
Abstract
A novel approach to treating heart failures was developed with the introduction of iPSC technology. Knowledge in regenerative medicine, developmental biology, and the identification of illnesses at the cellular level has exploded since the discovery of iPSCs. One of the most frequent causes of mortality associated with cardiovascular disease is the loss of cardiomyocytes (CMs), followed by heart failure. A possible treatment for heart failure involves restoring cardiac function and replacing damaged tissue with healthy, regenerated CMs. Significant strides in stem cell biology during the last ten years have transformed the in vitro study of human illness and enhanced our knowledge of the molecular pathways underlying human disease, regenerative medicine, and drug development. We seek to examine iPSC advancements in disease modeling, drug discovery, iPSC-Based cell treatments, and purification methods in this article.
Similar content being viewed by others
Background
Stem cell biology and regenerative medicine are new branches of sciences. For this reason, there has been a great interest in stem cells in the past decades due to their therapeutic application in regenerative medicine [1,2,3] Fathi, Farahzadi, et al.,. Although cardiac progenitor cells have been described to insist within the myocardium, the myocardium has no intrinsic regenerative capacity because of a lack of postnatal mitosis. For this reason, many studies have been investigated on all types of stem cells and even extracellular vesicles derived them [4].
Takahashi and Yamanaka started the project that would eventually become iPSC in 2006. By introducing the four transcriptional factors OCT3/4, c-MYC, SOX2, and KLF4 into mouse fibroblasts, they created mouse iPSCs [5]. iPSCs are somatic cells that have been genetically modified to resemble embryonic stem cells (ESCs) by expressing certain transcription factors [6]. There are various ways to induce pluripotency in cells, including nuclear transfer, exposure to factors expressed in pluripotent cells, and overexpression of certain transcription factors [7]. Human induced pluripotent stem cells (hiPSCs) and their derivatives provide a suitable supply of human cells and have been used in a number of scientific domains, including the modeling of illness and the identification of new drugs [8]. Worldwide, cardiovascular disease caused 17,921,000 fatalities in 2017. Some cardiac conditions, including long QT syndrome (LQTS), are linked to innate factors, such as mutations in the gene encoding a sodium ion channel, which lead to arrhythmias. Another condition called myocardial infarction (MI) causes the oxygen level in the cardiac muscles to fall, and instead of the CMs regenerating, the human heart forms an extensive scar that reduces blood flow and contractility [9]. Stem cells are one of the recommendations for treating the post-infarction heart since adult mammalian hearts do not have a large ability for regeneration in various injuries. Only cardiac progenitor cells seem to have the capacity to develop into highly functioning CMs and it is exceedingly difficult to acquire enough cardiac progenitor cells from a patient’s biopsy [10, 11]. Cellular metabolism plays a significant role in the maturation of iPSC-derived cardiac tissue models and the formation of CMs, and alterations in cellular metabolism lead to cardiac pathogenesis. iPSC-derived CMs (iPSC-CMs) vary from mature human CMs in several ways, including contractile function, electrophysiology, metabolism, and structure. iPSC-CMs resemble fetal CMs in several respects, such as the absence of pathways relevant to the adult phenotype and particular gene expression. In iPSC-CMs, aerobic glycolysis is the primary source of ATP generation, with limited assistance from oxidative phosphorylation. The use of iPSCs in clinic may be limited by these various metabolic characteristics. Due to these problems, researchers have created a number of methods for maturing iPSC-CMs, such as 3D culture, electrical pacing, extended culture, or adding hormones and fatty acids to the media used to grow cells. These methods improve the functional and structural maturity of iPSC-CMs [12]. According to studies, utilizing the aforementioned maturation techniques may boost the expression of genes in iPSC-CMs that control cardiac metabolism, including CD36, ACAT1, PDK4, ATP5, LPL, PPARA, and DGAT1. Additionally, fatty acid supplementation and extended culture were used to enhance the expression levels of the genes ESRRA and PPARGC1A, which are related to mitochondrial biogenesis, and ACADVL, PPARD, and SCD, which are related to fatty acid oxidation. PGK1, ALDOA, LDHA, HK1 and HK2 are among the glycolysis-related genes whose expression levels may be decreased by extended culture and fatty acid supplementation [13, 14].
Differentiation of iPSCs into cardiomyocytes
Differentiating CMs from hiPSCs is a crucial step in producing artificial cardiac muscle cells and tissues. Engineering efforts to purify the hiPSC-CMs and various techniques to stimulate hiPSC-CM development are investigated. In reality, ESCs and iPSCs develop in a similar manner and may become any kind of specialized human cell [15, 16]. Although there may be phenotypic differences between CMs produced from ESCs and iPSCs due to variations in gene expression. Based on in vivo heart development, researchers have created a variety of techniques to differentiate PSCs into CMs during the past two decades [17, 18]. Human ESCs (hESCs) may generate CMs on their own or with the aid of growth factor-directed methods. It is possible to enhance the production of CMs by treating hESCs with a variety of growth factors, including Vascular endothelial growth factor (VEGF), bone morphogenetic protein 4 (BMP-4), activin A, basic fibroblast growth factor (bFGF) and the peptide Wnt inhibitor dickkopf homolog 1 (DKK-1). Due to their resemblance to hESCs, the majority of these growth factors aid in the differentiation of iPSCs [19]. However, due to the fact that cardiac differentiation effectiveness often depends on cell line, these methods’ effectiveness in human ES cell lines varies. Additionally, using several growth factors is expensive. Due to their accessibility, potency, and capacity to penetrate cells, small molecules may be used to control the signaling pathways involved in the self-renewal and lineage differentiation of stem cells.
Signaling pathways involved in differentiation
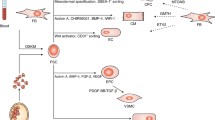
BMP-4 and Wnt/β-catenin signaling pathways were shown by Ren et al. to be able to trigger cardiac differentiation in hiPSCs. They found that pre-treatment with BMP-4 of hESCs and iPSCs, followed by post-treatment with a small molecule Wnt inhibitor, significantly boosts the formation of CMs with normal electrophysiological function and pharmacological responsiveness [20]. Wnt proteins have a role in embryonic development and control a variety of cellular functions in the adult organism, including cell division and proliferation, gene transcription, polarity, and migration. Wnt proteins also have a role in the development and differentiation of the heart. These proteins have the ability to activate both canonical (β-catenin dependent) and non-canonical (β-catenin independent) Wnt signaling, which are intracellular signaling pathways. Mesoderm development depends on canonical Wnt/ β-catenin signaling, which is weak during cardiac specification. Additionally, the proliferation of CMs depends on Wnt/β-catenin signaling [21,22,23]. Mazzotta et al. performed a research by using small-molecule inhibitors to analyze functions of canonical/non-canonical Wnt signaling in hESCs and they suggested that canonical signaling regulate mesoderm induction and non-canonical signaling modulate cardiovascular development [24]. Wnt family consists of 19 various members in mammals. Activation of Wnt/β-catenin signaling initiates with interaction between Wnt ligands and Frizzled receptor which is seven-pass transmembrane receptor and its co-receptor, LRP5/6 (low-density lipoprotein receptor-related protein). Destruction complex consists of 2 kinases, glycogen synthase kinase 3 (GSK3β) and casein kinase 1 (CK1), AXIN protein and the tumor suppressor gene product adenomatous polyposis coli (APC) [25, 26]. Activation of Wnt/ β-catenin signaling is depicted in Fig. 1 [25]. There are some other signaling pathways that adjust cardiac maturation. Activation of serine/threonine-protein kinase Akt, results in growth, differentiation and metabolism. In addition, PI3K/Akt signaling pathway is essential and is activated during CM differentiation. Inhibition of PI3K may lead to downregulation of Nkx-2.5 and GATA-4 which are cardiac marker genes. It also has been indicated that inhibition of the PI3K/Akt signaling pathway in the early cardiomyogenesis cause the suppression of Wnt/β-catenin signaling (Fathi, Valipour, et al., [27]). Yang et al. could differentiate CM cells from hESC embryoid bodies. They added different factors (activin A, BMP4, bFGF, VEGF, and DKK1) to serum-free media of hESC and genetare KDRlow/C-KITneg populations that have cardiac, endothelial, and vascular smooth muscle potential. By plating in monolayer culture, these KDRlow/C-KITneg cells differentiate and generate populations consisting of greater than 50% contracting CMs. These populations derived from the KDRlow/C-KITneg give rise to colonies that contain three lineages (CMs, endothelial cells and vascular smooth muscle cells) when plated in methyl cellulose cultures [28].

The graphical representation of Wnt/β-Catenin-dependent signaling. (A) When Wnt ligand is absent (off state), β catenin is phosphorylated by kinases (GSK3 and CK1) of the Destruction complex. Then, phosphorylated β-catenin ubiquitinate to be targeted for degradation in the proteasome. (B) Binding of Wnt ligand to Frizzled receptor and LRP5/6 (on state), allows the Destruction complex to decompose which induce the stabilization of β-catenin. β-catenin accumulates in cytoplasm and then is transferred to the nucleus and starts gene transcription. This figure adapted from Fig. 1 of reference number 25 [25]
As was previously indicated, early embryonic differentiation requiring Wnt for mesodermal specification; however, later Wnt signaling prevents cardiac specification, hence suppression of Wnt signaling may be required for the development of CMs [29]. This model is shown in Fig. 2 [30]. Using extrinsic stimuli like BMP4 and CHIR, WNT inhibition with XAV939 or IWR1, and enrichment of CMs by providing lactate as an energy source within 15 days, Kadari et al. were able to produce CMs from a variety of hiPSC lines. During the first two days of the induction phase, basal media containing insulin were administered. They demonstrate how the cells seem to be under a lot of stress when utilizing basal media without insulin right away. In order to reduce cell death and its detrimental effects on cardiac specification, they thus opted to maintain insulin for the first two days and stop using it during the specification phase [31]. According to research by Aguado et al., cells with high expression of the shelterin complex protein TRF1 and relatively long telomeres develop into CMs more quickly and effectively than cells with low expression. The differentiation of CMs produced from iPSCs with long telomeres was also enhanced by the use of ascorbic acid, but the differentiation of CMs produced from iPSCs with short telomeres was unaffected [32]. Ascorbic acid, growth hormones including BMP4 and FGF2, and small compounds that target the canonical Wnt signaling were shown to work well together to improve the differentiation efficiency and maturity of iPSC-CMs in a separate study by Yassa et al. [32].

In addition to mentioned signaling pathways, TGFβ family signaling is another one that involves in cardiac regeneration. TGFβ family have crucial roles in differentiation, apoptosis and proliferation. In addition, TGFβ has a role in various diseases such as cardiac hypertrophy and cardiac abnormality. This family include BMPs, TGFβ isoforms, inhibins/activins and growth and differentiation factors. As Dronkers et al. stated in their review article, TGFβ family can be split into two clusters of TGFβ and BMP. BMPs bind to receptor and cause to phosphorylation of SMAD1/5/8, while TGFβ isoforms function with activins toward phosphorylation of SMAD2/3. SMAD4 binds to this phosphorylated SMADS, creating a complex and then enters the nucleus which turn on TGFβ target genes. TGFβ family complex has 3 types of receptors include ALK 1–7 (activin-like kinase or type I receptors), ActR2A, ActR2B, TGFBRII and BMPRII (type II) and TGFβR3 (type III receptors). TGFβ can also send signals through other pathways so called “SMAD-independent pathways” such as Rho, PI3K/Akt and MAPK [33, 34]. TGFβ family signaling is shown in Fig. 3 [33].

