
Abstract
Radiation-induced brain injury (RBI) presents a significant challenge for patients undergoing radiation therapy for head, neck, and intracranial tumors. This review aims to elucidate the role of ferroptosis in RBI and its therapeutic implications. Specifically, we explore how ferroptosis can enhance the sensitivity of tumor cells to radiation while also examining strategies to mitigate radiation-induced damage to normal brain tissues. By investigating the mechanisms through which radiation increases cellular reactive oxygen species (ROS) and initiates ferroptosis, we aim to develop targeted therapeutic strategies that maximize treatment efficacy and minimize neurotoxicity. The review highlights key regulatory factors in the ferroptosis pathway, including glutathione peroxidase 4 (GPX4), cystine/glutamate antiporter system Xc- (System Xc-), nuclear factor erythroid 2-related factor 2 (NRF2), Acyl-CoA synthetase long-chain family member 4 (ACSL4), and others, and their interactions in the context of RBI. Furthermore, we discuss the clinical implications of modulating ferroptosis in radiation therapy, emphasizing the potential for selective induction of ferroptosis in tumor cells and inhibition in healthy cells. The development of advanced diagnostic tools and therapeutic strategies targeting ferroptosis offers a promising avenue for enhancing the safety and efficacy of radiation therapy, underscoring the need for further research in this burgeoning field.
Similar content being viewed by others
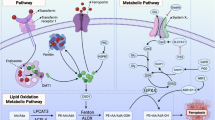


GPX4
Ferroptosis, a distinct form of regulated cell death, has emerged as a significant factor in radiation-induced brain injury [1]. Central to this process is GPX4 (glutathione peroxidase 4), a selenoprotein that acts as a primary defense against phospholipid peroxidation, a hallmark of ferroptosis [2]. GPX4 has the unique capability to degrade a spectrum of peroxides, from simple molecules to complex lipid peroxides. Its primary function is to counteract the accumulation of lethal ROS by converting cytotoxic phospholipid hydroperoxides into non-toxic phospholipid alcohols [3, 4]. Studies have highlighted that a reduction in GPX4 expression makes cells more susceptible to ferroptosis, whereas its upregulation offers resistance [5,6,7].
The active site of GPX4, enriched with selenocysteine, can be targeted by the GPX4 inhibitor RSL3 [8]. This interaction diminishes the antioxidative defenses of cells, leading to an increase in lipid ROS, and thereby initiating ferroptosis [7, 9, 10]. Notably, the synthesis of GPX4 is modulated by the mevalonate (MVA) pathway [11]. Any disruption in this pathway, especially in the maturation of selenocysteine tRNAs, can impair GPX4 functionality, promoting ferroptosis [12]. Additionally, glutathione, an essential tripeptide antioxidant, is vital for GPX4's function in neutralizing hydrogen peroxide [13,14,15]. Emerging research suggests that a decline in glutathione levels might precede GPX4 inactivation, further weakening the antioxidative defenses, escalating lipid ROS accumulation, and driving ferroptosis [16]. Given the susceptibility of the brain to radiation-induced oxidative stress, understanding the role of GPX4 in ferroptosis becomes paramount for developing potential therapeutic interventions.
System Xc-
System Xc- is a heterodimeric amino acid transporter embedded in the cell surface membrane, comprising member 2 of the solute carrier family 3 (SLC3A2) and member 11 of the solute carrier family 7 (SLC7A11) [17]. Among these, SLC7A11 stands out as the primary functional subunit, orchestrating the pivotal role of transporting cystine into the cell [18]. Once inside, cystine undergoes reduction to form cysteine in the cytoplasm, which then becomes a cornerstone in the synthesis of the antioxidant molecule, glutathione (GSH) [19,20,21,22].
The significance of System Xc- extends beyond mere cystine transport. It plays a central role in maintaining cellular redox homeostasis, ensuring that cells are equipped with adequate GSH to counteract phospholipid oxidative stress [23]. Intriguingly, when System Xc- is inhibited, cells respond with a compensatory transcriptional upregulation of SLC7A11 [10]. This adaptive response, however, comes with a caveat: it leads to a decline in intracellular GSH synthesis, which in turn impacts the biological activity of GPX4, a key enzyme in preventing lipid peroxidation and ferroptosis [24].
Radiation-induced brain injury often results from oxidative stress, where an imbalance between the production of reactive oxygen species (ROS) and the cell's ability to counteract or detoxify their harmful effects ensues [25]. Given the pivotal role of System Xc- in maintaining cellular redox homeostasis, its function becomes even more crucial in the context of radiation exposure. When cells are exposed to radiation, there is an upsurge in ROS production [26]. Without adequate GSH, which is synthesized from cysteine transported by System Xc-, cells become vulnerable to oxidative damage, leading to lipid peroxidation and, eventually, ferroptosis [27].
Furthermore, radiation can directly or indirectly modulate the expression and activity of System Xc-28. A decrease in its activity can exacerbate the oxidative stress induced by radiation, making neurons and other brain cells more susceptible to injury [29]. On the other hand, the compensatory upregulation of SLC7A11, as mentioned, might seem like a protective response, but it can be a double-edged sword. While it might enhance cystine uptake initially, the subsequent decline in GSH synthesis can leave cells defenseless against the onslaught of ROS, further promoting ferroptosis [30].
Given these insights, targeting System Xc- or its downstream pathways might offer a promising therapeutic strategy for mitigating radiation-induced brain injuries. By enhancing its activity or preventing its inhibition, we might be able to bolster the brain's defenses against radiation-induced oxidative stress and ferroptosis.
NRF2
NRF2 (nuclear factor erythroid 2-related factor 2) is a central transcriptional regulator in oxidative stress responses, playing a significant role in radiation-induced brain injury [31]. It governs a wide array of cellular processes, from antioxidant reactions to redox equilibrium, with a particular emphasis on lipid peroxidation and free iron accumulation processes [32]. Notably, many factors associated with ferroptosis, a form of cell death implicated in radiation-induced brain damage, have been identified as NRF2 target genes [33].
In the context of radiation exposure, the brain's vulnerability to oxidative stress is heightened. NRF2, sensing this oxidative milieu, becomes activated [34]. Under physiological conditions, NRF2 remains bound to its inhibitor, KEAP1, in the cytoplasm [35]. However, radiation-induced oxidative stress facilitates NRF2's dissociation from KEAP1, allowing it to translocate to the nucleus [36]. Here, it induces the expression of antioxidant genes, aiming to neutralize the surge in ROS. Yet, when ROS levels become overwhelming, leading to uncontrolled lipid peroxidation, NRF2 can shift its role, instigating ferroptosis as a primary cell death mechanism [37]. This process is particularly relevant in radiation-induced brain injuries, where the balance between protective and destructive pathways determines cellular fate.
Furthermore, NRF2's influence extends to the synthesis of various antioxidants, notably GSH, recognized as a downstream target gene of NRF2 [37]. Given the brain's susceptibility to radiation and the role of ferroptosis in mediating radiation-induced damage, NRF2 emerges as a potential therapeutic target. Modulating its activity could offer avenues for mitigating the detrimental effects of radiation on the brain, making it a focal point in contemporary neurobiological research [32, 38]. One such interaction is with the aforementioned System Xc- and GPX4. As NRF2 promotes the synthesis of GSH, a decline in System Xc- activity or GPX4 function can compromise NRF2's protective effects [32]. This interplay suggests that while NRF2 acts as a frontline defense against radiation-induced oxidative stress, its efficacy is contingent upon the proper functioning of other cellular pathways.
Additionally, recent studies have unveiled a connection between NRF2 and iron metabolism [39]. Iron accumulation, a hallmark of ferroptosis, can be modulated by NRF2-regulated genes involved in iron storage and transport [40]. This positions NRF2 as a potential modulator of iron homeostasis in the context of radiation-induced brain injury.
Moreover, the NRF2-KEAP1 pathway's pharmacological modulation has garnered attention. Compounds that can either stabilize NRF2 or inhibit KEAP1, thereby enhancing NRF2's antioxidative response, are under investigation for their potential neuroprotective effects against radiation-induced damage [41].
ACSL4
ACSL4, or acyl coenzyme A synthetase long chain member 4, is a crucial member of the long-chain acyl-CoA synthetase (ACSL) family [42]. Specializing in the metabolism of polyunsaturated fatty acids (PUFAs), ACSL4 has emerged as a key modulator of ferroptosis sensitivity [43]. Its role in determining phospholipid composition, and consequently, phospholipid peroxidation generation, has positioned ACSL4 as a potential biomarker for ferroptosis. [40, 41].
