
Abstract
The patient was a 27-year-old woman who was clinically diagnosed with tuberous sclerosis complex (TSC). She developed hypercalcemia and hypophosphatemia at age 23. In a detailed examination at age 26, she was diagnosed with primary hyperparathyroidism due to parathyroid adenoma. After undergoing parathyroidectomy, her hypercalcemia and hypophosphatemia rapidly normalized. Subsequent genetic testing revealed mutations of the TSC1 gene. TSC with concomitant parathyroid adenoma is extremely rare; only three cases have been reported worldwide. However, each of these cases was diagnosed clinically. Therefore, our case is the first to be diagnosed with genetic testing.
Similar content being viewed by others

Background
Tuberous sclerosis complex (TSC) is an autosomal dominant disorder that results from an inactivating mutation in one of two genes, TSC1 (on chromosome 9q34), which encodes hamartin, or TSC2 (on chromosome 16p13.3), which encodes tuberin.
Hamartin and tuberin form a complex that regulates cell proliferation and differentiation [1]. In TSC, hamartomas and neoplastic lesions form at various sites throughout the body. Lesions are often observed on the central nervous system, eyes, skin, kidneys, heart, or lungs; however, concomitant parathyroid adenoma is extremely rare and currently unreported in Japan. To date, only three cases have been reported in the entire world [2-4]. However, each of these cases was diagnosed clinically. Indeed, there is no reported case of TSC that was diagnosed with genetic testing. Here, we report our experience with a highly rare case of TSC with concomitant primary hyperparathyroidism due to parathyroid adenoma.
Case presentation
Clinical course
The patient was a 27-year-old woman who began having convulsive seizures at age 10. Her seizure types were consistent with the following categories: generalized tonic-clonic convulsion, absence seizure, and epileptic automatism.
She was found to have cortical tubers, subependymal nodules, retinal hamartoma, right ventricular cardiac rhabdomyoma, white leaf-shaped macules on the right thigh, and shagreen patches on the lower back. No intellectual disability was present, and she had no family history of TSC. She was clinically diagnosed with TSC. Although her epilepsy was initially intractable, the attacks eventually decreased and stopped occurring at age 17. A cyst was discovered on the right kidney at age 19, followed by multifocal micronodular pneumocyte hyperplasia and hepatic hemangioma at age 26. However, both were asymptomatic and did not show a tendency toward enlargement; therefore, the patient’s course is currently being observed while she receives only sodium valproate.
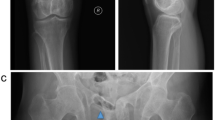
Hypercalcemia and hypophosphatemia developed and gradually progressed from age 23, according to regular annual testing (Table 1). She has shown no symptoms of hypercalcemia, such as fracture or renal stone. Bone mineral density (BMD) of the lumbar spine was normal (T-score: −0.7) and renal function was also normal (creatinine: 0.55 mg/dL). Thus, when the patient underwent detailed examination at age 26, she was diagnosed with asymptomatic primary hyperparathyroidism with increased parathyroid hormone activity (intact PTH) (Table 2). The results of tests of thyroid function, gastrin, insulin glucagon, vasoactive intestinal peptide, and prolactin were all normal. Cervical ultrasonography showed a mass shadow with a 1-cm diameter at a site corresponding to the right inferior pole of the thyroid gland. Technetium-99m methoxy-isobutyl isonitrile (Tc99m MIBI) scintigraphy demonstrated accumulation at the same site that had been identified through ultrasonography (Figures 1 and 2). She underwent minimally invasive parathyroidectomy (PTx). The resected parathyroid adenoma weighed 0.3 g, and we observed densely hyperplastic chief primary cells without fat cells. No atypia or evidence of infiltration was seen. Although a normal rim could not be observed, the sample was considered a parathyroid adenoma based on the enlargement of one gland (Figure 3). Following surgery, the hypercalcemia, hypophosphatemia, and the plasma intact PTH level rapidly normalized. These values currently remain at normal levels, 1 year after PTx (Table 3).

Cervical ultrasonography. A mass shadow with a 1-cm diameter at a site corresponding to the right inferior pole of the thyroid gland.

Technetium-99m methoxy-isobutyl isonitrile (Tc99m MIBI) scintigraphy. This image shows accumulation at the same site that had been identified through ultrasonography.