This figure depicts two clusters of TGFβ family signaling, BMP signaling and TGFβ signaling. BMPs (on the right site) bind to BMP type receptor or activin IIA or IIB which send signals via ALK1 or ALK 2, 3 ,6. As a result, SMAD 1, 5, 8 are phosphorylated and by joining SMAD4, this new made complex translocates to the nucleus. On left site, binding of TGFβ 1, 2, 3 to TGFβ receptor II and binding of Nodal, activing and inhibin to activing receptor IIA, respectively activate signaling through ALK5 and ALK4. This signaling leads to phosphorylation of SMAD 2, 3 and then SMAD4 binds to phosphorylated SMAD 2, 3. This complex enters the nucleus and cause gene transcription. This figure adapted from Fig. 1 of reference number 33 [33]
Drug discovery and disease modeling
In medication research, disease modeling, and even patient-specific cellular disease models, hiPSCs have become a popular platform. By enabling the generation of an endless supply of patient-specific human cells, iPSCs may revolutionize cardiology. Any human body cell type may be differentiated from somatic cells via iPSC reprogramming [35, 36]. From 2009 to 2018, it cost around USD 985 million to develop a new drug and release it into the market. Even though rigorous testing was conducted throughout the research period, 90% of medications fail to pass clinical trials. This is mostly due to the employment of unrelated cell culture techniques or inaccurate animal models that do not accurately mimic the human system. Because they offer human cell resources that are suited for research objectives, hiPSCs and their derivatives have been exploited in a broad range of scientific domains [8, 37, 38]. The iPSC can be developed into CMs while maintaining its genetic information and shares a genetic background with the initial reprogrammed cells [17]. The modeling of patient-specific iPSCs, a powerful tool for investigating illnesses and finding new drugs, as shown in Fig. 4 recently [39, 40]. The official and necessary procedure for releasing a new medicine into the market is overseen by the European Medicines Agency (EMA) in Europe and the Food and medicine Administration (FDA) in the USA, as Ovics et al. explained in a review study. Chemical and biological research, pre-clinical development, clinical trials, and one post-marketing phase are the three pre-marketing stages that make up drug development [41]. A wide range of cardiovascular models, such as LQTS, dilated cardiomyopathy, hypertrophic cardiomyopathy and arrhythmogenic right ventricular cardiomyopathy, were derived from patients’ iPSCs [8].

Various applications of iPSC-derived cardiomyocytes
Long QT syndromes
A dysfunctional ion channel is a genetic condition known as LQTS. LQTS is often defined by a lengthening of the QT interval on the ECG, which may result in cardiac arrest and is typically brought on by mental or emotional strain [42,43,44]. It has been discovered that diseases are brought on by mutations in the genes encoding for cardiac ion channels, ion channel subunits, or proteins that impact how an ion channel functions. 17 genes have been linked to congenital LQTS, according to research. Less than 5% of clinically confirmed instances of congenital LQTS have been linked to mutations in genes other than KCNQ1, KCHN2, and SCN5A, which account for more than 75% of cases [45]. Because of the underlying chromosomal abnormality, the LQTS subtype has been divided into many subgroups. LQTS type 1, type 2, and type 3 have distinct connections between genotypes and phenotypes that have been documented.
The potassium channel gene KCNQ1 on chromosome 11 is the basis for LQT1, while the KCNH2 gene on chromosome 7 is the basis for LQT2. Gain-of-function mutations in SCN5A on chromosome 3 are the basis for LQTS type 3. The action potential is prolonged as a result of an increase in the myocardial sodium current (INa), which occurs during the plateau period [46,47,48].
Role of iPSC research
The first LQTS iPSC-CM model was released by Moretti et al. and a number have since been published. From the dermal fibroblasts of two patients with LQT1 who had the R190Q mutation in the KCNQ1 gene, they created iPSC-CMs. Individual cells were identified as having a “ventricular,” “atrial,” or “nodal” phenotype based on the expression of cell-type-specific markers and recordings of action potentials in single cells. The “ventricular” and “atrial” action potential length was considerably extended in the mutant cells. Additionally, the IKs current was reduced by 70 to 80% in cells with the R190Q-KCNQ1 mutation, and the activation and deactivation characteristics of the channel were altered. Additionally, they demonstrated that beta-blockade decreased the phenotypic of mutant cells having an enhanced vulnerability to catecholamine-induced tachyarrhythmia [49]. Wang et al. used genetic modification techniques to overexpress the dominant negative gene mutation of KCNQ1 in order to create an iPSC-based model for the LQTS1 disease. They have shown that as compared to unedited control cells, editing iPSCs generated from cardiomyocytes display LQTS features and a lengthened action potential time. They demonstrated that the addition of nifedipine or pinacidil decreased the iPSC cardiomyocytes’ action potential length, strengthening the safety and effectiveness of isogenic iPSC lines in drug development [50]. In a different work, Garg et al. generated LQT2 iPSC-CM from T983I-KCNH2 variant peripheral blood mononuclear cells. In mutant cells, patch-clamp tests showed that action potential duration lengthening decreased the rapidly activating delayed rectifier K + current (IKr). In order to do this, they utilized a strong IKr activator called ICA-105,574, which improved IKr density and standardized action potential length in mutant cells [51]. This raised the possibility of medication for people with LQT2-specific disorders. Mesquita et al. developed iPSC- LQTS2 from two patients with c.1600 C > T in KCNH2, p.R534C in hERG mutation and inserted the same mutation in a control iPSC line, and then differentiated into CMs. Electrophysiology tests revealed that the mutant iPSC line’s action potential duration was much longer than that of the control cell line. Action potential lengthening was seen in the control cell line after treatment with E4031, an IKr inhibitor, whereas IKr was reduced in LQT2-iPSC [52]. In order to create a collection of isogenic iPSC lines with different KCNH2 mutations that produce LQT2, Brandao et al. (2020) genetically altered an iPSC line using CRISPR/Cas9 gene editing. They indicated that genetically modified hiPSC-CMs can have electrophysiological differences related to the KCNH2 mutation. Thus, it can be helpful in improving the diagnosis, prognosis and the classification of patients with congenital LQTS [53]. Wang et al. designed iPSC line of LQT patient harboring two mutations: a heterozygous KCNQ1 c.656G > A and a heterozygous TRPM4 c.479 C > T. They found that the QT interval in LQT patients was reduced by lidocaine, a sodium channel blocker, and verapamil, a calcium channel blocker. Verapamil dramatically decreased the number of ICD discharges. The relative decrease in QT interval after verapamil administration was similarly shown to be greater for LQT cardiomyocytes when compared to lidocaine [54].
Arrhythmogenic right ventricular cardiomyopathy
Inherent heart muscle disease called arrhythmogenic right ventricular cardiomyopathy (ARVC) increases the risk of ventricular arrhythmias and sudden cardiac death, especially in young people and athletes, as fatty fibrous tissue replaces the right ventricular myocardium as well as that of the left ventricle. It affects at least 1 in 1000 persons under the age of 35, and it may account for up to 10% of fatalities from undetected cardiac disease in this age range [55,56,57, 110]. ARVC is familial and frequently autosomal dominant in roughly 50% of cases. Changes in five desmosomal protein-encoding genes, including DSG2, DSC2, PKP2, and DSP, which may result in cell death and fragmentation, have been linked to ARVC. Desmosomal protein deficiency may alter electrocardiographic parameters, cardiomyocyte gap junction structure, and sodium channel function. These mutations, which cause the premature termination of related proteins, are categorized as insertions, deletions, or nonsense mutations [58,59,60,61]. Desmosomal proteins have a role in the Wnt/ β-catenin signaling pathway and are crucial for cardiac myogenesis. Desmosome protein alterations block Wnt/ β-catenin, which helps to generate mesoderm precursor adipose [62,63,64]. TGF-3 and TMEM43 are two non-desmosomal genes that have been found in ARVC. The first is for transmembrane protein 43, and the second is for transforming growth factor 3 [65, 66]. Desmosome gene mutations are present in more than 50% of the afflicted people, most often in the PKP2 gene, which codes for plakophilin-2. Reduced β- catenin activity and aberrant plakoglobin nuclear translocation are caused by PKP2 mutation in iPSC-CMs [62, 67].
Role of iPSC research
Ma et al. developed the first ARVC in vitro model with PKP2 gene mutation using patient iPSC-CMs [68]. From the fibroblasts of two ARVC patients with PKP2 mutations, Kim et al. created iPSC lines. By producing an adult-like metabolic energetics from an embryonic condition and activating the peroxisome proliferator-activated receptor-gamma, or PPARγ, they were able to produce an ARVC in-vitro model. In mutant PKP2 iPSC-CM, this model showed enhanced lipogenesis, apoptosis, and deficiencies in calcium handling [69]. By knocking down PKP2, Matthes et al. evaluated the role of the desmosomal protein PKP2 in epicardial and epicardium-derived cells, which led to increased lipid indicators, cell migration, and proliferation. Their study suggested that epicardium cells may be important for understanding the pathogenesis of ARVC [70]. In especially for ARVC, Capsi et al. (2013) emphasize the special potential of hiPSC technology to represent congenital cardiac diseases. They develop two ARVC patients with PKP2 mutations into hiPSC-derived CMs. In the ARVC model, there was a decrease in PKP2 and connexin-43 protein expression [71].The second most common ARVC variation is called desmoglein-2 (DSG2) [72]. An ARVC iPSC-CM model was created by Hawthorne et al. from a patient who had the DSG2 c.2358delA mutation. Less DSG2 mRNA, altered DSG2 protein distribution, atypical calcium regulation, and shorter action potentials were all seen in mutant cells. Additionally, elevated levels of pro-inflammatory cytokines have been seen, which may be connected to both conventional and non-canonical NF-B activation [73].