Elevated ACSL4 expression promotes the conversion of AA and other PUFAs, altering cellular phospholipid profiles and increasing cell susceptibility to ferroptosis [44]. This is particularly significant in the context of radiation-induced brain injury. In cerebral ischemia/reperfusion models, which often mirror radiation-induced damage effects, reducing ACSL4 levels has demonstrated protective effects against ischemic brain damage [45]. In contrast, its overexpression exacerbates the injury. Furthermore, ACSL4 can drive neuronal death by amplifying neuronal ferroptosis [46].
In radiation therapy, the significance of ACSL4 is accentuated. Considering radiation's potential to induce oxidative stress and lipid peroxidation, modulating ACSL4 can directly impact the severity of radiation-induced neuronal damage. As ACSL4 gains traction as a therapeutic target in oncology, its role in activating the ferroptosis mechanism presents a novel strategy for treating tumors, especially those resistant to conventional chemotherapy or adept at evading apoptosis [43]. In summary, ACSL4 not only offers a potential therapeutic avenue to induce tumor cell ferroptosis, but also serves as a pivotal determinant of ferroptosis sensitivity in radiation-induced brain injuries.
Coenzyme Q10
Coenzyme Q10, commonly referred to in its reduced form as ubiquinol, stands out as a potent lipophilic antioxidant [47]. Its role becomes particularly significant when considering the context of radiation-induced brain injuries [48]. As radiation therapy exposes the brain to oxidative stress, the presence of antioxidants like CoQ10 can play a pivotal role in mitigating the resultant damage [25]. Research has pinpointed CoQ10's ability to act as an endogenous inhibitor of ferroptosis, adeptly neutralizing the surge of free radicals produced post-radiation [49].
Furthermore, in the landscape of radiation-induced brain injuries, the significance of iron and its associated ferrous chelators cannot be understated. Agents such as deferoxamine (DFO), VK-28, deferiprone, ciclopirox, triethylenetetramine, ethylenediaminetetraacetic acid (EDTA), and chloroquine have been identified as potent inhibitors of ferroptosis [50, 51]. Their role in counteracting the detrimental effects of radiation-induced ferroptosis further underscores the importance of understanding and leveraging the protective mechanisms of compounds like CoQ10. In essence, CoQ10, in tandem with these chelators, offers a promising therapeutic strategy to combat the adverse outcomes of radiation therapy on the brain.
FSP1
FSP1, or Ferroptosis Suppressor Protein 1, emerges as a critical player in the context of radiation-induced brain injuries [52]. As a novel non-glutathione-dependent inhibitor of ferroptosis, FSP1 offers a unique mechanism of action. Specifically, through the FSP1-CoQ10-NAD (P)H pathway, it adeptly counters CoQ10-mediated ferroptosis, operating in a manner that runs parallel to the well-established GPX4 pathway [53].
In the aftermath of radiation exposure, the brain's delicate balance of oxidative and antioxidative processes can be severely disrupted [54]. Here, the role of NADPH becomes particularly salient. Recognized as a key biomarker for sensitivity to ferroptosis induction, NADPH functions as a reductase for GSH, ensuring that GSH remains in its reduced, active state [55]. Given the potential of radiation to deplete the brain's antioxidant reserves, the protective action of FSP1, in conjunction with NADPH, becomes invaluable. By bolstering the brain's defenses against ferroptosis, FSP1 offers a promising avenue for therapeutic interventions aimed at mitigating the detrimental effects of radiation on neural tissues.
P53
The tumor suppressor protein p53 has long been recognized for its canonical roles in cell cycle arrest, senescence, and apoptosis [56]. However, recent research has unveiled its profound influence on radiation-induced brain injuries, particularly through its modulation of ferroptosis [1]. Beyond its well-established functions, p53 has been found to play pivotal roles in metabolism, cell migration, invasion, stem cell processes, and notably, redox regulation [57, 58]. Its ability to suppress tumor progression by modulating ferroptosis offers a fresh perspective on its multifaceted roles in cellular processes [59, 60].
In the context of radiation-induced oxidative stress, p53 acts as a gatekeeper. It inhibits the transcription of SLC7A11, leading to a reduction in cystine uptake and GSH production, thereby amplifying the cell's vulnerability to ferroptosis [61]. This regulatory role of p53 becomes even more pronounced when considering that cells with activated p53 exhibit a compromised ability to counteract ROS [62, 63].
The brain, being a complex organ with intricate cellular interactions, is particularly susceptible to radiation-induced damage. Given its high content of PUFAs, it becomes a prime target for lipid peroxidation, a precursor to ferroptosis [64,65,66]. In this milieu, p53 emerges as a critical regulator. It can transactivate ferritin in response to ferroptosis triggers and inflammation, and by regulating p21, it can bolster the antioxidative defenses of the brain, particularly by inhibiting glutathione degradation and enhancing GPX4 activity [67].
In essence, while p53 was initially identified in the context of cancer, its profound implications in neurological diseases, especially radiation-induced brain injuries, cannot be overlooked [68]. Its ability to modulate ferroptosis, primarily by influencing the expression of its target genes, offers promising avenues for therapeutic interventions. As researchers continue to unravel the intricate mechanisms of p53 in the context of radiation-induced brain injuries, it stands as a beacon for potential diagnostic and therapeutic strategies. For a detailed understanding of the key regulatory factors in the ferroptosis-related pathway, including GPX4, System Xc-, NRF2, ACSL4, Coenzyme Q10, FSP1, and p53, refer to Fig. 1. This figure illustrates how each of these factors interacts within the ferroptosis pathway, highlighting their roles in modulating oxidative stress and cellular responses to radiation therapy.

Key regulatory factors in the ferroptosis-related pathway in radiation-induced brain injury
Radiation-induced brain injury in head and neck radiation therapy
With the increasing clinical adoption of linear accelerators and advanced radiosurgery technologies, such as CyberKnife, the incidence of radiation-induced brain injury (RBI) has seen a notable rise [69, 70]. Such injuries are primarily attributed to the direct or indirect effects of radiation on brain tissues [71]. For instance, vascular damage can lead to ischemic necrosis, and immune reactions also contribute to the injury [72]. Ionizing radiation generates reactive oxygen species in tumor cells, targeting their DNA and thereby offering therapeutic benefits against malignant tumors [73, 74]. However, this radiation can inadvertently harm the surrounding healthy cells, resulting in RBI [75]. This injury affects neurons, glial cells, and blood vessels in the brain, inducing molecular, cellular, and functional alterations [76].
Clinically, RBI presents a spectrum of consequences, ranging from cerebral edema and neuronal damage to inflammation and motor-cognitive impairments. Such complications not only hinder daily life, but also elevate the risk of mortality. Alarmingly, the medical community still grapples with the challenge of RBI, as there is no definitive treatment available. This gap in treatment is partly attributed to the absence of ideal therapeutic drugs and reliable clinical prognostic models.
In recent scientific explorations, ferroptosis, an iron-dependent regulated cell death mode, has emerged as a significant area of interest. Distinguished from traditional cell death pathways like apoptosis and autophagy, ferroptosis offers promising therapeutic avenues in cancer treatment and radioprotection [77, 78]. Building on the findings of Su et al. [79], this review discusses how ferroptosis can enhance the sensitivity of tumor cells to radiation, potentially improving outcomes in head and neck radiation therapy. By elucidating the intricate mechanisms of ferroptosis and its association with RBI, along with the regulatory factors involved, we aim to shed light on the potential applications of ferroptosis in RBI treatment. This exploration seeks to pave the way for innovative therapeutic strategies that not only mitigate the adverse effects of radiation on healthy brain tissue, but also enhance the efficacy of radiation therapy against tumors, marking a significant stride towards improving patient outcomes in head and neck cancer treatment.
Non-head and neck primary tumors and brain protection strategies
With the continuous advancement of radiation therapy techniques, the treatment strategies for non-head and neck primary tumors increasingly focus on protecting surrounding normal tissues, especially brain tissue. High-precision treatment technologies such as stereotactic radiotherapy (SRT) and proton therapy, by precisely controlling the distribution of radiation doses, effectively concentrate the treatment dose on the tumor area while minimizing damage to surrounding normal tissues [80, 81]. This is particularly important for the treatment of brain metastases, as brain tissue is highly sensitive to radiation, and inappropriate treatment can lead to severe neurotoxic effects [82, 83].
However, even with these advanced treatment technologies, whole brain radiation therapy (WBRT) inevitably causes some degree of damage to normal brain tissue when treating multiple brain metastases [84]. After WBRT, patients may experience neurotoxic effects ranging from mild cognitive decline to severe neurodegenerative changes [85]. Therefore, how to ensure treatment effectiveness while minimizing damage to normal brain tissue has become an important research direction in the field of radiation therapy.