Parathyroid adenoma resection sample. The sample weighed 0.3 g. We observed densely hyperplastic chief primary cells without fat cells. No atypia or evidence of infiltration is seen. Although a normal rim cannot be seen, the sample was considered a parathyroid adenoma based on the enlargement of one gland. A (Left): ×4; B (Right): ×10. Hematoxylin and eosin staining.
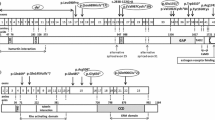
Previously, the patient had been clinically diagnosed with TSC. At this point, we decided that a genetic test should also be performed because new complications had been found, such as parathyroid adenoma. In the parathyroid adenoma, the TSC gene may have undergone loss of heterozygosity (LOH). A novel splicing mutation was revealed by direct sequencing analysis of the TSC1 gene extracted from peripheral blood. This gene abnormality occurred at the location of an important consensus sequence of the splicing site, and the probability of a gene splicing abnormality is high. Thus, we surmised that the mutation might be pathogenic (Figure 4). The results of the analysis of the TSC2 gene were normal. Genetic testing was performed by the Research Center for Bioscience and Technology at Tottori University.

Gene analysis results. A novel splicing mutation in the TSC1 gene. Mutated and normal nucleotides (A and G, respectively) were present in the patient.
Discussion
It is thought that the prevalence of TSC does not differ according to region or race [5,6]. Exact data on the prevalence of TSC in Japan are not available, but according to a local investigation, the prevalence is estimated to be at least about 1 in 6,000 persons. Thus, it is supposed that there are about 15,000 patients with TSC in Japan [6].
The incidence of primary hyperparathyroidism is considered to be 1 in 2,500 to 5,000 persons in Japan [7]. Asymptomatic cases, such as our case, are often detected because of the increasingly common use of blood tests, including those of calcium (Ca), phosphorous (P), and intact PTH.
TSC is an autosomal dominant disease that occurs because of abnormalities in the TSC1 or TSC2 tumor suppressor genes. Tumorigenesis in TSC is explained by Knudson’s two-hit mutational mechanism [8]. The first hit corresponds to a germline mutation inactivating one of the alleles of either TSC1 or TSC2, and the second hit, called LOH, is a somatic mutation inactivating the second allele [9,10]. In fact, LOH has been confirmed in the development of multiple hamartomas in a variety of organs and tissues in TSC patients [9,11-13].
TSC1 encodes hamartin and TSC2 encodes tuberin. TSC1 and TSC2 form a complex that regulates mammalian target of rapamycin (mTOR) activity in an inhibitory fashion. mTOR itself regulates cell growth and proliferation [14]. Thus, it is believed that mTOR increases if the hamartin-tuberin complex is functionally impaired, resulting in dysplasia, tumor formation, and angiogenesis due to accelerated cell proliferation, and thereby causing the systemic manifestation of various symptoms [10,15,16]. mTOR inhibitors have recently been found to be effective for subependymal giant cell astrocytoma, renal angiomyolipoma, and pulmonary lymphangioleiomyomatosis, which are severe manifestations of TSC [9,16,17].
Although cases of TSC rarely include concomitant neuroendocrine tumors of the pituitary gland, parathyroid, pancreas, or adrenal gland, such cases have been reported [3,18,19]. Because these cases are so infrequent, they tend to be viewed as coincidental. However, there are confirmed cases of TSC2 gene LOH with pancreatic neuroendocrine tumors accompanying TSC [18,19]. In addition, researchers investigating non-familial pancreatic neuroendocrine tumors have recently detected mutations of the TSC2 gene that affect the mTOR pathway in a manner that is similar to the mutations of phosphatase and tensin homolog deleted on chromosome 10 (PTEN) and phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha (PIK3CA) [20]. Note that these TSC2 gene mutations have not been detected as frequently as multiple endocrine neoplasia type 1 (MEN1) and death domain-associated protein/alpha thalassemia/mental retardation syndrome X-linked (DAXX/ATRX) gene mutations [20]. Therefore, neuroendocrine tumors are considered to be possible complications of TSC. In the present case, we attempted to investigate whether LOH had occurred in the resected parathyroid adenoma tissue; however, analysis was not possible due to the poor state of the sample. Although there are no reports of LOH of the TSC gene in parathyroid tissue, future cases are expected.