Dilated cardiomyopathy
The term “dilated cardiomyopathy” (or “DCM”) refers to a set of non-ischemic diseases of the heart muscle that are characterized by dilated left ventricles and dysfunctional contraction [74,75,76]. There are two types of the illness: familial/hereditary and non-familial. Familial factors account for 30–50% of DCM. Over 40 distinct genes, including TTN, TNNT2, PLN, MYH7, LMNA, and DES, have been identified as having genetic alterations [41, 77]. The TTN mutation, which is a frequent cause of dilated cardiomyopathy and occurs in around 25% of familial instances, is one such factor. The biggest known protein in humans, titin is a sarcomere protein encoded by the TTN gene [78, 79].
iPSC models
Hinson et al. demonstrated that sarcomere insufficiency, inconsistent cell signaling activation, and limited cell development were present in iPSC generations from DCM patients with TTN mutations. These cells responded to β-adrenergic stress with reduced effects [80]. Huang et al. used CRISPR-Cas9 technology to mimic hiPSC-CMs of DCM with A-band TTN mutations. This model showed aberrant sodium channel activity, sarcomere abnormalities, and contraction failure. With A-band TTN variations that improve contractility and lessen DCM symptoms, they also demonstrated the therapeutic potential of sarcomere modulators in DCM [81]. Patients with TTN mutations’ skin fibroblasts were used by Schick et al. to create iPSC-CMs. Mutated iPSC-CM showed defects in sarcomeric structure, diminished sensitivity to isoproterenol that mimicked β-adrenergic stimulation, and increased angiotensin-II levels [82]. Jeon et al. were able to create the hiPSC line YCMi007-A from a patient who had p. TNNT2 gene mutation causing Arg205Trp. These cells might be useful for the DCM model since they could develop into three germ layers and had a normal karyotype [83]. Perea-Gil et al. [101] used the CRISPR technique of gene editing to produce iPSC-CMs from a patient with the TNNT2- p.R183W mutation for an in vitro DCM model. They showed that the concomitant administration of the two kinase inhibitors Gö 6976 and SB 203,580 may boost the gene expression of the serine and glycine-encoding genes and restore contractile failure in cells with different mutations, including TNNT2, LMNA, and TTN. They also showed that DCM symptoms may be reduced by activating transcription factor 4 and downstream elements like a crucial enzyme in the serine biosynthesis pathway [84]. According to research by Dai et al., troponin connections with tropomyosin are dysfunctional in iPSC-CMs made from DCM patients with the TNNT2-R173W mutation using the CRISPR/Cas9 technique. This limits the binding of PKA to other proteins. This results in a deficiency in the MYH7-mediated and AMPK-dependent connections between sarcomeres and cytoskeleton filaments. They demonstrated that TNNT2-R173W mutant cells’ poor contractility could be repaired and sarcomere protein interactions improved by activating AMPK by A-769,662 [85]. Lamin A and lamin C are two major LMNA gene products that are expressed in the majority of differentiated somatic cells [86, 87]. Siu et al. produced two lines of DCM-derived iPSC-CMs from two patients. The LMNA exon 4 of one patient contained an R225X mutation, while the exon 4 of the second patient had a frame-shift mutation. They demonstrated that pharmacological blockade of the ERK2 pathway with MEK1/2 inhibitors, U0126 and selumetinib significantly reduced the apoptotic effects of electrical stimulation for mutant LMNA iPSCs [88]. Lee et al. modeled DCM with LMNA mutation using iPSC-CMs. Platelet-derived growth factor (PDGF) signaling pathway was activated in mutant cells and aberrant calcium transients were seen in mutant iPSC lines. Sunitinib and crenolanib are used to inhibit platelet-derived growth factor receptor- β (PDGFRB) expression in K117fs iPSC-CMs, which reduces the unique calcium transient and decreases CAMK2D levels. LMNA in DCM is brought on by the pathogenic mutation K117fs [89]. Three DCM patients’ peripheral blood mononuclear cells were transformed to produce three hiPSC lines by Lee et al. The c.1129 C > T LMNA mutation was present in all cases. All lines of iPSC have been shown to have normal morphology and high levels of pluripotent markers, making them suitable as an in vitro model for LMNA mutations in DCM [90].
Hypertrophic cardiomyopathy
The most prevalent congenital cardiomyopathy, hypertrophic cardiomyopathy (HCM), is characterized by left ventricular hypertrophy without a clear hemodynamic cause, such as severe hypertension or aortic stenosis. Adults are thought to have a prevalence of HCM that falls between 0.02 and 0.23% [91,92,93]. Most often, sarcomere mutations are the genetic basis of HCM. Heart failure, arrhythmias, and sudden cardiac death are all linked to HCM [94]. Myosin binding protein C (MYBPC3) and -myosin heavy chain (MYH7) mutations are the most common, followed by TNNI3 and TNNT2 mutations. However, 20 sarcomeric and myofilament-related proteins contain 450 mutations that have been linked to HCM [95,96,97]. Myosin heavy chain beta (MHC-β), which is produced in skeletal and heart muscles and is a crucial protein in the thick filament of the human heart and contributes to contraction, is encoded by MYH7 [41, 98, 99].
iPSC models
Han et al. produced iPSCs from an HCM patient with an Arginine442Glycine missense mutation in the MYH7 gene in more recent years. In HCM iPSC-CMs compared to control iPSC-CMs, electrophysiological abnormalities, disordered sarcomeres, and a substantial increase in genes important for cell proliferation were seen. They discovered that using verapamil plus the histone deacetylase inhibitor trichostatin A (TSA) abolished irregular heartbeats and reduced calcium irregularity in HCM CMs [100]. Ten family members with HCM, half of whom had the Arg663His missense mutation in the MYH7 gene, were used to create iPSC-CMs by Lan et al. The cellular expansion, calcium cycling dysregulation, contractile arrhythmia, and intracellular calcium increase were all characteristics of mutant cells. Verapamil, mexiletine, lidocaine, and ranolazine have all been shown via drug testing to restore unfavorable alterations in mutant cells [101]. Using CRISPR/Cas9 technology, Cohn et al. created four isogenic iPSC-CM models of the MYH7 and MYBPC3 mutations. Due to delayed relaxation kinetics, mutated cell line generated hypercontractility. They demonstrated that myofilament calcium delivery, variation location, and extracellular calcium level are not related to hypercontractility. Studies using RNA sequencing revealed p53 activation, oxidative stress, and cytotoxicity, all of which may be reversed by genetically altering p53. As a direct myosin inhibitor, verapamil and blebbistatin were also put to the test. Verapamil decreased twitch tension in R403Q+/- cardiomyocytes by 31.7% and blebbistatin decreased twitch force by 35.3% [102]. Escribá et al. created iPSC-CMs using CRISPR/Cas9 genome editing from two siblings who had HCM including the MYBPC3 mutation. Their research demonstrated that the MYBPC3 variation leads to mitochondrial bioenergetic dysfunction, which forces mitochondria to operate at greater efficiency in order to meet the escalating energy requirements of mutant cells. A seriously damaged person also showed abnormal excitation-contraction coupling [103]. Table 1 displays the stated models for disease modeling [49, 104,105,106].
Purification methods
Regenerative cell treatments need large amounts of pure PSCs. Animal models have shown several positive benefits of cardiac replacement treatment employing CMs derived from PSCs, including ESCs and iPSCs, to injured hearts. Thus, for the clinical application of PSC-CMs, 1 × 107–1 × 109 purified PSC-CMs are needed. Additionally, cell contamination should be avoided since it may result in unexpected events [107, 108]. There are several ways to clean up iPSCs. Here, we quickly go through two of the most popular techniques.
Antibody-based cell purification
The most effective approach for cell purification to date is antibody-based cell enrichment. Antibodies against cell type-specific surface proteins may tag the target cells and separate them using FACS or magnetic-activated cell sorting (MACS). The fundamental benefit of these approaches is their specificity and sensitivity. For PSC-CMs, researchers have created a few purification techniques. For instance, identifying CORIN, Signal-Regulatory Protein Alpha (SIRPA), and Vascular Cell Adhesion Molecule 1 (VCAM1) as cardiac-specific cell surface proteins. However, no research revealed surface markers that are exclusive to CMs. Because the MACS approach need specialized antibodies to the cellular surface proteins, it is challenging to utilize with cell types like CMs that lack certain cell surface proteins [109,110,111]. Rossler et al. compared the purification of hiPSC-CM cultures using lactate or MACS and their research revealed no considerable difference between these methods. They stated that purified cells via lactate or MACS had comparable features in structure, proteome, function and can be utilized in 3D cell cultures [112]. KDR (Flk-1) was previously employed in investigations to identify cardiac progenitor cells; however, since this receptor is expressed on a variety of mesoderm populations, it was unable to enrich solely hPSC-CMs [113, 114]. In addition, although markers like SIRPA and VCAM1 are expressed in other cell types, cell sorting utilizing antibodies should first allow enrichment of cardiomyocytes. Therefore, contamination from non-CMs or non-specific cells might reduce the effectiveness of the transplant [115]. Liew et al. performed a research and introduced a novel surface marker, JAK2, to purify and sort live cells of ventricular CMs. They isolated CMs which were SIRPA positive with expressed MLC2A and MLC2V proteins and their research demonstrated that isolating ventricular SIRPA+/JAK2 + cells can lead to higher purity [116].
miRNA-based methods
There are further purification techniques that rely on spotting miRNA (microRNA) activity in live target cells [115]. Non-coding RNA molecules called miRNAs are involved in the control of gene expression. RNA polymerase II and III carry out the transcription of miRNAs, producing precursors that go through a sequence of cleavage events to create mature miRNA. In other words, the majority of miRNAs are transcribed from DNA sequences as primary miRNAs and processed into precursor miRNAs and mature miRNAs [117, 118]. MiRNAs are typically 21–23 nucleotides in length and can bind to the 3′ untranslated regions (3’UTR) of messenger RNAs (mRNAs) to downregulate their target genes [119]. To quickly and effectively purify large numbers of PSC-derived cells, Tsujisaka et al. devised a technique that combines miRNA-responsive mRNA switch (miR-switch) with MACS (miR-switch-MACS). The miR-switch is a two-part synthetic mRNA. One is a protein-coding region, and the other is a complementary sequence to the target miRNA called a miRNA target site. Because of the contact between the target miRNA and the miR-switch, translation from the miR-switch is suppressed if the cell expresses the target miRNA. In contrast, the miR-switch is translated if the target miRNA is not expressed by the cell. To specifically suppress CD4 expression in iPSC-CMs, they created a miR-208a-responsive CD4-coding mRNA (miR-208a-CD4 switch) that encodes the CD4 cell surface protein. Because they could successfully filter iPSC-CMs among transfected cells (> 95% purity) and maintain more than 50% of iPSC-CMs based on the computation of the number of CMs before and after MACS, they employed 300 ng miR-208a-CD4 switch to harvest significant amounts of iPSC-CMs [108]. A powerful technique for cell purification based on endogenous miRNA activity was disclosed by Miki et al. (2015) They discovered numerous miRNA that were co-expressed on differentiation days 8 and 20 in hPSC-CMs, thus they created artificial mRNAs known as miRNA switches that can detect these miRNAs. They created artificial mRNAs called miRNA switches that code for fluorescent proteins that are marked with sequences that are targets of miRNAs produced by the cells of interest. CMs may be isolated and sorted from diverse cell types using synthetic miRNA switches. Because the fluorescent protein expression differential between CMs and non-CMs was greatest utilizing the miR-208a switch, they decided to employ miR-208a as a marker for CM-specific miRNA [120].