The application of dose fractionation schemes is one effective strategy to reduce neurotoxic effects [86]. By dividing the total radiation dose into multiple small fractions for treatment, not only can the therapeutic effect on the tumor be maintained, but normal tissues are also given more time for repair and recovery, thereby reducing the risk of long-term neurotoxicity [86, 87]. Recent studies have shown that using optimized dose fractionation schemes, combined with high-precision radiation therapy technologies, can better balance the relationship between treatment effects and side effects when treating non-head and neck primary tumors and their brain metastases [88, 89].
In summary, as radiation therapy technologies continue to develop and optimize, treatment strategies for non-head and neck primary tumors and their brain metastases are increasingly able to ensure treatment effectiveness while maximizing the protection of normal brain tissue and reducing neurotoxic effects. Future research needs to further explore and verify more protective strategies to improve patients' quality of life and treatment outcomes.
Radiation therapy techniques, treatment volumes, and dose fractionation in reducing brain exposure
The selection of radiation therapy techniques, the definition of treatment volumes, and the formulation of dose fractionation schemes play a crucial role in determining therapeutic outcomes and minimizing adverse effects, particularly in reducing radiation-induced brain injury. This section aims to elucidate the relationship between these factors and the primary tumor being treated with radiation therapy, focusing on the resulting dose to the brain and strategies to mitigate potential damage.
Radiation therapy techniques
Advancements in radiation therapy techniques, such as intensity-modulated radiation therapy (IMRT), stereotactic radiosurgery (SRS), and proton beam therapy, have significantly improved the precision of tumor targeting [90, 91]. These techniques allow for the delivery of high doses to the tumor while sparing surrounding normal tissues, including the brain [92, 93]. The choice of technique depends on the tumor's location, size, and proximity to critical structures.
Treatment volumes
The definition of treatment volumes is critical to ensure adequate tumor coverage and minimize exposure to healthy tissues. The use of advanced imaging modalities in treatment planning enables the delineation of target volumes with greater accuracy. For tumors located near the brain or with potential brain metastases, careful consideration of treatment volumes is essential to avoid unnecessary radiation to the brain [94,95,96].
Dose fractionation
Dose fractionation refers to the division of the total radiation dose into multiple smaller doses delivered over several sessions. This approach allows normal tissues more time to repair, reducing the risk of long-term side effects. The fractionation scheme is tailored based on the tumor type, location, and radiosensitivity, as well as the tolerance of surrounding normal tissues [97,98,99].
Relationship between treatment parameters and brain dose
The cumulative dose to the brain during radiation therapy is influenced by the chosen technique, treatment volume, and fractionation scheme. For non-head and neck primaries, minimizing brain exposure is a priority. Techniques such as IMRT and SRS, combined with meticulous planning and dose optimization, can significantly reduce the dose to the brain. Additionally, the use of protective measures, such as sparing organs at risk and employing neuroprotective agents, may further mitigate the risk of radiation-induced brain injury [100,101,102].
In conclusion, the integration of advanced radiation therapy techniques, precise treatment planning, and individualized dose fractionation schemes is paramount in minimizing brain exposure and protecting brain health while achieving optimal therapeutic outcomes. Ongoing research and technological advancements will continue to refine these strategies, enhancing the safety and efficacy of radiation therapy.
Discovery and characteristics of ferroptosis: exploring the potential of ferroptosis in cancer treatment
Ferroptosis is a unique regulated cell death mechanism, distinct from traditional cell death pathways such as apoptosis, necrosis, and autophagy [103]. This mechanism was initially identified during high-throughput screenings for potential anti-cancer drugs, which demonstrated the ability to target cancer cell lines through RAS transformation [10]. The defining features of ferroptosis include significant accumulation of lipid peroxides and reactive oxygen species (ROS) [104]. This groundbreaking discovery is credited to the Stockwell Laboratory at Columbia University in 2012, marking a significant advancement in the field [10]. Ferroptosis has been implicated in a variety of pathological conditions, including tumors, neurodegenerative diseases (e.g., Alzheimer's/Parkinson's disease), and ischemia–reperfusion injuries [105, 106].
Morphologically, cells undergoing ferroptosis exhibit characteristics such as cell membrane rupture, mitochondrial shrinkage, and increased cell membrane density [107]. From a biological perspective, ferroptosis is characterized by elevated ROS levels, iron accumulation, mitogen-activated protein kinase (MAPK) activation, decreased cysteine uptake, and glutathione (GSH) depletion [108, 109]. Immunologically, ferroptosis leads to the release of damage-associated molecular patterns, initiating inflammatory responses [110]. The intricate mechanisms of ferroptosis have become a research hotspot, especially in tumor studies focusing on ROS homeostasis.
The research by Su et al. further deepens our understanding of the role of ferroptosis in suppressing tumor progression [79]. Their work reveals the mechanisms by which ferroptosis suppresses tumor progression through increased ROS generation, causing iron overload, disrupting the antioxidant system, and promoting lipid peroxidation. These findings not only provide new strategies for the application of ferroptosis in cancer treatment but also offer new insights into improving the efficacy of radiotherapy.
By modulating the ferroptosis pathway, it might be possible to enhance tumor treatment effects while protecting normal tissues. These findings not only deepen our understanding of the role of ferroptosis in radiotherapy, but also provide a scientific basis for developing new therapeutic strategies. The work of Su et al. [79] emphasizes the importance of considering the ferroptosis pathway in designing radiotherapy plans, which is crucial for improving treatment outcomes and reducing therapy-related damage.
Ferroptosis mechanism in radiation-induced brain injury: dual role in enhancing tumor sensitivity and inducing damage in normal tissues.
Radiation-induced brain injury (RBI) is a multifaceted process, with recent studies, including those by Su et al [79], underscoring the critical role of ferroptosis in its development. Ferroptosis, an iron-dependent form of cell death, is triggered by the accumulation of intracellular lipid peroxides. Its regulation involves a complex interplay among iron metabolism, the cystine/glutamate antiporter (system Xc-)/GSH/GPX4 antioxidant pathway, and lipid peroxidation processes [111]. These pathways collectively modulate the cellular vulnerability to ferroptosis. The central nervous system, with its natural propensity for iron accumulation and high levels of polyunsaturated fatty acids and amino acids, is particularly prone to ferroptosis following radiation exposure [112, 113]. Key regulatory mechanisms include iron metabolism, glutathione peroxidase 4 (GPX4) enzyme activity, and lipid metabolism dynamics. Importantly, the ferroptosis suppressor protein 1 (FSP1)-coenzyme Q10 (CoQ10)-nicotinamide adenine dinucleotide (Phosphate) [NAD(P)H] and tetrahydrobiopterin (BH4)-dihydrofolate reductase (DHFR) pathways, though functioning independently, complement each other and, alongside GPX4 and glutathione, play crucial roles in preventing phospholipid peroxidation and, consequently, ferroptosis [114,115,116].
Integrating insights from Su et al [79], this section explores the dual role of ferroptosis in both enhancing the sensitivity of tumor cells to radiation and contributing to radiation-induced damage in normal tissues. The potential to modulate the ferroptosis pathway offers promising therapeutic avenues to maximize treatment benefits. By understanding these mechanisms, there is potential to develop strategies that not only amplify the therapeutic effects of radiation on tumor cells but also mitigate the adverse effects on normal brain tissues, thereby improving the overall outcomes of radiotherapy.
To further elucidate the complex role of ferroptosis in radiation-induced brain injury (RBI), we have summarized the key regulatory mechanisms of ferroptosis and its dual impact on tumor cells and normal brain tissues in the following figure (Fig. 2: the role of ferroptosis in radiation-induced brain injury). As shown in Fig. 2, ferroptosis affects the outcomes of radiation therapy through various biochemical pathways. These include iron metabolism, the System Xc-/GSH/GPX4 antioxidant pathway, and lipid peroxidation processes. Iron metabolism contributes to the accumulation of lipid peroxides, a key step in ferroptosis. The System Xc-/GSH/GPX4 pathway plays a critical role in protecting cells from oxidative damage by reducing lipid peroxides. However, when this pathway is inhibited, it leads to increased susceptibility to ferroptosis. By modulating these mechanisms, we can develop therapeutic strategies that enhance the sensitivity of tumor cells to radiation while mitigating radiation-induced damage to normal brain tissues, thereby improving the overall outcomes of radiotherapy.

The role of ferroptosis in radiation-induced brain injury
Ferroptosis and patient data in radiation-induced brain injury
In recent years, the role of ferroptosis in radiation-induced brain injury (RBI) has garnered widespread attention. Clinical studies and patient data have revealed the critical role of ferroptosis in the development of RBI, especially following radiation therapy in patients with head and neck tumors [117]. Research indicates that radiation not only increases the accumulation of iron in the brain, but also promotes the production of lipid peroxidation, both of which are major drivers of ferroptosis [118].
