjogren–Larsson syndrome: molecular genetics and biochemical pathogenesis of fatty aldehyde dehydrogenase deficiency. Mol Genet Metab. 2007;90(1):1–9.
Rizzo WB. Genetics and prospective therapeutic targets for Sjogren–Larsson syndrome. Expert Opin Orphan Drugs. 2016;4(4):395–406.
Haug S, Braun-Falco M. Restoration of fatty aldehyde dehydrogenase deficiency in Sjogren–Larsson syndrome. Gene Ther. 2006;13(13):1021–6.
Samuelov L, Sarig O, Harmon RM, Rapaport D, Ishida-Yamamoto A, Isakov O, et al. Desmoglein 1 deficiency results in severe dermatitis, multiple allergies and metabolic wasting. Nat Genet. 2013;45(10):1244–8.
McAleer MA, Pohler E, Smith FJ, Wilson NJ, Cole C, MacGowan S, et al. Severe dermatitis, multiple allergies, and metabolic wasting syndrome caused by a novel mutation in the N-terminal plakin domain of desmoplakin. J Allergy Clin Immunol. 2015;136(5):1268–76.
Valentin F, Wiegmann H, Tarinski T, Nikolenko H, Traupe H, Liebau E, et al. Development of a pathogenesis-based therapy for peeling skin syndrome type 1. Br J Dermatol. 2021;184(6):1123–31.
Richard G, Rouan F, Willoughby CE, Brown N, Chung P, Ryynanen M, et al. Missense mutations in GJB2 encoding connexin-26 cause the ectodermal dysplasia keratitis–ichthyosis–deafness syndrome. Am J Hum Genet. 2002;70(5):1341–8.
van Steensel MA, van Geel M, Nahuys M, Smitt JH, Steijlen PM. A novel connexin 26 mutation in a patient diagnosed with keratitis–ichthyosis–deafness syndrome. J Invest Dermatol. 2002;118(4):724–7.
Caceres-Rios H, Tamayo-Sanchez L, Duran-Mckinster C, de la Luz OM, Ruiz-Maldonado R. Keratitis, ichthyosis, and deafness (KID syndrome): review of the literature and proposal of a new terminology. Pediatr Dermatol. 1996;13(2):105–13.
Messmer EM, Kenyon KR, Rittinger O, Janecke AR, Kampik A. Ocular manifestations of keratitis–ichthyosis–deafness (KID) syndrome. Ophthalmology. 2005;112(2):e1-6.
Yoneda K. Inherited ichthyosis: syndromic forms. J Dermatol. 2016;43(3):252–63.
Bergqvist C, Abdallah B, Hasbani DJ, Abbas O, Kibbi AG, Hamie L, et al. CHILD syndrome: a modified pathogenesis-targeted therapeutic approach. Am J Med Genet A. 2018;176(3):733–8.
Fontao L, Laffitte E, Briot A, Kaya G, Roux-Lombard P, Fraitag S, et al. Infliximab infusions for Netherton syndrome: sustained clinical improvement correlates with a reduction of thymic stromal lymphopoietin levels in the skin. J Invest Dermatol. 2011;131(9):1947–50.
Roda A, Mendonca-Sanches M, Travassos AR, Soares-de-Almeida L, Metze D. Infliximab therapy for Netherton syndrome: a case report. JAAD Case Rep. 2017;3(6):550–2.
Mazereeuw-Hautier J, Hernandez-Martin A, O’Toole EA, Bygum A, Amaro C, Aldwin M, et al. Management of congenital ichthyoses: European guidelines of care, part two. Br J Dermatol. 2019;180(3):484–95.
Paller AS, Renert-Yuval Y, Suprun M, Esaki H, Oliva M, Huynh TN, et al. An IL-17-dominant immune profile is shared across the major orphan forms of ichthyosis. J Allergy Clin Immunol. 2017;139(1):152–65.
Czarnowicki T, He H, Leonard A, Malik K, Magidi S, Rangel S, et al. The major orphan forms of ichthyosis are characterized by systemic T-cell activation and Th-17/Tc-17/Th-22/Tc-22 polarization in blood. J Invest Dermatol. 2018;138(10):2157–67.