Stem cell-based therapy
Any treatment for diseases employing various types of viable human stem cells, such as adult stem cells, iPSCs, and ESCs, is referred to as stem cell-based therapy. Two methods for iPSC-based cell treatment are autologous and allogeneic cell transplantation. In allogeneic transplantation, iPSCs donate from someone other than the patient, employing human leukocyte antigen (HLA) that match donor in order to prevent immunological rejection by the host. In autologous transplantation, the patient’s own iPSCs develop into target cells. The immune system in humans is significantly regulated by HLA [8, 121]. In 2014, the first transplantation of autologous iPSC-derived cells was performed on a Japanese woman in her seventies suffering from age-related macular degeneration. A degeneration of the retinal epithelium, a layer of cells that supports photoreceptors necessary for vision, is the cause of age-related macular degeneration. Following surgery, the patient did not encounter any severe complications. During the first year, there was no evidence of immunological rejection of the transplanted iPSC-derived retinal pigment epithelium cells, and the patient’s visual acuity did not increase or decrease without further treatment [122, 123]. There are several obstacles that make the autologous approach difficult to use. For instance, safety testing for selecting the finest iPSC line from 30 distinct lines is estimated to cost US$500,000, and the total cost per patient is approximately $1,000,000. In addition, autologous transplantation is a lengthy procedure. There are more obstacles for autologous stem cell therapies than for allogeneic transplantation. During allogeneic therapy, the cell product may be extensively characterized and examined over a lengthy period of preclinical evaluation before being administered to a variety of patients. But in the autologous approach, the available time for this evaluation is much shorter [124]. In a rat model of MI, Guan et al. evaluated how well hiPSC-CM transplantation improved myocardial function. They discovered that animals had reduced mortality than the control group four weeks after receiving an iPSC-CM transplant. When iPSC-CMs were put into the control group instead of cell-free fluid, this model of left ventricular dysfunction was attenuated. Their study showed that hiPSC-CM grafts survived in infarcted rat hearts for 4 weeks and restored myocardial function [125]. Li et al. investigated the effects of secreted exosomes from hiPSC-derived endothelial cells on survival of CMs after MI and they suggested that these exosomes could reduce the injury of glucose and oxygen deprivation, improve the Ca2+ homeostasis and restore heart function [126]. Ye et al. injected three types of hiPSC-derived cells (CMs, endothelial cells and smooth muscle cells) into injured hearts of a porcine model with MI and indicated engraftment of all three lineages cells at the site of injury after almost 4th week of injection. Their study showed enhancements in contractile performance, myocardial wall stress, metabolism and no development of arrhythmias [127]. The immaturity and heterogeneity of heart cells are two challenges for iPSC-based treatment. Regarding these problems, better methods to deal with the variability of the iPSC-CMs have been established, along with separation strategies based on particular cardiac markers, such as VCAM1. The population of cardiac cells in the CM consists of nodal-like, atrial, pacemaker, ventricular, and functional cells [76]. In a study, Funakoshi et al. [39] examined the engraftment efficiency of CMs at various stages of development in numerous days of mouse mesodermal cells and found that day 20 had the highest engraftment ratio. They administered day 20 cm to immune-deficient animals with infarcted hearts, and they saw a significant improvement in function and echocardiogram of the heart, demonstrating the therapeutic potential of these cells [128]. One of the issues with iPSC transplants is arrhythmias that has been observed in non-human primates [129]. Marchiano et al. distinguished ion channel genes that are expressed after hPSC-CM transplantation which lead to automaticity and arrhythmias. They utilized CRISPR/Cas9 technology to overexpress gene KCNJ2, knock out CACNA1H, HCN4, SLC8A1 genes and created adult like CMs that have decreased automaticity and contracted in response to exterior stimulations. These cells were transplanted in vivo and engrafted into host CMs which indicate no arrhythmias. They manifested that automaticity and risk of arrhythmias can be reduced by inducing the expression of adult like ion genes in PSC-CMs [130].
Challenges and future perspectives of iPSCs in vitro models
The creation of iPSC lines was a significant development in the area of regenerative medicine. These cells may develop into a variety of cell types, including vascular endothelial cells, smooth muscle cells, and CMs, making them an innovative and trustworthy model system for studying the molecular and genetic causes of human disorders [76]. iPSC-CMs are a viable alternative to primary cells and animal models since they express a number of cardiac-specific genes, have a human genome, and possess key characteristics of human CMs. Although iPSC-CMs provide opportunities for drug development and testing of therapeutic approaches, these models have several limitations. Imperfect replication of adult disease phenotype and intricate intercellular communication of disease, is the primary limitation of iPSC-CMs which is because of their embryonic structure and lack of intercellular interactions between various cell types. Among the embryonic structures of iPSC-CMs are short sarcomeres, variable calcium ion concentration, and less mitochondria. Moreover, the complexity of certain diseases involving multiple genes, heterogeneous diseases, and individuals carrying distinct gene mutations are obstacles for iPSC technology. Consequently, it may be impossible to produce a single compound for a spectrum of diseases using a single patient-derived cell line. In addition, it is challenging to simulate the physiological state of the heart and the intercellular interactions of different cell types using a 2D iPSC-CMs culture. As a result, efforts are being made to develop 3D culture systems [8, 131]. Standard 2D cell culture is unable to replicate the complex physical and environmental interactions that occur in vivo. Therefore, they are unable to develop some cardiac disease phenotypes. ECM scaffolds, cellular components, and fluidic microenvironments may all be obtained from 3D culture models. Hydrogel scaffold-based models with biowires, engineered cardiac/heart tissues, HoC, and designed human myocardium are examples of 3D cardiac models. Scaffold-free 3D models include cardioids, cardiac spheroids, and cell sheets as another category. Researchers have taken a variety of steps to undertake morphological and functional maturation of iPSC-CM, to enhance CMs models, and to learn the underlying processes in addition to developing disease-treating therapies. Although 3D models may replicate disease-specific models that haven’t been replicated in 2D models, it is essential to reach a full CM maturation in the development of novel medications, therefore perhaps patient-specific iPSC models couldn’t totally replace animal models or conventional cell line assays. [132]. Maturation is a difficult process. A variety of elements, including as mechanical cues, electric signals, biochemical adhesion, and cell to cell contacts, influence the growth of CMs in vivo. It is impossible to produce a significant number of CMs for regenerative medicine quickly and at high capacity. As a result, it is anticipated that additional techniques will be introduced to supplement the ones now in use, resulting in improved iPSC-CM maturation induction and cell transplantation [133]. Overall based on the review article written by Gill et al. hurdles and challenges for the clinical translation of stem cell therapy can be summarized in heterogeneity of stem cells, uncontrolled differentiation, migration, survival of transplanted stem cells and immunogenicity of these cells after transplantation [134] and for more detailed information it is suggested to read the mentioned article.
Conclusion
These days, iPSC technology has developed into an interesting tool for scientists to model diseases, offering new prospects in the area of modeling cardiac disease that make it feasible to research complicated cardiac arrhythmia syndromes and to reproduce patient phenotypic in vitro models. The development of iPSC technology seems to be useful for making notable advancements in understanding the fundamental principles of cardiomyogenesis, and it may aid in future biomedical research by facilitating the testing of novel medications and potentially significant therapeutic applications. One of the most significant advancements for iPSC technology is specifically giving a new source for individualized cardiac myocytes in the future and effective methods for cell differentiation towards chosen somatic cell types.
Data availability
Not applicable.
Abbreviations
- iPSC:
-
Induced pluripotent stem cell
- CMs:
-
Cardiomyocytes
- PSCs:
-
Pluripotent stem cells
- hPSC:
-
Human Pluripotent stem cells
- PSC:
-
CMs-PSC-derived CMs
- iPSC:
-
CMs-iPSC-derived CMs
- hiPSCs:
-
Human Induced pluripotent stem cells from
- ESCs:
-
Embryonic stem cells
- bFGF:
-
Basic fibroblast growth factor
- MI:
-
Myocardial infarction
- VEGF:
-
Vascular endothelial growth factor
- DKK:
-
1-Peptide Wnt inhibitor dickkopf homolog 1
- BMP:
-
4-Bone morphogenetic protein 4
- Wnt:
-
Wingless-related integration site
- XAV939:
-
Wnt Signaling Inhibitor
- TRF1:
-
Telomere Repeat binding Factor 1
- LRP5/6:
-
Low-density lipoprotein receptor-related protein
- TGF:
-
β-Transforming growth factor beta
- FGF2:
-
Fibroblast growth factor 2
- GSK3β:
-
Glycogen synthase kinase 3
- CK1:
-
Casein kinase 1
- APC:
-
Adenomatous polyposis coli
- PI3K:
-
Phosphoinositide 3-kinase
- Akt:
-
Protein kinase B
- BMPs:
-
Bone morphogenetic proteins
- SMAD:
-
Small mother against decapentaplegic
- ALK:
-
Activin-like kinase
- ActR2A:
-
Activin receptor type-2 A
- ActR2B:
-
Activin receptor type-2B
- TGFBRII:
-
Transforming growth factor beta receptor 2
- BMPR2:
-
Bone morphogenetic protein receptor type 2
- TGFBR3:
-
Transforming growth factor beta receptor III
- MAPK:
-
Mitogen-activated protein kinase
- PKP2:
-
Plakophilin 2
- TTN:
-
Titin
- TNNT2:
-
Troponin T2, cardiac type
- PLN:
-
Phospholamban
- LMNA:
-
Lamin A/C
- DES:
-
Desmin
- PKA:
-
Protein kinase A
- AMPK:
-
AMP-activated protein kinase
- CAMK2D:
-
Calcium/calmodulin dependent protein kinase II delta
- MEK:
-
Mitogen-activated protein kinase kinase
- ERK:
-
Extracellular signal-regulated kinase
- TNNI3:
-
Troponin I3, cardiac type
- FACS:
-
Fluorescence-activated cell sorting
- MACS:
-
Magnetic-activated cell sorting
- SIRPA:
-
Signal-Regulatory Protein Alpha
- VCAM1:
-
Vascular Cell Adhesion Molecule 1
- miRNA:
-
MicroRNA
- mRNAs:
-
Messenger RNAs
- 3'UTR:
-
3′ untranslated regions
- FDA:
-
Food and medicine Administration
- EMA:
-
European Medicines Agency
- LQTS:
-
Long QT syndrome
- INa:
-
Sodium current
- IKr:
-
Potassium current
- IKs:
-
Delayed rectifier K + current
- CRISPR:
-
Clustered regularly interspaced short palindromic repeats
- Cas9:
-
CRISPR-associated protein 9
- ARVC:
-
Arrhythmogenic right ventricular cardiomyopathy
- PPARγ:
-
Peroxisome proliferator-activated receptor-gamma
- DSG2:
-
Desmoglein-2
- DCM:
-
Dilated cardiomyopathy
- PDGF:
-
Platelet-derived growth factor
- PDGFRB:
-
Platelet-derived growth factor receptor-β
- HCM:
-
Hypertrophic cardiomyopathy
- MYBPC3:
-
Myosin binding protein C
- MYH7:
-
Myosin heavy chain 7
- MHC:
-
β-Myosin heavy chain beta
- TSA:
-
Trichostatin A
- HLA:
-
Human leukocyte antigen
- JAK2:
-
Janus kinase 2
- KDR:
-
Kinase insert domain receptor (KDR is also known Flk1)
- Flk1:
-
Fetal Liver Kinase 1
- KIT:
-
KIT proto-oncogene receptor tyrosine kinase
- MLC2A:
-
Myosin light chain 2 A
- MLC2V:
-
Myosin light chain 2 V
References
Fathi E, Farahzadi R. Mesenchymal stem cells as a cell-based therapeutic strategy targeting the telomerase activity of KG1 Acute myeloid leukemia cells. Acta Medica Iranica. 2022;60(2 SE–Articles). https://doi.org/10.18502/acta.v60i2.8817.
Fathi E, Farahzadi R, Sheervalilou R, Sanaat Z, Vietor I. A general view of CD33 + leukemic stem cells and CAR-T cells as interesting targets in acute myeloblatsic leukemia therapy. Blood Res. 2020;55(1):10–6. https://doi.org/10.5045/br.2020.55.1.10.
Fathi E, Azarbad S, Farahzadi R, Javanmardi S, Vietor I. Effect of rat bone marrow derived-mesenchymal stem cells on Granulocyte differentiation of mononuclear cells as Preclinical Agent in Cellbased Therapy. Curr Gene Ther. 2022;22(2):152–61. https://doi.org/10.2174/1566523221666210519111933.
Farahzadi R, Fathi E, Valipour B, Ghaffary S. Stem cells-derived exosomes as cardiac regenerative agents. Int J Cardiol Heart Vasculature. 2024;52:101399. https://doi.org/10.1016/j.ijcha.2024.101399.
Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–76. https://doi.org/10.1016/j.cell.2006.07.024.
Betters E, Murdoch B, Leung AW, García-Castro MI. (2014). Chapter 18 - Human Neural Crest Cells and Stem Cell-Based Models (P. A. B. T.-N. C. C. Trainor, editor; pp. 395–412). Academic Press. https://doi.org/10.1016/B978-0-12-401730-6.00019-3.
Amabile G, Meissner A. Induced pluripotent stem cells: current progress and potential for regenerative medicine. Trends Mol Med. 2009;15(2):59–68. https://doi.org/10.1016/j.molmed.2008.12.003.
Nicholson MW, Ting CY, Chan DZH, Cheng YC, Lee YC, Hsu CC, Huang CY, Hsieh PCH. Utility of iPSC-Derived cells for Disease modeling, Drug Development, and cell therapy. Cells. 2022;11(11). https://doi.org/10.3390/cells11111853.
Takahashi F, Patel P, Kitsuka T, Arai K. The exciting realities and possibilities of iPS-Derived cardiomyocytes. Bioeng (Basel Switzerland). 2023;10(2). https://doi.org/10.3390/bioengineering10020237.
Uygur A, Lee RT. Mechanisms of Cardiac Regeneration. Dev Cell. 2016;36(4):362–74. https://doi.org/10.1016/j.devcel.2016.01.018.
Lewandowski J, Rozwadowska N, Kolanowski TJ, Malcher A, Zimna A, Rugowska A, Fiedorowicz K, Łabędź W, Kubaszewski Ł, Chojnacka K, Bednarek-Rajewska K, Majewski P, Kurpisz M. The impact of in vitro cell culture duration on the maturation of human cardiomyocytes derived from induced pluripotent stem cells of myogenic origin. Cell Transplant. 2018;27(7):1047–67. https://doi.org/10.1177/0963689718779346.
Vučković S, Dinani R, Nollet EE, Kuster DWD, Buikema JW, Houtkooper RH, Nabben M, van der Velden J, Goversen B. Characterization of cardiac metabolism in iPSC-derived cardiomyocytes: lessons from maturation and disease modeling. Stem Cell Res Therapy. 2022;13(1):1–19. https://doi.org/10.1186/s13287-022-03021-9.
Correia C, Koshkin A, Duarte P, Hu D, Carido M, Sebastião MJ, Gomes-Alves P, Elliott DA, Domian IJ, Teixeira AP, Alves PM, Serra M. 3D aggregate culture improves metabolic maturation of human pluripotent stem cell derived cardiomyocytes. Biotechnol Bioeng. 2018;115(3):630–44. https://doi.org/10.1002/bit.26504.
Feyen DAM, McKeithan WL, Bruyneel AAN, Spiering S, Hörmann L, Ulmer B, Zhang H, Briganti F, Schweizer M, Hegyi B, Liao Z, Pölönen R-P, Ginsburg KS, Lam CK, Serrano R, Wahlquist C, Kreymerman A, Vu M, Amatya PL, Mercola M. Metabolic maturation media improve physiological function of human iPSC-Derived cardiomyocytes. Cell Rep. 2020;32(3):107925. https://doi.org/10.1016/j.celrep.2020.107925.
Kingham E, Bone ROCO, Development H, Cells S, Development H, Sciences D, So S, Kingdom U. Embryonic and Induced Pluripotent Stem cells:understanding, creating, Regenerative Medicine. ACS Nano. 2013;7(3):1867–81.
Huang Y, Wang T, López MEU, Hirano M, Hasan A, Shin SR. Recent advancements of human iPSC derived cardiomyocytes in drug screening and tissue regeneration. Microphysiological Syst. 2020;4(September):2–2. https://doi.org/10.21037/mps-20-3.
Funakoshi S, Yoshida Y. Recent progress of iPSC technology in cardiac diseases. Arch Toxicol. 2021;95(12):3633–50. https://doi.org/10.1007/s00204-021-03172-3.
Batalov I, Feinberg AW. Differentiation of cardiomyocytes from human pluripotent stem cells using monolayer culture. Biomark Insights. 2015;10:71–6. https://doi.org/10.4137/BMI.S20050.
Ding VMY, Ling L, Natarajan S, Yap MGS, Cool SM, Choo ABH. FGF-2 modulates wnt signaling in undifferentiated hESC and iPS cells through activated PI3-K/GSK3beta signaling. J Cell Physiol. 2010;225(2):417–28. https://doi.org/10.1002/jcp.22214.
Ren Y, Lee MY, Schliffke S, Paavola J, Amos PJ, Ge X, Ye M, Zhu S, Senyei G, Lum L, Ehrlich BE, Qyang Y. Small molecule wnt inhibitors enhance the efficiency of BMP-4-directed cardiac differentiation of human pluripotent stem cells. J Mol Cell Cardiol. 2011;51(3):280–7. https://doi.org/10.1016/j.yjmcc.2011.04.012.
Gessert S, Kühl M. The multiple phases and faces of wnt signaling during cardiac differentiation and development. Circul Res. 2010;107(2):186–99. https://doi.org/10.1161/CIRCRESAHA.110.221531.
Willems E, Spiering S, Davidovics H, Lanier M, Xia Z, Dawson M, Cashman J, Mercola M. Small-molecule inhibitors of the wnt pathway potently promote cardiomyocytes from human embryonic stem cell-derived mesoderm. Circul Res. 2011;109(4):360–4. https://doi.org/10.1161/CIRCRESAHA.111.249540.
Guo Y, Dorn T, Kühl SJ, Linnemann A, Rothe M, Pfister AS, Vainio S, Laugwitz KL, Moretti A, Kühl M. The wnt inhibitor Dkk1 is required for maintaining the normal cardiac differentiation program in Xenopus laevis. Dev Biol. 2019;449(1):1–13. https://doi.org/10.1016/j.ydbio.2019.02.009.
Mazzotta S, Neves C, Bonner RJ, Bernardo AS, Docherty K, Hoppler S. Distinctive roles of canonical and noncanonical wnt signaling in human embryonic Cardiomyocyte Development. Stem Cell Rep. 2016;7(4):764–76. https://doi.org/10.1016/j.stemcr.2016.08.008.
de Jaime-Soguero A, De Oliveira WAA, Lluis F. The pleiotropic effects of the canonical wnt pathway in early development and pluripotency. Genes. 2018;9(2):1–23. https://doi.org/10.3390/genes9020093.
Silva-García O, Valdez-Alarcón JJ, Baizabal-Aguirre VM. Wnt/β-catenin signaling as a molecular target by pathogenic bacteria. Front Immunol. 2019;10(SEP):1–14. https://doi.org/10.3389/fimmu.2019.02135.
Fathi E, Valipour B, Vietor I, Farahzadi R. An overview of the myocardial regeneration potential of cardiac c-Kit + progenitor cells via PI3K and MAPK signaling pathways. Future Cardiol. 2020;16(3):199–209. https://doi.org/10.2217/fca-2018-0049.
Yang L, Soonpaa MH, Adler ED, Roepke TK, Kattman SJ, Kennedy M, Henckaerts E, Bonham K, Abbott GW, Linden RM, Field LJ, Keller GM. Human cardiovascular progenitor cells develop from a KDR + embryonic-stem-cell-derived population. Nature. 2008;453(7194):524–8. https://doi.org/10.1038/nature06894.
Ueno S, Weidinger G, Osugi T, Kohn AD, Golob JL, Pabon L, Reinecke H, Moon RT, Murry CE. Biphasic role for Wnt/beta-catenin signaling in cardiac specification in zebrafish and embryonic stem cells. Proc Natl Acad Sci USA. 2007;104(23):9685–90. https://doi.org/10.1073/pnas.0702859104.
Ozhan G, Weidinger G. Wnt/β-catenin signaling in heart regeneration. Cell Regeneration. 2015;4(1):4:3. https://doi.org/10.1186/s13619-015-0017-8.
Kadari A, Mekala S, Wagner N, Malan D, Köth J, Doll K, Stappert L, Eckert D, Peitz M, Matthes J, Sasse P, Herzig S, Brüstle O, Ergün S, Edenhofer F. Robust generation of cardiomyocytes from human iPS cells requires precise modulation of BMP and WNT signaling. Stem Cell Reviews Rep. 2015;11(4):560–9. https://doi.org/10.1007/s12015-014-9564-6.
Aguado T, Gutiérrez FJ, Aix E, Schneider RP, Giovinazzo G, Blasco MA, Flores I. Telomere length defines the Cardiomyocyte differentiation potency of Mouse Induced Pluripotent Stem cells. Stem Cells. 2017;35(2):362–73. https://doi.org/10.1002/stem.2497.
Dronkers E, Wauters MMM, Goumans MJ, Smits AM. Epicardial TGFβ and BMP signaling in cardiac regeneration: what lesson can we learn from the developing heart? Biomolecules. 2020;10(3). https://doi.org/10.3390/biom10030404.
Saadat S, Noureddini M, Mahjoubin-Tehran M, Nazemi S, Shojaie L, Aschner M, Maleki B, Abbasi-kolli M, Moghadam R, Alani H, B., Mirzaei H. Pivotal role of TGF-β/Smad signaling in Cardiac Fibrosis: non-coding RNAs as effectual players. Front Cardiovasc Med. 2021;7(January):1–18. https://doi.org/10.3389/fcvm.2020.588347.
Moretti A, Laugwitz K-L, Dorn T, Sinnecker D, Mummery C. Pluripotent stem cell models of human heart disease. Cold Spring Harbor Perspect Med. 2013;3(11). https://doi.org/10.1101/cshperspect.a014027.
Paik DT, Chandy M, Wu JC. Patient and Disease-Specific Induced Pluripotent Stem cells for Discovery of Personalized Cardiovascular drugs and therapeutics. Pharmacol Rev. 2020;72(1):320–42. https://doi.org/10.1124/pr.116.013003.
Carvajal-Vergara X, Sevilla A, D’Souza SL, Ang Y-S, Schaniel C, Lee D-F, Yang L, Kaplan AD, Adler ED, Rozov R, Ge Y, Cohen N, Edelmann LJ, Chang B, Waghray A, Su J, Pardo S, Lichtenbelt KD, Tartaglia M, Lemischka IR. Patient-specific induced pluripotent stem-cell-derived models of LEOPARD syndrome. Nature. 2010;465(7299):808–12. https://doi.org/10.1038/nature09005.
Sala L, Gnecchi M, Schwartz PJ. Long QT syndrome modelling with Cardiomyocytes Derived from Human-induced pluripotent stem cells. Arrhythmia Electrophysiol Rev. 2019;8(2):105–10. https://doi.org/10.15420/aer.2019.1.1.
Al Abbar A, Ngai SC, Nograles N, Alhaji SY, Abdullah S. Induced Pluripotent Stem cells: reprogramming platforms and applications in cell replacement therapy. BioResearch Open Access. 2020;9(1):121–36. https://doi.org/10.1089/biores.2019.0046.
Huang J, Feng Q, Wang L, Zhou B. Human pluripotent stem cell-derived Cardiac cells: application in Disease modeling, cell therapy, and Drug Discovery. Front Cell Dev Biology. 2021;9(April):1–8. https://doi.org/10.3389/fcell.2021.655161.