In patients undergoing radiation therapy, markers associated with ferroptosis, such as increased lipid peroxidation products and iron content, have been significantly correlated with cognitive decline and neurodegenerative changes [11, 119, 120]. These findings underscore the potential role of ferroptosis in long-term brain damage induced by radiation, providing important clues for the development of new therapeutic strategies.
Moreover, interventions targeting the ferroptosis pathway have shown potential in animal models to alleviate RBI [117, 118]. The use of ferroptosis inhibitors, such as fatty acid inhibitors and iron chelators, has been able to reduce brain damage and improve cognitive functions [121, 122]. These research outcomes support the role of ferroptosis in RBI and offer promising targets for future clinical interventions.
However, translating these findings into clinical applications remains challenging. Understanding the specific role of ferroptosis in different patient populations and how to precisely modulate this process to minimize damage to healthy brain tissue is key to future research. Additionally, developing non-invasive biomarkers to monitor ferroptosis and assess treatment efficacy will be crucial for improving the therapeutic management of patients with RBI.
By delving deeper into the connection between ferroptosis and RBI, as well as how this process affects clinical outcomes for patients, we can take significant steps toward developing more effective treatment strategies to mitigate the side effects of radiation therapy and improve patients' quality of life.
Iron metabolism and its role in ferroptosis
Iron metabolism plays a pivotal role in various physiological processes, and any aberration in its regulation can have profound implications, including the onset of ferroptosis [123]. Iron, a redox-active metal, is crucial for generating free radicals and propagating lipid peroxidation [124]. It is sourced either from intestinal absorption or the degradation of red blood cells [125]. Once internalized, iron undergoes oxidation and assimilation into cells, facilitated by a myriad of essential proteins and enzymes [126]. Disruptions in iron's intake, transport, storage, or utilization can activate the Fenton reaction, leading to a surge of ROS and hydroxyl radicals [127]. These radicals target the polyunsaturated phospholipids in the cell membrane, initiating peroxidation reactions that culminate in membrane disruption and cell death [128]. Consequently, elements of iron metabolism are viewed as potential inducers of ferroptosis.
In the context of radiation-induced brain injury, when GPX4 is compromised, iron catalyzes the creation of hydroxyl radicals, amplifying lipid peroxidation [129]. The brain tissue, with its high oxygen consumption, becomes especially susceptible to oxidative stress and radical-induced damage. While radiotherapy directly damages DNA, it also indirectly inflicts cellular harm by producing a plethora of ROS [130, 131]. These ROS, in the presence of brain iron, generate additional oxidants, such as malondialdehyde (MDA) [132]. Radiation further impairs the body's radical-clearing capability, evident from the reduced SOD activity and elevated MDA levels [133].
Given the intricate relationship between iron metabolism and ferroptosis, especially in the backdrop of radiation-induced brain injuries, it becomes imperative to delve deeper into this domain. Current ferroptosis-targeted treatments are primarily in the animal testing phase, awaiting clinical validation. Harnessing the potential of iron metabolism and ferroptosis could pave the way for innovative therapeutic strategies, offering a promising avenue for research and clinical applications.
Lipid metabolism and ferroptosis
Lipid metabolism, a cornerstone of cellular processes, plays a pivotal role in determining cell fate, especially in the context of ferroptosis [134]. Central to this is the role of polyunsaturated fatty acids (PUFAs), which are instrumental in phospholipid peroxide accumulation [135]. Due to their inherent susceptibility to phospholipid peroxidation, PUFAs' cellular content and localization largely determine the extent of phospholipid peroxidation, making them indispensable for ferroptosis [136].
Under normal metabolic conditions, reactive oxygen species (ROS) maintain cellular homeostasis and signaling [137]. However, in pathological scenarios, excessive ROS accumulation can precipitate cell death [138]. Current research underscores lipid peroxides as critical intermediates in various diseases, including inflammation, cancer, and neurodegenerative disorders [139]. The initiation of phospholipid peroxidation by OH- results in lipid radicals, which subsequently interact with PUFAs, generating phospholipid peroxides that drive ferroptosis [49]. This intricate relationship suggests that ferroptosis might be a direct consequence of lipid peroxidation.
Further insights reveal that phosphatidylethanolamines (PE) rich in arachidonic acid (AA) or its derivatives are the primary lipids causing cellular ferritin denaturation. Enzymes like Acyl-CoA synthetase long-chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3) are instrumental in PE biosynthesis, activating PUFAs and modulating their transmembrane dynamics [140]. By inhibiting ACSL4 and LPCAT3 expression, phospholipid peroxide accumulation can be curtailed, thereby mitigating ferroptosis [141]. Moreover, radiation-induced phospholipid peroxidation can also trigger ferroptosis, closely linked to DNA damage [142]. This interplay suggests that DNA damage, stemming from phospholipid peroxidation-induced iron deposition, might be a primary instigator. Given the profound implications of lipid metabolism in radiation-induced brain injuries, it becomes imperative to delve deeper into this domain, offering a promising avenue for research and therapeutic interventions [9].
System Xc-/GPX4 pathway and amino acid metabolism
The System Xc-/GPX4 pathway, pivotal in amino acid metabolism, plays a crucial role in determining cell fate, especially in the context of ferroptosis [111]. Central to this is the cystine/glutamate antiporter, known as System Xc-17. Comprising the heavy chain SLC3A2 and the light chain SLC7A11, its primary function is to facilitate the import of extracellular cystine into cells while exporting intracellular glutamate [59]. Once internalized, cystine is reduced to cysteine, a vital substrate for GSH synthesis [143]. A decline in SLC7A11 expression can result in diminished GSH synthesis, thereby amplifying lipid peroxidation [10].
Glutathione peroxidase 4 (GPX4) is a linchpin in counteracting ferroptosis due to its anti-lipid peroxidation activity [144]. While GPXs are integral in maintaining the oxidation–antioxidation equilibrium, GPX4 is especially critical in preventing ferroptosis [145]. Its unique ability to reduce lipid hydroperoxides is reliant on GSH [111]. A suppression in System Xc-'s activity affects cellular cystine uptake, leading to a drop in GSH synthesis, impacting GPX4's function, and accelerating ferroptosis [5]. Notably, both erastin and sorafenib induce ferroptosis by inhibiting System Xc-'s activity [146].
Dixon and his team's pioneering work established a link between ferroptosis and acute brain injury. Their experiments revealed that high glutamate concentrations were cytotoxic to rat hippocampal slices, but this toxicity was alleviated with ferrostatin-1 or liproxstatin [147]. Further studies indicated that brain white matter damage is associated with elevated lipid peroxidation, consistent with ferroptosis mechanisms [148].
The tumor-suppressor gene, p53, plays a pivotal role in this context. It can curtail the transcription of SLC7A11, influencing System Xc-'s activity [65]. Moreover, p53 can indirectly elevate intracellular GSH levels by regulating p21, boosting GPX4 synthesis, and subsequently inhibiting ferroptosis [149]. In radiation therapy, while SLC7A11 expression is suppressed, ACSL4 is enhanced, potentially exacerbating lipid peroxidation damage [150]. Given these findings, the exploration of ferroptosis inhibitors in radiation-induced brain injuries becomes paramount, offering a promising avenue for research and therapeutic interventions.
Iron metabolism and radiation-induced brain injury
Iron metabolism plays a pivotal role in various physiological processes, and its dysregulation can have profound implications, especially in the context of radiation-induced brain injuries [151]. In 2015, a groundbreaking study using a glioma mouse model shed light on the role of iron ions in enhancing the efficacy of radiation therapy [152]. The study revealed that iron not only stimulates glioma growth but also, when combined with iron chelators, can effectively counteract this tumor growth [153]. This discovery paved the way for subsequent research, which further emphasized the potential of ferroptosis as a promising approach in cancer therapy. The mechanism involves the acceleration of the Fenton reaction, leading to an increase in ROS production, ultimately inducing cancer cell death [154]. Building on this, Gao et al. (2019) [155] demonstrated the potential of ibuprofen in triggering ferroptosis in glioblastoma cells by inhibiting the NRF2 signaling pathway, presenting a novel therapeutic strategy against GBM. Furthermore, ACSL4 has been identified as a key player in inhibiting glioma cell proliferation by activating apoptosis [43].
The implications of dysregulated iron metabolism extend beyond cancer. It has been associated with various neurological conditions, including post-stroke hemorrhage and pulmonary thromboembolism [156, 157]. Within the central nervous system, the degradation of hemoglobin leads to the deposition of heme iron, a primary form of iron storage in humans [158]. This accumulation is closely linked to a plethora of neurological disorders, emphasizing the disruption of cellular iron homeostasis in the CNS [159]. Given the profound impact of iron-mediated cell death mechanisms on neurological health, it becomes imperative to explore therapeutic interventions targeting these pathways.