Malik K, He H, Huynh TN, Tran G, Mueller K, Doytcheva K, et al. Ichthyosis molecular fingerprinting shows profound TH17 skewing and a unique barrier genomic signature. J Allergy Clin Immunol. 2019;143(2):604–18.
Paller AS. Pathogenesis-based therapy with repurposed biologics for monogenic inflammatory skin disorders. JAMA Dermatol. 2020;156(8):839–41.
Patel DD, Lee DM, Kolbinger F, Antoni C. Effect of IL-17A blockade with secukinumab in autoimmune diseases. Ann Rheum Dis. 2013;72(Suppl 2):ii116-23.
Luchsinger I, Knopfel N, Theiler M, des Bonnet Claustres M, Barbieux C, Schwieger-Briel A, et al. Secukinumab therapy for Netherton syndrome. JAMA Dermatol. 2020;156(8):907–11.
Blanchard SK, Prose NS. Successful use of secukinumab in Netherton syndrome. JAAD Case Rep. 2020;6(6):577–8.
Paul C. Ixekizumab or secukinumab in psoriasis: what difference does it make? Br J Dermatol. 2018;178(5):1003–5.
Barbieux C, Des Claustres MB, De la Brassinne M, Bagot M, Bourrat E, Hovnanian A. Duality of Netherton syndrome manifestations and response to ixekizumab. J Am Acad Dermatol. 2021;84(5):1476–80.
Croxtall JD. Ustekinumab: a review of its use in the management of moderate to severe plaque psoriasis. Drugs. 2011;71(13):1733–53.
Volc S, Maier L, Gritsch A, Aichelburg MC, Volc-Platzer B. Successful treatment of Netherton syndrome with ustekinumab in a 15-year-old girl. Br J Dermatol. 2020;183(1):165–7.
Wohlrab J, Kreft B. Treatment of pityriasis rubra pilaris with ustekinumab. Br J Dermatol. 2010;163(3):655–6.
Eytan O, Sarig O, Sprecher E, van Steensel MA. Clinical response to ustekinumab in familial pityriasis rubra pilaris caused by a novel mutation in CARD14. Br J Dermatol. 2014;171(2):420–2.
Lwin SM, Hsu CK, Liu L, Huang HY, Levell NJ, McGrath JA. Beneficial effect of ustekinumab in familial pityriasis rubra pilaris with a new missense mutation in CARD14. Br J Dermatol. 2018;178(4):969–72.
Steuer AB, Cohen DE. Treatment of Netherton syndrome with dupilumab. JAMA Dermatol. 2020;156(3):350–1.
Andreasen TH, Karstensen HG, Duno M, Lei U, Zachariae C, Thyssen JP. Successful treatment with dupilumab of an adult with Netherton syndrome. Clin Exp Dermatol. 2020;45(7):915–7.
Wang J, Yu L, Zhang S, Wang C, Li Z, Li M, et al. Successful treatment of Netherton syndrome with dupilumab: a case report and review of the literature. J Dermatol. 2022;49(1):165–7.
Sussmuth K, Traupe H, Loser K, Stander S, Kessel C, Wittkowski H, et al. Response to dupilumab in two children with Netherton syndrome: improvement of pruritus and scaling. J Eur Acad Dermatol Venereol. 2021;35(2):e152–5.
Murase C, Takeichi T, Taki T, Yoshikawa T, Suzuki A, Ogi T, et al. Successful dupilumab treatment for ichthyotic and atopic features of Netherton syndrome. J Dermatol Sci. 2021;102(2):126–9.
Aktas M, Salman A, Apti Sengun O, Comert Ozer E, Hosgoren Tekin S, Akin Cakici O, et al. Netherton syndrome: temporary response to dupilumab. Pediatr Dermatol. 2020;37(6):1210–1.