Ovics P, Regev D, Baskin P, Davidor M, Shemer Y, Neeman S, Ben-Haim Y, Binah O. Drug development and the use of induced pluripotent stem cell-derived cardiomyocytes for disease modeling and drug toxicity screening. Int J Mol Sci. 2020;21(19):1–42. https://doi.org/10.3390/ijms21197320.
Crotti L, Celano G, Dagradi F, Schwartz PJ. Congenital long QT syndrome. Orphanet J Rare Dis. 2008;3(1):1–16. https://doi.org/10.1186/1750-1172-3-18.
Yang C, Al-Aama J, Stojkovic M, Keavney B, Trafford A, Lako M, Armstrong L. Concise Review: Cardiac Disease modeling using Induced Pluripotent Stem cells. Stem Cells. 2015;33(9):2643–51. https://doi.org/10.1002/stem.2070.
Wallace E, Howard L, Liu M, O’Brien T, Ward D, Shen S, Prendiville T. Long QT syndrome: Genetics and Future Perspective. Pediatr Cardiol. 2019;40(7):1419–30. https://doi.org/10.1007/s00246-019-02151-x.
Yu Y, Deschenes I, Zhao M-T. Precision medicine for long QT syndrome: patient-specific iPSCs take the lead. Expert Rev Mol Med. 2023;25:e5. Available from: https://www.cambridge.org/core/article/precision-medicine-for-long-qt-syndrome-patientspecific-ipscs-take-the-lead/EC165F1E34476BF19A3D7F4BBFC0D388.
Van Langen IM, Birnie E, Alders M, Jongbloed RJ, Le Marec H, Wilde AAM. (2003). The use of genotype-phenotype correlations in mutation analysis for the long QT syndrome. In Journal of medical genetics (Vol. 40, Issue 2, pp. 141–145). https://doi.org/10.1136/jmg.40.2.141.
Schwartz PJ, Crotti L, Insolia R. Long-QT syndrome: from genetics to management. Circ Arrhythm Electrophys. 2012;5(4):868–77. https://doi.org/10.1161/CIRCEP.111.962019.
Wilde AAM, Amin AS, Postema PG. Diagnosis, management and therapeutic strategies for congenital long QT syndrome. Heart. 2022;108(5):332–8. https://doi.org/10.1136/heartjnl-2020-318259.
Moretti A, Bellin M, Welling A, Jung CB, Lam JT, Bott-Flügel L, Dorn T, Goedel A, Höhnke C, Hofmann F, Seyfarth M, Sinnecker D, Schömig A, Laugwitz K-L. Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N Engl J Med. 2010;363(15):1397–409. https://doi.org/10.1056/NEJMoa0908679.
Wang Y, Liang P, Lan F, Wu H, Lisowski L, Gu M, Hu S, Kay MA, Urnov FD, Shinnawi R, Gold JD, Gepstein L, Wu JC. Genome editing of isogenic human induced pluripotent stem cells recapitulates long QT phenotype for drug testing. J Am Coll Cardiol. 2014;64(5):451–9. https://doi.org/10.1016/j.jacc.2014.04.057.
Garg P, Oikonomopoulos A, Chen H, Li Y, Lam CK, Sallam K, Perez M, Lux RL, Sanguinetti MC, Wu JC. Genome editing of Induced Pluripotent Stem cells to Decipher Cardiac Channelopathy variant. J Am Coll Cardiol. 2018;72(1):62–75. https://doi.org/10.1016/j.jacc.2018.04.041.
Mesquita FCP, Arantes PC, Kasai-Brunswick TH, Araujo DS, Gubert F, Monnerat G, Silva dos Santos D, Neiman G, Leitão IC, Barbosa RAQ, Coutinho JL, Vaz IM, dos Santos MN, Borgonovo T, Cruz FES, Miriuka S, Medei EH, de Carvalho C, A. C., Carvalho AB. R534C mutation in hERG causes a trafficking defect in iPSC-derived cardiomyocytes from patients with type 2 long QT syndrome. Sci Rep. 2019;9(1):19203. https://doi.org/10.1038/s41598-019-55837-w.
Brandão KO, van den Brink L, Miller DC, Grandela C, van Meer BJ, Mol MPH, de Korte T, Tertoolen LGJ, Mummery CL, Sala L, Verkerk AO, Davis RP. Isogenic sets of hiPSC-CMs harboring distinct KCNH2 mutations Differ functionally and in susceptibility to Drug-Induced Arrhythmias. Stem Cell Rep. 2020;15(5):1127–39. https://doi.org/10.1016/j.stemcr.2020.10.005.
Wang F, Han Y, Sang W, Wang L, Liang X, Wang L, Xing Q, Guo Y, Zhang J, Zhang L, Zukela T, Xiaokereti J, Lu Y, Zhou X, Tang B, Li Y. In Vitro Drug Screening using iPSC-Derived cardiomyocytes of a long QT-Syndrome patient carrying KCNQ1 & TRPM4 dual mutation: an experimental personalized treatment. Cells. 2022;11(16). https://doi.org/10.3390/cells11162495.
Sen-Chowdhry S, Morgan RD, Chambers JC, McKenna WJ. Arrhythmogenic cardiomyopathy: etiology, diagnosis, and treatment. Annu Rev Med. 2010;61(1):233–53. https://doi.org/10.1146/annurev.med.052208.130419.
Basso C, Bauce B, Corrado D, Thiene G. Pathophysiology of arrhythmogenic cardiomyopathy. Nat Reviews Cardiol. 2012;9(4):223–33. https://doi.org/10.1038/nrcardio.2011.173.
Li KHC, Bazoukis G, Liu T, Li G, Wu WKK, Wong SH, Wong WT, Chan YS, Wong MCS, Wassilew K, Vassiliou VS, Tse G. Arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/D) in clinical practice. J Arrhythmia. 2018;34(1):11–22. https://doi.org/10.1002/joa3.12021.
Watkins H, Ashrafian H, Redwood C. Inherited cardiomyopathies. N Engl J Med. 2011;364(17):1643–56. https://doi.org/10.1056/NEJMra0902923.
Corrado D, Van Tintelen PJ, McKenna WJ, Hauer RNW, Anastastakis A, Asimaki A, Basso C, Bauce B, Brunckhorst C, Bucciarelli-Ducci C, Duru F, Elliott P, Hamilton RM, Haugaa KH, James CA, Judge D, Link MS, Marchlinski FE, Mazzanti A, Calkins H. (2020). Arrhythmogenic right ventricular cardiomyopathy: Evaluation of the current diagnostic criteria and differential diagnosis. European Heart Journal, 41(14), 1414-1427b. https://doi.org/10.1093/eurheartj/ehz669.
Musunuru K. An overview of Genome Editing in Cardiovascular and Metabolic diseases. Adv Exp Med Biol. 2023;1396:3–16. https://doi.org/10.1007/978-981-19-5642-3_1.
Pan Z, Ebert A, Liang P. Human-induced pluripotent stem cells as models for rare cardiovascular diseases: from evidence-based medicine to precision medicine. Pflugers Archiv Eur J Physiol. 2021;473(7):1151–65. https://doi.org/10.1007/s00424-020-02486-y.
Garcia-Gras E, Lombardi R, Giocondo MJ, Willerson JT, Schneider MD, Khoury DS, Marian AJ. Suppression of canonical Wnt/beta-catenin signaling by nuclear plakoglobin recapitulates phenotype of arrhythmogenic right ventricular cardiomyopathy. J Clin Investig. 2006;116(7):2012–21. https://doi.org/10.1172/JCI27751.
Djouadi F, Lecarpentier Y, Hébert J-L, Charron P, Bastin J, Coirault C. A potential link between peroxisome proliferator-activated receptor signalling and the pathogenesis of arrhythmogenic right ventricular cardiomyopathy. Cardiovascular Res. 2009;84(1):83–90. https://doi.org/10.1093/cvr/cvp183.
Lombardi R, Dong J, Rodriguez G, Bell A, Leung TK, Schwartz RJ, Willerson JT, Brugada R, Marian AJ. Genetic fate mapping identifies second heart field progenitor cells as a source of adipocytes in arrhythmogenic right ventricular cardiomyopathy. Circul Res. 2009;104(9):1076–84. https://doi.org/10.1161/CIRCRESAHA.109.196899.
Beffagna G, Occhi G, Nava A, Vitiello L, Ditadi A, Basso C, Bauce B, Carraro G, Thiene G, Towbin JA, Danieli GA, Rampazzo A. Regulatory mutations in transforming growth factor-beta3 gene cause arrhythmogenic right ventricular cardiomyopathy type 1. Cardiovascular Res. 2005;65(2):366–73. https://doi.org/10.1016/j.cardiores.2004.10.005.
Merner ND, Hodgkinson KA, Haywood AFM, Connors S, French VM, Drenckhahn J-D, Kupprion C, Ramadanova K, Thierfelder L, McKenna W, Gallagher B, Morris-Larkin L, Bassett AS, Parfrey PS, Young T-L. Arrhythmogenic right ventricular cardiomyopathy type 5 is a fully penetrant, lethal arrhythmic disorder caused by a missense mutation in the TMEM43 gene. Am J Hum Genet. 2008;82(4):809–21. https://doi.org/10.1016/j.ajhg.2008.01.010.
Awad MM, Calkins H, Judge DP. Mechanisms of disease: molecular genetics of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Nat Clin Pract Cardiovasc Med. 2008;5(5):258–67. https://doi.org/10.1038/ncpcardio1182.
Ma D, Wei H, Lu J, Ho S, Zhang G, Sun X, Oh Y, Tan SH, Ng ML, Shim W, Wong P, Liew R. Generation of patient-specific induced pluripotent stem cell-derived cardiomyocytes as a cellular model of arrhythmogenic right ventricular cardiomyopathy. Eur Heart J. 2012;34(15):1122–33. https://doi.org/10.1093/eurheartj/ehs226.
Kim C, Wong J, Wen J, Wang S, Wang C, Spiering S, Kan NG, Forcales S, Puri PL, Leone TC, Marine JE, Calkins H, Kelly DP, Judge DP, Chen H-SV. Studying arrhythmogenic right ventricular dysplasia with patient-specific iPSCs. Nature. 2013;494(7435):105–10. https://doi.org/10.1038/nature11799.
Matthes SA, Taffet S, Delmar M. Plakophilin-2 and the migration, differentiation and transformation of cells derived from the epicardium of neonatal rat hearts. Cell Communication Adhes. 2011;18(4):73–84. https://doi.org/10.3109/15419061.2011.621561.
Caspi O, Huber I, Gepstein A, Arbel G, Maizels L, Boulos M, Gepstein L. Modeling of arrhythmogenic right ventricular cardiomyopathy with human induced pluripotent stem cells. Circ Cardiovasc Genet. 2013;6(6):557–68. https://doi.org/10.1161/CIRCGENETICS.113.000188.
Groeneweg JA, Bhonsale A, James CA, te Riele AS, Dooijes D, Tichnell C, Murray B, Wiesfeld ACP, Sawant AC, Kassamali B, Atsma DE, Volders PG, de Groot NM, de Boer K, Zimmerman SL, Kamel IR, van der Heijden JF, Russell SD, Cramer J, Calkins M, H. Clinical presentation, Long-Term Follow-Up, and outcomes of 1001 Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy Patients and family members. Circ Cardiovasc Genet. 2015;8(3):437–46. https://doi.org/10.1161/CIRCGENETICS.114.001003.