Ferroptosis and radiation-induced neuroinflammation
The intricate interplay between ferroptosis and radiation-induced neuroinflammation has garnered significant attention, especially in the context of secondary brain injuries [160]. Such injuries, characteristic of various neurological disorders like cerebral hemorrhage, are predominantly driven by oxidative stress, inflammatory cascades, and cellular death [161]. Central to this is the excessive accumulation of ROS, which instigates lipid peroxidation, leading to extensive cellular and tissue damage [162, 163]. A seminal study in 2017 delved into the therapeutic potential of ferroptosis inhibitors, including Fer-1, deferoxamine, and vitamin E analogs [164], in mitigating cell toxicity induced by cerebral hemorrhage. The outcomes were promising, with these inhibitors significantly attenuating cell toxicity. Moreover, in murine models of collagenase-induced cerebral hemorrhage, Fer-1 exhibited profound neuroprotective effects, enhancing neurological function [165].
Further research endeavors have elucidated the complex relationship between ferroptosis and secondary brain damage post-cerebral hemorrhage [166]. In specific rat models, the onset of ferroptosis post-hemorrhage was observed to instigate a potent inflammatory response [167]. Intriguingly, Fer-1 intervention not only substantially reduced inflammatory markers like ROS, IL-1β, and TNFα, but also fostered significant neurological recovery in ischemic rats [168].
The pivotal role of GPX4, a central regulator of ferroptosis, has been highlighted in various animal studies [169]. Prior to GPX4 ablation, mice manifested pronounced cognitive deficits and neuronal damage, especially in the hippocampus [170]. Detailed hippocampal analysis unveiled a spike in lipid peroxidation, accompanied by escalated neuroinflammation. These revelations underscore the critical role of ferroptosis in hippocampal degeneration, emphasizing the need for further research in this domain [171].
Ferroptosis and radiation-induced vascular damage
The intricate relationship between radiation-induced brain injuries and ferroptosis is increasingly recognized, especially in the context of vascular damage [172]. Such injuries predominantly arise from vascular damage, manifesting within a day post-radiation and often progressing to severe tissue necrosis due to oxygen deprivation [173]. This vascular compromise triggers cellular swelling, necrosis, and a consequent surge in ROS production [174]. The ensuing inflammatory cascade, marked by cytokine and chemokine release, sets the stage for platelet thrombosis, fibrinoid necrosis, blood–brain barrier breaches, and the onset of cerebral edema [175].
The aftermath of radiation, particularly the intracerebral hemorrhage resulting from vascular rupture and blood infiltration into brain tissue, is deeply intertwined with ferroptosis [176]. The iron released from hemoglobin intensifies ROS production, aggravating neuronal damage [165]. Notably, Ferrostatin-1 and Liproxstatin-1 have exhibited potential in curbing cell death in the hippocampal region of brain slices exposed to hemoglobin or free iron [165]. Hemoglobin-associated iron depletes GSH reserves, compromising GPX4 functionality in hippocampal cells. In a collagenase-induced vascular injury model, the application of iron porphyrin inhibitors either directly at the injury site or distally significantly reduced cellular and overall damage, improving neurological outcomes [165]. Moreover, augmenting intracellular cysteine levels can boost GSH synthesis [177]. The cell-permeable cysteine derivative, N-acetyl-cysteine (NAC), has shown promise in safeguarding brain tissue from hemorrhage-induced cell death, underscoring the idea that post-hemorrhage ferroptosis is driven by GSH synthesis deficits [56]. The mounting evidence solidifies the profound link between radiation-induced brain injury pathogenesis and ferroptosis, emphasizing the need for further exploration in this domain.
Ferroptosis and radiation-induced glial cell damage
The intricate relationship between radiation therapy and glial cell damage in the brain is increasingly recognized, especially in regions like the hippocampus and temporal lobe [178]. Radiation therapy directly impairs oligodendrocytes, leading to their abnormal proliferation and subsequent peripheral or central demyelination [179]. Clinically, such damage manifests as acute encephalomyelitis with accompanying neurological symptoms [180]. Microglial cells, although constituting a minor population in the central nervous system, play a pivotal role in mediating immune responses [181]. Radiation therapy can disrupt the normal functioning of these cells, causing them to release an excess of inflammatory cytokines, thereby initiating an inflammatory cascade that results in neuronal tissue damage [182].
Furthermore, hemoglobin and its by-products have been identified as significant contributors to secondary brain injuries [183]. Iron ions, derived from hemoglobin degradation, accumulate in the hematoma and adjacent brain tissues post-injury, leading to iron overload [184, 185]. Elevated concentrations of these ions can detrimentally affect neighboring glial cells and neurons by promoting lipid peroxidation and free radical generation, causing oxidative damage [186, 187]. Laboratory studies support this notion: in vitro cultured microglial cells, when exposed to FeCl2, underwent activated morphological changes [188]. A significant increase in OX6-positive stained cells was observed, highlighting the direct role of iron ions in microglial activation [189]. This growing body of evidence underscores the profound connection between radiation therapy, ferroptosis, and glial cell damage, emphasizing the need for further exploration in this domain [190].
Ferroptosis, free radical damage, and radiation-induced brain injury
The nexus between autoimmunity and radiation-induced brain injury is a burgeoning field of research [191]. A pronounced increase in the expression of vascular endothelial growth factor (VEGF) has been identified as a linchpin in radiation-induced brain injuries, a perspective fortified by empirical evidence [192]. This autoimmune cascade culminates in the release of pro-inflammatory cytokines, which are also pivotal to the pathophysiological mechanisms observed in various neurodegenerative ailments [193]. The precise orchestration of this response, however, remains shrouded in mystery.
Free radicals and their impact: Under physiological conditions, free radicals are maintained at a minimal threshold, playing instrumental roles in immune responses and signal transduction [138]. However, when cells are exposed to ionizing radiation, it catalyzes a reaction with intracellular water molecules, leading to an overproduction of reactive oxygen species (ROS) [194]. This radiation exposure also compromises mitochondrial integrity, further amplifying the generation of free radicals, including both ROS and reactive nitrogen species (RNS) [195]. These radicals, in their quest for electron stability, can inflict significant cellular damage [196].
Iron and its dual role: Iron, pivotal for myriad biochemical processes, can also manifest its toxic side [197]. Specifically, ferrous iron, when reacting with hydrogen peroxide, yields the highly reactive hydroxyl radical via the Fenton reaction [198]. An imbalance in iron homeostasis can catalyze an overproduction of free radicals, setting off a cascade of pathological events [199]. The body's robust antioxidant defense mechanism, however, can counteract this [200]. Yet, the central nervous system, with its relatively diminished glutathione reserves, is particularly susceptible to oxidative challenges [201, 202], potentially leading to ferroptosis.
Antioxidant therapies: Groundbreaking research underscores the detrimental role of ROS overproduction in the nervous system post-radiation, marking it as a pivotal contributor to radiation-induced brain injury [203]. Consequently, antioxidant therapies emerge as a beacon of hope for mitigating post-radiation cognitive deficits [204]. In addressing radiation-induced brain injuries, a cornerstone approach is the neutralization or curtailment of oxygen free radicals, safeguarding neural structures, a principle echoed across numerous scientific investigations [173].
Clinical prognostic model for radiation brain injury
Understanding the intricate relationship between ferroptosis and radiation-induced brain injury provides a promising avenue for the development of clinical prognostic models. Such models, grounded in the molecular mechanisms of ferroptosis, can offer invaluable insights into patient outcomes post-radiation therapy.
Recent advancements in genomics and transcriptomics have enabled the identification of specific ferroptosis-related gene signatures that can serve as potential prognostic markers. For instance, the expression levels of genes like SLC7A11, GPX4, and ACSL4, which are central to the ferroptosis pathway, can be correlated with the severity of radiation-induced brain damage and the likelihood of recovery [205].
Furthermore, integrating these molecular markers with clinical data, such as the extent of brain injury, patient age, and previous medical history, can refine the accuracy of these prognostic models. Such comprehensive models can predict the risk of severe complications post-radiation, enabling clinicians to tailor therapeutic interventions more effectively.
Moreover, the therapeutic targeting of ferroptosis pathways, especially SLC7A11, offers a dual advantage. While it can potentiate the eradication of tumor cells, it also holds the promise of safeguarding healthy brain tissues from radiation-induced damage. This selective targeting can significantly reduce the collateral damage often seen in radiation therapies, thereby improving the quality of life for patients post-treatment [206].