Conclusions
Our case suggests that it is possible for primary hyperparathyroidism to be complicated with TSC, even though this presentation is extremely rare.
Consent
Written informed consent was obtained from the patient for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Abbreviations
- 1-25(OH)2D:
-
1,25-dihydroxyvitamin D3
- 25(OH)D:
-
25-hydroxyvitamin D
- ATRX:
-
alpha thalassemia/mental retardation syndrome X-linked
- BMD:
-
bone mineral density
- Ca:
-
calcium
- DAXX:
-
death domain-associated protein
- LOH:
-
loss of heterozygosity
- MEN1:
-
multiple endocrine neoplasia type 1
- mTOR:
-
mammalian target of rapamycin
- P:
-
phosphorous
- PIK3CA:
-
phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic alpha
- PTEN:
-
phosphatase and tensin homolog deleted on chromosome 10
- PTH:
-
parathyroid hormone
- PTHrp:
-
parathyroid hormone-related protein-C
- PTx:
-
parathyroidectomy
- Tc99m MIBI:
-
technetium-99m methoxy-isobutyl isonitrile
- VIP:
-
vasoactive intestinal peptide
References
Curatolo P, Maria BL. Tuberous sclerosis. Handb Clin Neurol. 2013;111:323–31.
Yin W, Zhu DM, Wang DS, Wang DX, Wu EH, Zhu XY. Parathyroid adenoma with primary hyperparathyroidism and tuberous sclerosis. A case report. Chin Med J. 1984;97:599–602.
Ilgren EB, Westmoreland D. Tuberous sclerosis: unusual association in four cases. J Clin Pathol. 1984;37:272–8.
Mortensen LS, Rungby J. Tuberous sclerosis and parathyroid adenoma. J Clin Pathol. 1991;44:961–2.
National Institute of Neurological Disorders and Stroke (NINDS). National Institute of Health (NIH). Tuberous Sclerosis Fact Sheet. 2015. http://www.ninds.nih.gov/disorders/tuberous_sclerosis. Accessed 17 Feb 2015.
Japan Intractable Research Foundation. Japan Intractable Diseases Information Center. 2014. http://www.nanbyou.or.jp. Accessed 17 Feb 2015. [in Japanese]
Obara T. Diagnosis and treatment of primary-hyperparathyroidism. Nihon Naibunpi Gakkai Zasshi. 1992;68:1167–76 [in Japanese].
Knudson Jr AG. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A. 1971;68:820–3.
Sepp T, Yates JR, Green AJ. Loss of heterozygosity in tuberous sclerosis hamartomas. J Med Genet. 1996;33:962–4.
Ouyang T, Zhang N, Benjamin T, Wang L, Jiao J, Zhao Y, et al. Subependymal giant cell astrocytoma: current concepts, management, and future directions. Childs Nerv Syst. 2014;30:561–70.
Green AJ, Smith M, Yates JR. Loss of heterozygosity on chromosome 16p13.3 in hamartomas from tuberous sclerosis patients. Nat Genet. 1994;6:193–6.
Carbonara C, Longa L, Grosso E, Borrone C, Garrè MG, Brisigotti M, et al. 9q34 loss of heterozygosity in a tuberous sclerosis astrocytoma suggests a growth suppressor-like activity also for the TSC1 gene. Hum Mol Genet. 1994;3:1829–32.
Henske EP, Scheithauer BW, Short MP, Wollmann R, Nahmias J, Hornigold N, et al. Allelic loss is frequent in tuberous sclerosis kidney lesions but rare in brain lesions. Am J Hum Genet. 1996;59:400–6.
Huang J, Manning BD. The TSC1-TSC2 complex: a molecular switchboard controlling cell growth. Biochem J. 2008;412:179–90.
Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med. 2006;355:1345–56.
Curatolo P, Moavero R. mTOR Inhibitors in tuberous sclerosis complex. Curr Neuropharmacol. 2012;10:404–15.
Budde K, Gaedeke J. Tuberous sclerosis complex-associated angiomyolipomas: focus on mTOR inhibition. Am J Kidney Dis. 2012;59:276–83.
Dworakowska D, Grossman AB. Are neuroendocrine tumours a feature of tuberous sclerosis? A systematic review. Endocr Relat Cancer. 2009;16:45–58.
Díaz Díaz D, Ibarrola C, Goméz Sanz R, Pérez Hurtado B, Salazar Tabares J, Colina Ruizdelgado F. Neuroendocrine tumor of the pancreas in a patient with tuberous sclerosis: a case report and review of the literature. Int J Surg Pathol. 2012;20:390–5.
Jiao Y, Shi C, Edil BH, de Wilde RF, Klimstra DS, Maitra A, et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science. 2011;331:1199–203.
Acknowledgement
We thank the staffs of the Research Center for Bioscience and Technology at Tottori University for providing us genetic testing.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
YI was involved in the acquisition of data and the preparation of the figures. All authors have read and approved the final manuscript.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Shinzato, Y., Ikehara, Y. A case of tuberous sclerosis complex with concomitant primary hyperparathyroidism due to parathyroid adenoma: a case report. World J Surg Onc 13, 106 (2015). https://doi.org/10.1186/s12957-015-0520-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12957-015-0520-y