Hawthorne RN, Blazeski A, Lowenthal J, Kannan S, Teuben R, DiSilvestre D, Morrissette-McAlmon J, Saffitz JE, Boheler KR, James CA, Chelko SP, Tomaselli G, Tung L. Altered Electrical, Biomolecular, and immunologic phenotypes in a Novel patient-derived stem cell model of Desmoglein-2 Mutant ARVC. J Clin Med. 2021;10(14). https://doi.org/10.3390/jcm10143061.
Lakdawala NK, Winterfield JR, Funke BH. Dilated cardiomyopathy. Circulation: Arrhythmia Electrophysiol. 2013;6(1):228–37. https://doi.org/10.1161/CIRCEP.111.962050.
Schultheiss H-P, Fairweather D, Caforio ALP, Escher F, Hershberger RE, Lipshultz SE, Liu PP, Matsumori A, Mazzanti A, McMurray J, Priori SG. Dilated cardiomyopathy. Nat Reviews Disease Primers. 2019;5(1):32. https://doi.org/10.1038/s41572-019-0084-1.
Parrotta EI, Lucchino V, Scaramuzzino L, Scalise S, Cuda G. Modeling cardiac disease mechanisms using induced pluripotent stem cell-derived cardiomyocytes: Progress, promises and challenges. Int J Mol Sci (Vol. 2020;21:1–30. https://doi.org/10.3390/ijms21124354.
Japp AG, Gulati A, Cook SA, Cowie MR, Prasad SK. The diagnosis and evaluation of dilated cardiomyopathy. J Am Coll Cardiol. 2016;67(25):2996–3010. https://doi.org/10.1016/j.jacc.2016.03.590.
Herman DS, Lam L, Taylor MRG, Wang L, Teekakirikul P, Christodoulou D, Conner L, DePalma SR, McDonough B, Sparks E, Teodorescu DL, Cirino AL, Banner NR, Pennell DJ, Graw S, Merlo M, Di Lenarda A, Sinagra G, Bos JM, Seidman CE. Truncations of titin causing dilated cardiomyopathy. N Engl J Med. 2012;366(7):619–28. https://doi.org/10.1056/NEJMoa1110186.
Mestroni L, Brun F, Spezzacatene A, Sinagra G, Taylor MR. GENETIC CAUSES OF DILATED CARDIOMYOPATHY. Prog Pediatr Cardiol. 2014;37(1–2):13–8. https://doi.org/10.1016/j.ppedcard.2014.10.003.
Hinson JT, Chopra A, Nafissi N, Polacheck WJ, Benson CC, Swist S, Gorham J, Yang L, Schafer S, Sheng CC, Haghighi A, Homsy J, Hubner N, Church G, Cook SA, Linke WA, Chen CS, Seidman JG, Seidman CE. HEART DISEASE. Titin mutations in iPS cells define sarcomere insufficiency as a cause of dilated cardiomyopathy. Sci (New York N Y). 2015;349(6251):982–6. https://doi.org/10.1126/science.aaa5458.
Huang G, Bisaria A, Wakefield DL, Yamawaki TM, Luo X, Zhang JA, Vigneault P, Wang J, Reagan JD, Oliverio O, Zhou H, Li CM, Vila OF, Wang S, Malik FI, Hartman JJ, Hale CM. Titin-truncating variants in hiPSC cardiomyocytes induce pathogenic proteinopathy and sarcomere defects with preserved core contractile machinery. Stem Cell Rep. 2023;18(1):220–36. https://doi.org/10.1016/j.stemcr.2022.11.008.
Schick R, Mekies LN, Shemer Y, Eisen B, Hallas T, Ben Jehuda R, Ben-Ari M, Szantai A, Willi L, Shulman R, Gramlich M, Pane LS, My I, Freimark D, Murgia M, Santamaria G, Gherghiceanu M, Arad M, Moretti A, Binah O. Functional abnormalities in induced pluripotent stem cell-derived cardiomyocytes generated from titin-mutated patients with dilated cardiomyopathy. PLoS ONE. 2018;13(10):e0205719. https://doi.org/10.1371/journal.pone.0205719.
Jeon S-B, Kim H, Chun K-H, Oh J, Kwon C, Choi H-K, Kim S, Kim H-P, Kim I-C, Yoo J-Y, Park SW, Kang S-M, Lee S-H. Human induced pluripotent stem cell line YCMi007-A generated from a dilated cardiomyopathy patient with a heterozygous dominant c.613C > T (p. Arg205Trp) variant of the TNNT2 gene. Stem Cell Res. 2023;67:103048. https://doi.org/10.1016/j.scr.2023.103048.
Perea-Gil I, Seeger T, Bruyneel AAN, Termglinchan V, Monte E, Lim EW, Vadgama N, Furihata T, Gavidia AA, Ataam A, Bharucha J, Martinez-Amador N, Ameen N, Nair M, Serrano P, Kaur R, Feyen B, Diecke DAM, Snyder S, Karakikes MP, I. Serine biosynthesis as a novel therapeutic target for dilated cardiomyopathy. Eur Heart J. 2022;43(36):3477–89. https://doi.org/10.1093/eurheartj/ehac305.
Dai Y, Amenov A, Ignatyeva N, Koschinski A, Xu H, Soong PL, Tiburcy M, Linke WA, Zaccolo M, Hasenfuss G, Zimmermann WH, Ebert A. Troponin destabilization impairs sarcomere-cytoskeleton interactions in iPSC-derived cardiomyocytes from dilated cardiomyopathy patients. Sci Rep. 2020;10(1):1–15. https://doi.org/10.1038/s41598-019-56597-3.
Lin F, Worman HJ. Structural organization of the human gene encoding nuclear lamin A and nuclear lamin C. J Biol Chem. 1993;268(22):16321–6.
Hutchison CJ, Worman HJ. A-type lamins: guardians of the soma? Nat Cell Biol. 2004;6(11):1062–7. https://doi.org/10.1038/ncb1104-1062.
Siu C-W, Lee Y-K, Ho JC-Y, Lai W-H, Chan Y-C, Ng K-M, Wong L-Y, Au K-W, Lau Y-M, Zhang J, Lay KW, Colman A, Tse H-F. Modeling of lamin A/C mutation premature cardiac aging using patient-specific induced pluripotent stem cells. Aging. 2012;4(11):803–22. https://doi.org/10.18632/aging.100503.
Lee J, Termglinchan V, Diecke S, Itzhaki I, Lam CK, Garg P, Lau E, Greenhaw M, Seeger T, Wu H, Zhang JZ, Chen X, Gil IP, Ameen M, Sallam K, Rhee J-W, Churko JM, Chaudhary R, Chour T, Wu JC. Activation of PDGF pathway links LMNA mutation to dilated cardiomyopathy. Nature. 2019;572(7769):335–40. https://doi.org/10.1038/s41586-019-1406-x.
Lee C, Cho S, Lai C, Shenoy S, Vagelos R, Wu JC. Generation of three iPSC lines from dilated cardiomyopathy patients carrying a pathogenic LMNA variant. Stem Cell Res. 2022;59:102638. https://doi.org/10.1016/j.scr.2021.102638.
Elliott PM, Anastasakis A, Borger MA, Borggrefe M, Cecchi F, Charron P, Hagege AA, Lafont A, Limongelli G, Mahrholdt H, McKenna WJ, Mogensen J, Nihoyannopoulos P, Nistri S, Pieper PG, Pieske B, Rapezzi C, Rutten FH, Tillmanns C, Watkins H. 2014 ESC guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the diagnosis and management of hypertrophic cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J. 2014;35(39):2733–79. https://doi.org/10.1093/eurheartj/ehu284.
Geske JB, Ommen SR, Gersh BJ. Hypertrophic cardiomyopathy: clinical update. JACC: Heart Fail. 2018;6(5):364–75. https://doi.org/10.1016/j.jchf.2018.02.010.
Eschenhagen T, Carrier L. Cardiomyopathy phenotypes in human-induced pluripotent stem cell-derived cardiomyocytes-a systematic review. Pflug Arch: Eur J Physiol. 2019;471(5):755–68. https://doi.org/10.1007/s00424-018-2214-0.
Ho CY, Day SM, Ashley EA, Michels M, Pereira AC, Jacoby D, Cirino AL, Fox JC, Lakdawala NK, Ware JS, Caleshu CA, Helms AS, Colan SD, Girolami F, Cecchi F, Seidman CE, Sajeev G, Signorovitch J, Green EM, Olivotto I. Genotype and lifetime burden of Disease in Hypertrophic Cardiomyopathy: insights from the Sarcomeric Human Cardiomyopathy Registry (SHaRe). Circulation. 2018;138(14):1387–98. https://doi.org/10.1161/CIRCULATIONAHA.117.033200.
Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, Moss AJ, Seidman CE, Young JB. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; quality of Care and Outcomes Research and Functi. Circulation. 2006;113(14):1807–16. https://doi.org/10.1161/CIRCULATIONAHA.106.174287.
Maron BJ, Maron MS. Hypertrophic cardiomyopathy. Lancet (London England). 2013;381(9862):242–55. https://doi.org/10.1016/S0140-6736(12)60397-3.
Wolf CM. Hypertrophic cardiomyopathy: Genetics and clinical perspectives. Cardiovasc Diagnosis Therapy. 2019;9(Suppl 2):S388–415. https://doi.org/10.21037/CDT.2019.02.01.
Bouvagnet P, Mairhofer H, Leger JO, Puech P, Leger JJ. Distribution pattern of alpha and beta myosin in normal and diseased human ventricular myocardium. Basic Res Cardiol. 1989;84(1):91–102. https://doi.org/10.1007/BF01907006.
Nakao K, Minobe W, Roden R, Bristow MR, Leinwand LA. Myosin heavy chain gene expression in human heart failure. J Clin Investig. 1997;100(9):2362–70. https://doi.org/10.1172/JCI119776.
Han L, Li Y, Tchao J, Kaplan AD, Lin B, Li Y, Mich-Basso J, Lis A, Hassan N, London B, Bett GCL, Tobita K, Rasmusson RL, Yang L. Study familial hypertrophic cardiomyopathy using patient-specific induced pluripotent stem cells. Cardiovascular Res. 2014;104(2):258–69. https://doi.org/10.1093/cvr/cvu205.
Lan F, Lee AS, Liang P, Sanchez-Freire V, Nguyen PK, Wang L, Han L, Yen M, Wang Y, Sun N, Abilez OJ, Hu S, Ebert AD, Navarrete EG, Simmons CS, Wheeler M, Pruitt B, Lewis R, Yamaguchi Y, Wu JC. Abnormal calcium handling properties underlie familial hypertrophic cardiomyopathy pathology in patient-specific induced pluripotent stem cells. Cell Stem Cell. 2013;12(1):101–13. https://doi.org/10.1016/j.stem.2012.10.010.
Cohn R, Thakar K, Lowe A, Ladha FA, Pettinato AM, Romano R, Meredith E, Chen YS, Atamanuk K, Huey BD, Hinson JT. A contraction stress model of hypertrophic cardiomyopathy due to Sarcomere mutations. Stem Cell Rep. 2019;12(1):71–83. https://doi.org/10.1016/j.stemcr.2018.11.015.
Escribá R, Larrañaga-Moreira JM, Richaud-Patin Y, Pourchet L, Lazis I, Jiménez-Delgado S, Morillas-García A, Ortiz-Genga M, Ochoa JP, Carreras D, Pérez GJ, de la Pompa JL, Brugada R, Monserrat L, Barriales-Villa R, Raya A. iPSC-Based modeling of variable clinical presentation in hypertrophic cardiomyopathy. Circul Res. 2023;108–19. https://doi.org/10.1161/circresaha.122.321951.