In conclusion, as research continues to unravel the complexities of ferroptosis in the context of radiation-induced brain injury, the integration of these findings into clinical prognostic models will undoubtedly revolutionize the management and treatment of patients undergoing radiation therapy. The future holds immense promise for the development of more precise, personalized, and effective therapeutic strategies, all anchored in our understanding of ferroptosis and its role in radiation brain injury.
Clinical implications of promoting and inhibiting ferroptosis in radiation brain injury
The dual nature of ferroptosis in the clinical setting underscores its potential as a novel therapeutic strategy in the treatment of radiation brain injury (RBI). On one hand, the ability to selectively induce ferroptosis in tumor cells, particularly those resistant to conventional treatments, presents a promising avenue to enhance the efficacy of radiation therapy [207, 208]. This approach, as highlighted by the findings of Su et al [79], could ensure maximal tumor cell death and potentially overcome the challenge of radiation resistance observed in certain tumors.
Conversely, the importance of inhibiting ferroptosis in surrounding healthy cells cannot be overstated. Such inhibition offers neuroprotection, especially crucial when tumors are located in or near vital brain regions [113, 209]. It also serves to shield the brain from the long-term neurodegenerative outcomes associated with chronic activation of ferroptosis [32, 210]. The strategic administration of ferroptosis inhibitors alongside radiation therapy could act as a safeguard for healthy brain tissue against the collateral damage of radiation [211, 212].
However, the clinical application of ferroptosis modulation faces significant challenges, particularly in achieving specificity of action. A critical question remains: how can we ensure that ferroptosis is promoted exclusively in tumor cells while being inhibited in healthy ones? The answer may lie in the development of advanced drug delivery systems, such as nanoparticles, which offer the promise of targeted drug delivery to tumor sites.
Furthermore, while the immediate benefits of modulating ferroptosis are clear, the long-term effects of such modulation, both beneficial and detrimental, require further exploration. The findings of Su et al [79] contribute to a deeper understanding of ferroptosis and its implications in RBI, paving the way for the development of new therapeutic strategies that leverage the regulation of ferroptosis for improved patient outcomes.
In summary, the exploration of ferroptosis in the context of radiation therapy, inspired by the work of Su et al., opens up new therapeutic possibilities. By promoting ferroptosis in tumor cells and inhibiting it in normal tissues, we can potentially protect against RBI while enhancing the treatment efficacy against tumors. Continued research and a nuanced understanding of ferroptosis mechanisms are essential for realizing the full potential of this approach in clinical practice, offering hope for improved outcomes and brighter prognoses for patients undergoing radiation therapy.
As shown in Fig. 3, ferroptosis affects the outcomes of radiation therapy through several key biochemical pathways, including iron metabolism, the System Xc-/GSH/GPX4 antioxidant pathway, and lipid peroxidation processes. This figure provides a detailed analysis of these pathways and highlights potential therapeutic targets such as GPX4, System Xc-, NRF2, ACSL4, Coenzyme Q10, and FSP1. Understanding these mechanisms allows for the development of therapeutic strategies that enhance tumor sensitivity to radiation while protecting healthy brain tissue from radiation-induced damage.

Detailed analysis of ferroptosis in radiation therapy
Summary and outlook: expanding the impact of ferroptosis in radiation therapy
The exploration of the relationship between iron metabolism, ferroptosis, and radiation-induced brain injury has become a pivotal area of interest in neuro-oncological research. The accumulation of iron in the central nervous system, a common feature in numerous neurological disorders, has highlighted the critical need to understand the mechanisms of cellular iron homeostasis within the CNS [213]. Ferroptosis, characterized by its dual nature, introduces both challenges and opportunities in radiation therapy, presenting novel therapeutic strategies for managing brain tumors and extending to other cancer types, including head and neck primaries.
Recent advancements have illuminated the potential of targeting ferroptosis to alleviate radiation-induced brain injury. The application of ferroptosis inhibitors, such as liproxstatin-1, in animal models has demonstrated promising results in reducing brain damage following radiation therapy [214, 215]. These findings open a new therapeutic pathway that could improve the safety and effectiveness of radiation treatments across a spectrum of cancers. Specifically, for patients with head and neck cancers—who frequently undergo radiation therapy as a part of their treatment regimen—the risk of inadvertent brain exposure to radiation and subsequent ferroptosis-induced brain injury is a significant concern. A deeper understanding of ferroptosis mechanisms in these contexts is essential for devising protective strategies that safeguard the brain while effectively targeting the primary tumor.
While ferroptosis inhibitors have shown promise in alleviating radiation-induced brain injury, it is crucial to address the potential conflict with radiotherapy's mechanism of inducing ferroptosis in tumor tissues. This dual nature of ferroptosis presents a unique challenge and opportunity in cancer treatment. One potential strategy to overcome this barrier is the temporal separation of treatments. Ferroptosis inhibitors could be administered in a specific time window after radiotherapy to protect normal brain tissues while allowing sufficient time for radiotherapy to exert its effects on tumor cells. Another approach could involve the development of targeted delivery systems that direct ferroptosis inhibitors specifically to normal tissues, minimizing their impact on tumor cells.
Ferroptosis represents a vulnerability in cancer due to the unique metabolic and oxidative stress conditions within the tumor microenvironment. Tumor cells often exhibit altered iron metabolism and increased reactive oxygen species (ROS) levels, making them more susceptible to ferroptosis. By selectively inducing ferroptosis in tumor cells while protecting normal tissues, it is possible to exploit this vulnerability for therapeutic benefit. This approach requires a nuanced understanding of the differential regulation of ferroptosis in tumor versus normal cells, and the development of precise therapeutic regimens that maximize efficacy while minimizing collateral damage.
Additionally, fractionation of radiotherapy, which involves dividing the total radiation dose into multiple smaller doses administered over several sessions, has been shown to improve therapeutic outcomes and reduce neurotoxicity. Fractionated radiotherapy allows healthy tissues to recover between sessions, thereby minimizing neurotoxicity and other radiation-induced damages. For patients with primary head and neck cancers, fractionation may reduce the risk of inadvertent brain exposure and subsequent ferroptosis-induced brain injury, offering a promising approach to enhancing treatment efficacy while preserving neurological function.
However, the path to clinical application is fraught with challenges. The diversity of tumor types and the complex nature of brain tissue responses to radiation underscore the limitations of a universal treatment approach. Personalized treatment strategies, informed by the genetic and metabolic profiles of both the tumor and the surrounding tissues, are likely required to fully exploit the benefits of ferroptosis modulation in radiation therapy.
Moreover, the development of non-invasive diagnostic tools for the early detection of ferroptosis in brain tissue could markedly enhance patient prognosis following radiation therapy. The integration of advanced imaging techniques with biomarker analysis promises to reveal new insights into ferroptosis dynamics in vivo, facilitating timely interventions to prevent or reduce brain injury.
As we look forward, embracing a multidisciplinary approach that combines knowledge from neurobiology, oncology, pharmacology, and radiology will be vital for deepening our understanding of ferroptosis and its implications for radiation-induced brain injury. Through collaborative research efforts, we can hasten the development of innovative therapeutics and diagnostic technologies, ultimately elevating patient outcomes and quality of life.
In conclusion, the journey to comprehend and utilize ferroptosis in the context of radiation-induced brain injury is only beginning, yet it holds immense promise. By continuing to investigate this intriguing field, our ultimate aim is to enhance the safety and efficacy of radiation therapy, thereby making significant strides in the battle against brain tumors, head and neck cancers, and other neurological conditions. This endeavor not only aligns with the scientific pursuit of knowledge, but also directly addresses the concerns raised by the reviewers, ensuring that the discussion is both comprehensive and relevant to the broader context of cancer treatment.
Availability of data and materials
No datasets were generated or analyzed during the current study.
Abbreviations
- RBI:
-
Radiation-induced brain injury
- ROS:
-
Reactive oxygen species
- FWBI:
-
Fractionated whole brain irradiation
- AD:
-
Alzheimer's disease
- PD:
-
Parkinson's disease
- MAPK:
-
Mitogen-activated protein kinase
- GSH:
-
Glutathione
- GPX4:
-
Glutathione peroxidase
- FSP1:
-
Ferroptosis suppressor protein 1
- CoQ 10:
-
Coenzyme Q10
- NAD (P)H:
-
Nicotinamide adenine dinucleotide (Phosphate)
- BH4:
-
Tetrahydrobiopterin
- DHFR:
-
Dihydrofolate reductase
- SOD:
-
Superoxide dismutase
- MDA:
-
Malondialdehyde
- PUFAs:
-
Polyunsaturated fatty acids
- PE:
-
Phosphatidylethanolamines
- AA:
-
Arachidonic acid
- ACSL4:
-
Acyl-CoA synthetase long-chain family member 4
- LPCAT3:
-
Lysophosphatidylcholine acyltransferase 3
- System Xc-:
-
Cystine/glutamate antiporter
- SLC3A2:
-
Solute carrier family 3 member 2
- SLC7A11:
-
Solute carrier family 7 member 11
- NRF2:
-
Nuclear factor erythroid 2-related factor
- GBM:
-
Glioblastoma multiforme
- CNS:
-
Central nervous system
- IL-1β:
-
Interleukin-1 beta
- TNFα:
-
Tumor necrosis factor alpha
- NAC:
-
N-Acetyl-cysteine
- FeCl2:
-
Iron(II) chloride
- VEGF:
-
Vascular endothelial growth factor
- RNS:
-
Reactive nitrogen species
- MVA:
-
Mevalonate
- KEAP1:
-
Kelch-like ECH-associated protein 1
- DFO:
-
Deferoxamine
- EDTA:
-
Ethylenediaminetetraacetic acid
References
Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. 2021;22:266–82. https://doi.org/10.1038/s41580-020-00324-8.