Poulton C, Gration D, Murray K, Baynam G, Halbert A. Autosomal recessive congenital ichthyosis due to homozygous variants in NIPAL4 with a dramatic response to ustekinumab. Pediatr Dermatol. 2019;36(6):1002–3.
Haiges D, Fischer J, Horer S, Has C, Schempp CM. Biologic therapy targeting IL-17 ameliorates a case of congenital ichthyosiform cornification disorder. J Dtsch Dermatol Ges. 2019;17(1):70–2.
Hernandez-Martin A, Kennedy-Batalla R, Canedo E, Bernaldo-de-Quiros E, Carazo-Gallego B, Vera A, et al. Imbalance in T-Helper 17 cells and targeted therapy in an infant with SAM-like syndrome. N Engl J Med. 2019;381(22):2176–8.
Paller AS, Czarnowicki T, Renert-Yuval Y, Holland K, Huynh T, Sadlier M, et al. The spectrum of manifestations in desmoplakin gene (DSP) spectrin repeat 6 domain mutations: immunophenotyping and response to ustekinumab. J Am Acad Dermatol. 2018;78(3):498-505 e2.
Lefferdink R, Rangel SM, Chima M, Ibler E, Pavel AB, Kim H, et al. Secukinumab responses vary across the spectrum of congenital ichthyosis in adults. Arch Dermatol Res. 2022.
Walker AL, Bingham RP, Edgar EV, Ferrie A, Holmes DS, Liddle J, et al. Structure guided drug design to develop kallikrein 5 inhibitors to treat Netherton syndrome. Bioorg Med Chem Lett. 2019;29(12):1454–8.
Torres T, Filipe P. Small molecules in the treatment of psoriasis. Drug Dev Res. 2015;76(5):215–27.
Li R, Hadi S, Guttman-Yassky E. Current and emerging biologic and small molecule therapies for atopic dermatitis. Expert Opin Biol Ther. 2019;19(4):367–80.
Furio L, Pampalakis G, Michael IP, Nagy A, Sotiropoulou G, Hovnanian A. KLK5 inactivation reverses cutaneous hallmarks of netherton syndrome. PLoS Genet. 2015;11(9):e1005389.
Wang S, Olt S, Schoefmann N, Stuetz A, Winiski A, Wolff-Winiski B. SPINK5 knockdown in organotypic human skin culture as a model system for Netherton syndrome: effect of genetic inhibition of serine proteases kallikrein 5 and kallikrein 7. Exp Dermatol. 2014;23(7):524–6.
Kasparek P, Ileninova Z, Zbodakova O, Kanchev I, Benada O, Chalupsky K, et al. KLK5 and KLK7 ablation fully rescues lethality of Netherton syndrome-like phenotype. PLoS Genet. 2017;13(1):e1006566.
Chen X, Riley BT, de Veer SJ, Hoke DE, Van Haeften J, Leahy D, et al. Potent, multi-target serine protease inhibition achieved by a simplified beta-sheet motif. PLoS ONE. 2019;14(1):e0210842.
Mazereeuw-Hautier J, Cope J, Ong C, Green A, Hovnanian A, Harper JI. Topical recombinant alpha1-antitrypsin: a potential treatment for Netherton syndrome? Arch Dermatol. 2006;142(3):396–8.
Chateau M. A phase I/II, multicenter, randomized, double-blind, placebo within-patient controlled, first-in-human (FIH) Proof of Concept (PoC) study to evaluate the safety and efficacy of topically applied SXR1096 cream in patients with Netherton syndrome (NS): Sixera Pharma; 2021. http://sixerapharma.com/2021/12/07/15398/. Accessed 9 Jand 2022.
Inc AT. Aldeyra therapeutics announces first patient enrolled in Sjögren–Larsson syndrome pivotal phase 3 Clinical trial 2018. https://ir.aldeyra.com/news-releases/news-release-details/aldeyra-therapeutics-announces-first-patient-enrolled-sjogren-0. Accessed 15 Feb 2022.
Lachmann RH. Enzyme replacement therapy for lysosomal storage diseases. Curr Opin Pediatr. 2011;23(6):588–93.