Harding D, Chong MHA, Lahoti N, Bigogno CM, Prema R, Mohiddin SA, Marelli-Berg F. Dilated cardiomyopathy and chronic cardiac inflammation: Pathogenesis, diagnosis and therapy. J Intern Med. 2023;293(1):23–47. https://doi.org/10.1111/joim.13556.
Sayed N, Liu C, Wu J. Translation of Human-Induced pluripotent stem cells. J Am Coll Cardiol. 2016;67:2161–76. https://doi.org/10.1016/j.jacc.2016.01.083.
Harvey PA, Leinwand LA. The cell biology of disease: cellular mechanisms of cardiomyopathy. J Cell Biol. 2011;194(3):355–65. https://doi.org/10.1083/jcb.201101100.
Chong JJH, Yang X, Don CW, Minami E, Liu Y-W, Weyers JJ, Mahoney WM, Van Biber B, Cook SM, Palpant NJ, Gantz JA, Fugate JA, Muskheli V, Gough GM, Vogel KW, Astley CA, Hotchkiss CE, Baldessari A, Pabon L, Murry CE. Human embryonic-stem-cell-derived cardiomyocytes regenerate non-human primate hearts. Nature. 2014;510(7504):273–7. https://doi.org/10.1038/nature13233.
Tsujisaka Y, Hatani T, Okubo C, Ito R, Kimura A, Narita M, Chonabayashi K, Funakoshi S, Lucena-Cacace A, Toyoda T, Osafune K, Kimura T, Saito H, Yoshida Y. Purification of human iPSC-derived cells at large scale using microRNA switch and magnetic-activated cell sorting. Stem Cell Rep. 2022;17(7):1772–85. https://doi.org/10.1016/j.stemcr.2022.05.003.
Dubois NC, Craft AM, Sharma P, Elliott DA, Stanley EG, Elefanty AG, Gramolini A, Keller G. SIRPA is a specific cell-surface marker for isolating cardiomyocytes derived from human pluripotent stem cells. Nat Biotechnol. 2011;29(11):1011–8. https://doi.org/10.1038/nbt.2005.
Uosaki H, Fukushima H, Takeuchi A, Matsuoka S, Nakatsuji N, Yamanaka S, Yamashita JK. Efficient and scalable purification of cardiomyocytes from human embryonic and induced pluripotent stem cells by VCAM1 surface expression. PLoS ONE. 2011;6(8):e23657. https://doi.org/10.1371/journal.pone.0023657.
Ban K, Bae S, Yoon Y-S. Current strategies and challenges for Purification of cardiomyocytes derived from human pluripotent stem cells. Theranostics. 2017;7(7):2067–77. https://doi.org/10.7150/thno.19427.
Rossler KJ, de Lange WJ, Mann MW, Aballo TJ, Melby JA, Zhang J, Kim G, Bayne EF, Zhu Y, Farrell ET, Kamp TJ, Ralphe JC, Ge Y. Lactate- and immunomagnetic-purified hiPSC-derived cardiomyocytes generate comparable engineered cardiac tissue constructs. JCI Insight. 2024;9(1). https://doi.org/10.1172/jci.insight.172168.
Ema M, Takahashi S, Rossant J. Deletion of the selection cassette, but not cis-acting elements, in targeted Flk1-lacZ allele reveals Flk1 expression in multipotent mesodermal progenitors. Blood. 2006;107(1):111–7. https://doi.org/10.1182/blood-2005-05-1970.
Kattman SJ, Witty AD, Gagliardi M, Dubois NC, Niapour M, Hotta A, Ellis J, Keller G. Stage-specific optimization of activin/nodal and BMP signaling promotes cardiac differentiation of mouse and human pluripotent stem cell lines. Cell Stem Cell. 2011;8(2):228–40. https://doi.org/10.1016/j.stem.2010.12.008.
Miki K, Endo K, Takahashi S, Funakoshi S, Takei I, Katayama S, Toyoda T, Kotaka M, Takaki T, Umeda M, Okubo C, Nishikawa M, Oishi A, Narita M, Miyashita I, Asano K, Hayashi K, Osafune K, Yamanaka S, Yoshida Y. Efficient detection and purification of cell populations using synthetic MicroRNA switches. Cell Stem Cell. 2015a;16(6):699–711. https://doi.org/10.1016/j.stem.2015.04.005.
Liew LC, Poh BM, An O, Ho BX, Lim CYY, Pang JKS, Beh LY, Yang HH, Soh BS. JAK2 as a surface marker for enrichment of human pluripotent stem cells-derived ventricular cardiomyocytes. Stem Cell Res Therapy. 2023;14(1):1–14. https://doi.org/10.1186/s13287-023-03610-2.
Macfarlane L-A, Murphy PR. MicroRNA: Biogenesis, function and role in Cancer. Curr Genom. 2010;11(7):537–61. https://doi.org/10.2174/138920210793175895.
O’Brien J, Hayder H, Zayed Y, Peng C. Overview of microRNA biogenesis, mechanisms of actions, and circulation. Front Endocrinol. 2018;9(AUG):1–12. https://doi.org/10.3389/fendo.2018.00402.
Li C, Nong W, Zhao S, Lin X, Xie Y, Cheung M-Y, Xiao Z, Wong AYP, Chan TF, Hui JHL, Lam H-M. Differential microRNA expression, microRNA arm switching, and microRNA:long noncoding RNA interaction in response to salinity stress in soybean. BMC Genomics. 2022;23(1):65. https://doi.org/10.1186/s12864-022-08308-y.
Miki K, Endo K, Takahashi S, Funakoshi S, Takei I, Katayama S, Toyoda T, Kotaka M, Takaki T, Umeda M, Okubo C, Nishikawa M, Oishi A, Narita M, Miyashita I, Asano K, Hayashi K, Osafune K, Yamanaka S, Yoshida Y. Efficient detection and purification of cell populations using synthetic MicroRNA switches. Cell Stem Cell. 2015b;16(6):699–711. https://doi.org/10.1016/j.stem.2015.04.005.
Bai X. Stem cell-based Disease modeling and cell therapy. Cells. 2020;9(10):12–4. https://doi.org/10.3390/cells9102193.
Cyranoski D. Japanese woman is first recipient of next-generation stem cells. Nature. 2014. https://doi.org/10.1038/nature.2014.15915.
Oikonomopoulos A, Kitani T, Wu JC. Pluripotent stem cell-derived cardiomyocytes as a platform for cell therapy applications: Progress and hurdles for clinical translation. Mol Ther. 2018;26(7):1624–34. https://doi.org/10.1016/j.ymthe.2018.02.026.
Blair NF, Barker RA. (2016). Making it personal: the prospects for autologous pluripotent stem cell-derived therapies. In Regenerative medicine (Vol. 11, Issue 5, pp. 423–425). https://doi.org/10.2217/rme-2016-0057.
Guan X, Xu W, Zhang H, Wang Q, Yu J, Zhang R, Chen Y, Xia Y, Wang J, Wang D. Transplantation of human induced pluripotent stem cell-derived cardiomyocytes improves myocardial function and reverses ventricular remodeling in infarcted rat hearts. Stem Cell Res Ther. 2020;11. https://doi.org/10.1186/s13287-020-01602-0.
Li H, Wang L, Ma T, Liu Z, Gao L. Exosomes secreted by endothelial cells derived from human induced pluripotent stem cells improve recovery from myocardial infarction in mice. Stem Cell Res Therapy. 2023;14(1):1–20. https://doi.org/10.1186/s13287-023-03462-w.
Ye L, Chang Y-H, Xiong Q, Zhang P, Zhang L, Somasundaram P, Lepley M, Swingen C, Su L, Wendel JS, Guo J, Jang A, Rosenbush D, Greder L, Dutton JR, Zhang J, Kamp TJ, Kaufman DS, Ge Y, Zhang J. Cardiac repair in a porcine model of acute myocardial infarction with human induced pluripotent stem cell-derived cardiovascular cells. Cell Stem Cell. 2014;15(6):750–61. https://doi.org/10.1016/j.stem.2014.11.009.
Funakoshi S, Miki K, Takaki T, Okubo C, Hatani T, Chonabayashi K, Nishikawa M, Takei I, Oishi A, Narita M, Hoshijima M, Kimura T, Yamanaka S, Yoshida Y. Enhanced engraftment, proliferation, and therapeutic potential in heart using optimized human iPSC-derived cardiomyocytes. Sci Rep. 2016;6(December 2015):1–14. https://doi.org/10.1038/srep19111.
Nakamura K, Neidig LE, Yang X, Weber GJ, El-Nachef D, Tsuchida H, Dupras S, Kalucki FA, Jayabalu A, Futakuchi-Tsuchida A, Nakamura DS, Marchianò S, Bertero A, Robinson MR, Cain K, Whittington D, Tian R, Reinecke H, Pabon L, Murry CE. Pharmacologic therapy for engraftment arrhythmia induced by transplantation of human cardiomyocytes. Stem Cell Rep. 2021;16(10):2473–87. https://doi.org/10.1016/j.stemcr.2021.08.005.
Marchiano S, Nakamura K, Reinecke H, Neidig L, Lai M, Kadota S, Perbellini F, Yang X, Klaiman JM, Blakely LP, Karbassi E, Fields PA, Fenix AM, Beussman KM, Jayabalu A, Kalucki FA, Futakuchi-Tsuchida A, Weber GJ, Dupras S, Murry CE. Gene editing to prevent ventricular arrhythmias associated with cardiomyocyte cell therapy. Cell Stem Cell. 2023;30(4):396–e4149. https://doi.org/10.1016/j.stem.2023.03.010.
Zhu K, Bao X, Wang Y, Lu T, Zhang L. Human induced pluripotent stem cell (hiPSC)-derived cardiomyocyte modelling of cardiovascular diseases for natural compound discovery. Biomed Pharmacotherapy. 2023;157(October 2022):113970. https://doi.org/10.1016/j.biopha.2022.113970.
Tani H, Tohyama S. Human Engineered Heart tissue models for Disease modeling and Drug Discovery. Front Cell Dev Biology. 2022;10(March):1–21. https://doi.org/10.3389/fcell.2022.855763.
Wu P, Deng G, Sai X, Guo H, Huang H, Zhu P. Maturation strategies and limitations of induced pluripotent stem cell-derived cardiomyocytes. Biosci Rep. 2021;41(6). https://doi.org/10.1042/BSR20200833.
Gill JK, Rehsia SK, Verma E, Sareen N, Dhingra S. Stem cell therapy for cardiac regeneration: past, present, and future. Can J Physiol Pharmacol. 2024;102(3):161–79. https://doi.org/10.1139/cjpp-2023-0202.
Funding
This project was financially funded by a research grant from the Tabriz University of Medical Sciences, Tabriz, Iran (Pazhoohan ID: 73124).
Author information
Authors and Affiliations
Contributions
S.P. F as well as R. F. had main contribution in design, and manuscript writing; E. F. cooperated in final editing and critical review of the manuscript; B. V. collaborated in drawing the figures.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Farboud, S.P., Fathi, E., Valipour, B. et al. Toward the latest advancements in cardiac regeneration using induced pluripotent stem cells (iPSCs) technology: approaches and challenges. J Transl Med 22, 783 (2024). https://doi.org/10.1186/s12967-024-05499-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-024-05499-8