Weaver K, Skouta R. The selenoprotein glutathione peroxidase 4: from molecular mechanisms to novel therapeutic opportunities. Biomedicines. 2022. https://doi.org/10.3390/biomedicines10040891.
Ursini F, Maiorino M, Valente M, Ferri L, Gregolin C. Purification from pig liver of a protein which protects liposomes and biomembranes from peroxidative degradation and exhibits glutathione peroxidase activity on phosphatidylcholine hydroperoxides. Biochim Biophys Acta. 1982;710:197–211. https://doi.org/10.1016/0005-2760(82)90150-3.
Brigelius-Flohe R, Maiorino M. Glutathione peroxidases. Biochim Biophys Acta. 2013;1830:3289–303. https://doi.org/10.1016/j.bbagen.2012.11.020.
Friedmann Angeli JP, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol. 2014;16:1180–91. https://doi.org/10.1038/ncb3064.
Yang CZ, et al. Elevated level of serum growth differentiation factor 15 is associated with oral leukoplakia and oral squamous cell carcinoma. J Oral Pathol Med. 2014;43:28–34. https://doi.org/10.1111/jop.12091.
Liang C, Zhang X, Yang M, Dong X. Recent progress in ferroptosis inducers for cancer therapy. Adv Mater. 2019;31: e1904197. https://doi.org/10.1002/adma.201904197.
Eaton JK, Furst L, Cai LL, Viswanathan VS, Schreiber SL. Structure-activity relationships of GPX4 inhibitor warheads. Bioorg Med Chem Lett. 2020;30: 127538. https://doi.org/10.1016/j.bmcl.2020.127538.
Yang WS, Stockwell BR. Ferroptosis: death by lipid peroxidation. Trends Cell Biol. 2016;26:165–76. https://doi.org/10.1016/j.tcb.2015.10.014.
Dixon SJ, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–72. https://doi.org/10.1016/j.cell.2012.03.042.
Yan HF, et al. Ferroptosis: mechanisms and links with diseases. Signal Transduct Target Ther. 2021;6:49. https://doi.org/10.1038/s41392-020-00428-9.
Yu H, Guo P, Xie X, Wang Y, Chen G. Ferroptosis, a new form of cell death, and its relationships with tumourous diseases. J Cell Mol Med. 2017;21:648–57. https://doi.org/10.1111/jcmm.13008.
Ursini F, Maiorino M. Lipid peroxidation and ferroptosis: the role of GSH and GPx4. Free Radic Biol Med. 2020;152:175–85. https://doi.org/10.1016/j.freeradbiomed.2020.02.027.
Lu SC. Glutathione synthesis. Biochim Biophys Acta. 2013;1830:3143–53. https://doi.org/10.1016/j.bbagen.2012.09.008.
Lu SC. Regulation of glutathione synthesis. Mol Aspects Med. 2009;30:42–59. https://doi.org/10.1016/j.mam.2008.05.005.
Yant LJ, et al. The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radic Biol Med. 2003;34:496–502. https://doi.org/10.1016/s0891-5849(02)01360-6.
Parker JL, et al. Molecular basis for redox control by the human cystine/glutamate antiporter system xc(). Nat Commun. 2021;12:7147. https://doi.org/10.1038/s41467-021-27414-1.
Li S, et al. The role of SLC7A11 in cancer: friend or foe? Cancers. 2022. https://doi.org/10.3390/cancers14133059.
Sato H, Tamba M, Ishii T, Bannai S. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J Biol Chem. 1999;274:11455–8. https://doi.org/10.1074/jbc.274.17.11455.
McBean GJ. The transsulfuration pathway: a source of cysteine for glutathione in astrocytes. Amino Acids. 2012;42:199–205. https://doi.org/10.1007/s00726-011-0864-8.
Chen L, et al. Erastin sensitizes glioblastoma cells to temozolomide by restraining xCT and cystathionine-gamma-lyase function. Oncol Rep. 2015;33:1465–74. https://doi.org/10.3892/or.2015.3712.
Bridges RJ, Natale NR, Patel SA. System xc(-) cystine/glutamate antiporter: an update on molecular pharmacology and roles within the CNS. Br J Pharmacol. 2012;165:20–34. https://doi.org/10.1111/j.1476-5381.2011.01480.x.
Koppula P, Zhang Y, Zhuang L, Gan B. Amino acid transporter SLC7A11/xCT at the crossroads of regulating redox homeostasis and nutrient dependency of cancer. Cancer Commun. 2018. https://doi.org/10.1186/s40880-018-0288-x.
Dixon SJ, Stockwell BR. The role of iron and reactive oxygen species in cell death. Nat Chem Biol. 2014;10:9–17. https://doi.org/10.1038/nchembio.1416.
Pizzino G, et al. Oxidative stress: harms and benefits for human health. Oxid Med Cell Longev. 2017;2017:8416763. https://doi.org/10.1155/2017/8416763.
Azzam EI, Jay-Gerin JP, Pain D. Ionizing radiation-induced metabolic oxidative stress and prolonged cell injury. Cancer Lett. 2012;327:48–60. https://doi.org/10.1016/j.canlet.2011.12.012.
Li FJ, et al. System X(c) (-)/GSH/GPX4 axis: An important antioxidant system for the ferroptosis in drug-resistant solid tumor therapy. Front Pharmacol. 2022;13: 910292. https://doi.org/10.3389/fphar.2022.910292.
Lu Z, et al. The potential of ferroptosis combined with radiotherapy in cancer treatment. Front Oncol. 2023;13:1085581. https://doi.org/10.3389/fonc.2023.1085581.
Salim S. Oxidative stress and the central nervous system. J Pharmacol Exp Ther. 2017;360:201–5. https://doi.org/10.1124/jpet.116.237503.
Liu X, et al. SLC7A11/GPX4 inactivation-mediated ferroptosis contributes to the pathogenesis of triptolide-induced cardiotoxicity. Oxid Med Cell Longev. 2022;2022:3192607. https://doi.org/10.1155/2022/3192607.
Bayo Jimenez MT, et al. Protective actions of nuclear factor erythroid 2-related factor 2 (NRF2) and downstream pathways against environmental stressors. Free Radic Biol Med. 2022;187:72–91. https://doi.org/10.1016/j.freeradbiomed.2022.05.016.
Song X, Long D. Nrf2 and ferroptosis: a new research direction for neurodegenerative diseases. Front Neurosci. 2020;14:267. https://doi.org/10.3389/fnins.2020.00267.
Zhang Q, et al. Atorvastatin induces mitochondria-dependent ferroptosis via the modulation of Nrf2-xCT/GPx4 axis. Front Cell Dev Biol. 2022;10: 806081. https://doi.org/10.3389/fcell.2022.806081.
Ngo V, Duennwald ML. Nrf2 and oxidative stress: a general overview of mechanisms and implications in human disease. Antioxidants. 2022. https://doi.org/10.3390/antiox11122345.
Baird L, Yamamoto M. The molecular mechanisms regulating the KEAP1-NRF2 pathway. Mol Cell Biol. 2020. https://doi.org/10.1128/MCB.00099-20.
Kaspar JW, Niture SK, Jaiswal AK. Nrf 2:INrf2 (Keap1) signaling in oxidative stress. Free Radic Biol Med. 2009;47:1304–9. https://doi.org/10.1016/j.freeradbiomed.2009.07.035.
Ma Q. Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol. 2013;53:401–26. https://doi.org/10.1146/annurev-pharmtox-011112-140320.
Jelic MD, Mandic AD, Maricic SM, Srdjenovic BU. Oxidative stress and its role in cancer. J Cancer Res Ther. 2021;17:22–8. https://doi.org/10.4103/jcrt.JCRT_862_16.
Kerins MJ, Ooi A. The roles of NRF2 in modulating cellular iron homeostasis. Antioxid Redox Signal. 2018;29:1756–73. https://doi.org/10.1089/ars.2017.7176.