Aufenvenne K, Larcher F, Hausser I, Duarte B, Oji V, Nikolenko H, et al. Topical enzyme-replacement therapy restores transglutaminase 1 activity and corrects architecture of transglutaminase-1-deficient skin grafts. Am J Hum Genet. 2013;93(4):620–30.
Fischer J. Autosomal recessive congenital ichthyosis. J Invest Dermatol. 2009;129(6):1319–21.
Plank R, Yealland G, Miceli E, Lima Cunha D, Graff P, Thomforde S, et al. Transglutaminase 1 replacement therapy successfully mitigates the autosomal recessive congenital ichthyosis phenotype in full-thickness skin disease equivalents. J Invest Dermatol. 2019;139(5):1191–5.
Grond S, Eichmann TO, Dubrac S, Kolb D, Schmuth M, Fischer J, et al. PNPLA1 deficiency in mice and humans leads to a defect in the synthesis of omega-O-acylceramides. J Invest Dermatol. 2017;137(2):394–402.
Mauldin EA, Crumrine D, Casal ML, Jeong S, Opalka L, Vavrova K, et al. Cellular and metabolic basis for the ichthyotic phenotype in NIPAL4 (Ichthyin)-deficient canines. Am J Pathol. 2018;188(6):1419–29.
Paller AS, van Steensel MA, Rodriguez-Martin M, Sorrell J, Heath C, Crumrine D, et al. Pathogenesis-based therapy reverses cutaneous abnormalities in an inherited disorder of distal cholesterol metabolism. J Invest Dermatol. 2011;131(11):2242–8.
Sandoval KR, Machado MCR, Oliveira ZNP, Nico MMS. CHILD syndrome: successful treatment of skin lesions with topical lovastatin and cholesterol lotion. An Bras Dermatol. 2019;94(3):341–3.
Yu X, Chen L, Yang Z, Gu Y, Zheng W, Wu Z, et al. An excellent response to topical therapy of four congenital hemidysplasia with ichthyosiform erythroderma and limb defects syndrome patients with an increased concentration of simvastatin ointment. J Eur Acad Dermatol Venereol. 2020;34(1):e8–11.
Kallis P, Bisbee E, Garganta C, Schoch JJ. Rapid improvement of skin lesions in CHILD syndrome with topical 5% simvastatin ointment. Pediatr Dermatol. 2022;39(1):151–2.
Bajawi SM, Jafarri SA, Buraik MA, Al Attas KM, Hannani HY. Pathogenesis-based therapy: cutaneous abnormalities of CHILD syndrome successfully treated with topical simvastatin monotherapy. JAAD Case Rep. 2018;4(3):232–4.
Mavilio F, Pellegrini G, Ferrari S, Di Nunzio F, Di Iorio E, Recchia A, et al. Correction of junctional epidermolysis bullosa by transplantation of genetically modified epidermal stem cells. Nat Med. 2006;12(12):1397–402.
Hirsch T, Rothoeft T, Teig N, Bauer JW, Pellegrini G, De Rosa L, et al. Regeneration of the entire human epidermis using transgenic stem cells. Nature. 2017;551(7680):327–32.
Bilousova G. Gene therapy for skin fragility diseases: the new generation. J Invest Dermatol. 2019;139(8):1634–7.
Gorell E, Nguyen N, Lane A, Siprashvili Z. Gene therapy for skin diseases. Cold Spring Harb Perspect Med. 2014;4(4):a015149.
Chulpanova DS, Shaimardanova AA, Ponomarev AS, Elsheikh S, Rizvanov AA, Solovyeva VV. Current strategies for the gene therapy of autosomal recessive congenital ichthyosis and other types of inherited ichthyosis. Int J Mol Sci. 2022;23(5):2506.
Danner E, Bashir S, Yumlu S, Wurst W, Wefers B, Kuhn R. Control of gene editing by manipulation of DNA repair mechanisms. Mamm Genome. 2017;28(7–8):262–74.
Porto EM, Komor AC, Slaymaker IM, Yeo GW. Base editing: advances and therapeutic opportunities. Nat Rev Drug Discov. 2020;19(12):839–59.
Freiberg RA, Choate KA, Deng H, Alperin ES, Shapiro LJ, Khavari PA. A model of corrective gene transfer in X-linked ichthyosis. Hum Mol Genet. 1997;6(6):927–33.
Haug S, Braun-Falco M. Adeno-associated virus vectors are able to restore fatty aldehyde dehydrogenase-deficiency. Implications for gene therapy in Sjogren–Larsson syndrome. Arch Dermatol Res. 2005;296(12):568–72.
March OP, Lettner T, Klausegger A, Ablinger M, Kocher T, Hainzl S, et al. Gene editing-mediated disruption of epidermolytic ichthyosis-associated KRT10 alleles restores filament stability in keratinocytes. J Invest Dermatol. 2019;139(8):1699-710 e6.
Choate KA, Medalie DA, Morgan JR, Khavari PA. Corrective gene transfer in the human skin disorder lamellar ichthyosis. Nat Med. 1996;2(11):1263–7.
Freedman JC, Parry TJ, Zhang P, Majumdar A, Krishnan S, Regula LK, et al. Preclinical evaluation of a modified herpes simplex virus type 1 vector encoding human TGM1 for the treatment of autosomal recessive congenital ichthyosis. J Invest Dermatol. 2021;141(4):874-82 e6.
Gurevich I, Agarwal P, Zhang P, Dolorito JA, Oliver S, Liu H, et al. In vivo topical gene therapy for recessive dystrophic epidermolysis bullosa: a phase 1 and 2 trial. Nat Med. 2022;28(4):780–8.
Dang L, Zhou X, Zhong X, Yu W, Huang S, Liu H, et al. Correction of the pathogenic mutation in TGM1 gene by adenine base editing in mutant embryos. Mol Ther. 2022;30(1):175–83.
Lee MY, Wang HZ, White TW, Brooks T, Pittman A, Halai H, et al. Allele-specific small interfering RNA corrects aberrant cellular phenotype in keratitis–ichthyosis–deafness syndrome keratinocytes. J Invest Dermatol. 2020;140(5):1035–44.
Di WL, Lwin SM, Petrova A, Bernadis C, Syed F, Farzaneh F, et al. Generation and clinical application of gene-modified autologous epidermal sheets in Netherton syndrome: lessons learned from a phase 1 trial. Hum Gene Ther. 2019;30(9):1067–78.
Pasmooij AM, Jonkman MF, Uitto J. Revertant mosaicism in heritable skin diseases: mechanisms of natural gene therapy. Discov Med. 2012;14(76):167–79.
Gostynski A, Pasmooij AM, Jonkman MF. Successful therapeutic transplantation of revertant skin in epidermolysis bullosa. J Am Acad Dermatol. 2014;70(1):98–101.
Acknowledgements
Not applicable
Funding
None.
Author information
Authors and Affiliations
Contributions
MJ: conceptualization, investigation, literature search, analysis, data curation, visualization, writing original draft and review. JC: conceptualization, investigation, literature search, analysis, data curation, visualization, writing original draft and review. NJ: investigation, writing original draft and review. JMH: supervision, conceptualization, writing original draft and review. AG: supervision, methodology, conceptualization, analysis, visualization, writing original draft and review, project administration. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
AG: speakers fee by Abbvie for a presentation about genodermatosis on a Dutch dermatology congress, including data about treatment of ichthyosis by IL-17 inhibitors; stocks AstraZeneca, Abott Laboratories, Abbvie, GSK, Incyte Corp, Krystal Biotech Inc, Sanofi, Johnson and Johnson, Novartis, Pfizer, UCB; Novartis is providing secukinumab free of charge for four patients with ichthyosis treated in my centre. The other authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Joosten, M.D.W., Clabbers, J.M.K., Jonca, N. et al. New developments in the molecular treatment of ichthyosis: review of the literature. Orphanet J Rare Dis 17, 269 (2022). https://doi.org/10.1186/s13023-022-02430-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-022-02430-6