Doll S, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13:91–8. https://doi.org/10.1038/nchembio.2239.
Yuan H, Li X, Zhang X, Kang R, Tang D. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem Biophys Res Commun. 2016;478:1338–43. https://doi.org/10.1016/j.bbrc.2016.08.124.
Kuwata H, et al. Long-chain acyl-CoA synthetase 4 participates in the formation of highly unsaturated fatty acid-containing phospholipids in murine macrophages. Biochim Biophys Acta Mol Cell Biol Lipids. 2019. https://doi.org/10.1016/j.bbalip.2019.07.013.
Cheng J, et al. ACSL4 suppresses glioma cells proliferation via activating ferroptosis. Oncol Rep. 2020;43:147–58. https://doi.org/10.3892/or.2019.7419.
Jia B, Li J, Song Y, Luo C. ACSL4-mediated ferroptosis and its potential role in central nervous system diseases and injuries. Int J Mol Sci. 2023. https://doi.org/10.3390/ijms241210021.
Xie R, Li J, Zhao H. The underlying mechanisms involved in the protective effects of ischemic postconditioning. Cond Med. 2018;1:73–9.
Li Y, et al. Ischemia-induced ACSL4 activation contributes to ferroptosis-mediated tissue injury in intestinal ischemia/reperfusion. Cell Death Differ. 2019;26:2284–99. https://doi.org/10.1038/s41418-019-0299-4.
Suarez-Rivero JM, et al. Coenzyme Q(10) analogues: benefits and challenges for therapeutics. Antioxidants. 2021. https://doi.org/10.3390/antiox10020236.
Cirilli I, et al. Role of coenzyme Q(10) in health and disease: an update on the last 10 years (2010–2020). Antioxidants. 2021. https://doi.org/10.3390/antiox10081325.
Stockwell BR, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171:273–85. https://doi.org/10.1016/j.cell.2017.09.021.
Han C, et al. Ferroptosis and its potential role in human diseases. Front Pharmacol. 2020;11:239. https://doi.org/10.3389/fphar.2020.00239.
Du Y, Guo Z. Recent progress in ferroptosis: inducers and inhibitors. Cell Death Discov. 2022;8:501. https://doi.org/10.1038/s41420-022-01297-7.
Zheng J, Conrad M. The metabolic underpinnings of ferroptosis. Cell Metab. 2020;32:920–37. https://doi.org/10.1016/j.cmet.2020.10.011.
Karni-Schmidt O, Lokshin M, Prives C. The roles of MDM2 and MDMX in cancer. Annu Rev Pathol. 2016;11:617–44. https://doi.org/10.1146/annurev-pathol-012414-040349.
Huang TT, Zou Y, Corniola R. Oxidative stress and adult neurogenesis–effects of radiation and superoxide dismutase deficiency. Semin Cell Dev Biol. 2012;23:738–44. https://doi.org/10.1016/j.semcdb.2012.04.003.
Levine AJ. The many faces of p53: something for everyone. J Mol Cell Biol. 2019;11:524–30. https://doi.org/10.1093/jmcb/mjz026.
Karuppagounder SS, et al. N-acetylcysteine targets 5 lipoxygenase-derived, toxic lipids and can synergize with prostaglandin E(2) to inhibit ferroptosis and improve outcomes following hemorrhagic stroke in mice. Ann Neurol. 2018;84:854–72. https://doi.org/10.1002/ana.25356.
Simabuco FM, et al. p53 and metabolism: from mechanism to therapeutics. Oncotarget. 2018;9:23780–823. https://doi.org/10.18632/oncotarget.25267.
Levine AJ, Puzio-Kuter AM, Chan CS, Hainaut P. The role of the p53 protein in stem-cell biology and epigenetic regulation. Cold Spring Harb Perspect Med. 2016. https://doi.org/10.1101/cshperspect.a026153.
Koppula P, Zhuang L, Gan B. Cystine transporter SLC7A11/xCT in cancer: ferroptosis, nutrient dependency, and cancer therapy. Protein Cell. 2021;12:599–620. https://doi.org/10.1007/s13238-020-00789-5.
Zhang Z, et al. Activation of ferritinophagy is required for the RNA-binding protein ELAVL1/HuR to regulate ferroptosis in hepatic stellate cells. Autophagy. 2018;14:2083–103. https://doi.org/10.1080/15548627.2018.1503146.
Tang X, et al. Research progress on SLC7A11 in the regulation of cystine/cysteine metabolism in tumors. Oncol Lett. 2022;23:47. https://doi.org/10.3892/ol.2021.13165.
Magtanong L, Ko PJ, Dixon SJ. Emerging roles for lipids in non-apoptotic cell death. Cell Death Differ. 2016;23:1099–109. https://doi.org/10.1038/cdd.2016.25.
Anthonymuthu TS, Kenny EM, Lamade AM, Kagan VE, Bayir H. Oxidized phospholipid signaling in traumatic brain injury. Free Radic Biol Med. 2018;124:493–503. https://doi.org/10.1016/j.freeradbiomed.2018.06.031.
Venkatesh D, et al. MDM2 and MDMX promote ferroptosis by PPARalpha-mediated lipid remodeling. Genes Dev. 2020;34:526–43. https://doi.org/10.1101/gad.334219.119.
Jiang L, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520:57–62. https://doi.org/10.1038/nature14344.
Ou Y, Wang SJ, Li D, Chu B, Gu W. Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc Natl Acad Sci U S A. 2016;113:E6806–12. https://doi.org/10.1073/pnas.1607152113.
Venkatesh D, Stockwell BR, Prives C. p21 can be a barrier to ferroptosis independent of p53. Aging (Albany NY). 2020;12:17800–14. https://doi.org/10.18632/aging.103961.
Wang QQ, et al. Ionizing radiation-induced brain cell aging and the potential underlying molecular mechanisms. Cells. 2021. https://doi.org/10.3390/cells10123570.
Ding C, Saw CB, Timmerman RD. Cyberknife stereotactic radiosurgery and radiation therapy treatment planning system. Med Dosim. 2018;43:129–40. https://doi.org/10.1016/j.meddos.2018.02.006.
Wang Y, et al. A new strategy of CyberKnife treatment system based radiosurgery followed by early use of adjuvant bevacizumab treatment for brain metastasis with extensive cerebral edema. J Neurooncol. 2014;119:369–76. https://doi.org/10.1007/s11060-014-1488-0.
Balentova S, Adamkov M. Molecular, cellular and functional effects of radiation-induced brain injury: a review. Int J Mol Sci. 2015;16:27796–815. https://doi.org/10.3390/ijms161126068.
Hu X, De Silva TM, Chen J, Faraci FM. Cerebral vascular disease and neurovascular injury in ischemic stroke. Circ Res. 2017;120:449–71. https://doi.org/10.1161/CIRCRESAHA.116.308427.
Zou Z, Chang H, Li H, Wang S. Induction of reactive oxygen species: an emerging approach for cancer therapy. Apoptosis. 2017;22:1321–35. https://doi.org/10.1007/s10495-017-1424-9.
Dayal R, Singh A, Pandey A, Mishra KP. Reactive oxygen species as mediator of tumor radiosensitivity. J Cancer Res Ther. 2014;10:811–8. https://doi.org/10.4103/0973-1482.146073.
Makranz C, et al. Short report: plasma based biomarkers detect radiation induced brain injury in cancer patients treated for brain metastasis: a pilot study. PLoS ONE. 2023;18: e0285646. https://doi.org/10.1371/journal.pone.0285646.
Qin D, et al. Traumatic brain injury: ultrastructural features in neuronal ferroptosis, glial cell activation and polarization, and blood-brain barrier breakdown. Cells. 2021. https://doi.org/10.3390/cells10051009.
Zhang S, et al. The regulatory effects and the signaling pathways of natural bioactive compounds on ferroptosis. 2021. Foods. https://doi.org/10.3390/foods10122952.
Lee J, Roh JL. Unleashing ferroptosis in human cancers: targeting ferroptosis suppressor protein 1 for overcoming therapy resistance. Antioxidants. 2023. https://doi.org/10.3390/antiox12061218.
Su J, et al. Cooperation effects of radiation and ferroptosis on tumor suppression and radiation injury. Front Cell Dev Biol. 2022;10: 951116. https://doi.org/10.3389/fcell.2022.951116.
Pacelli R, et al. Technological evolution of radiation treatment: implications for clinical applications. Semin Oncol. 2019;46:193–201. https://doi.org/10.1053/j.seminoncol.2019.07.004.
Garibaldi C, et al. Recent advances in radiation oncology. Ecancermedicalscience. 2017;11:785. https://doi.org/10.3332/ecancer.2017.785.
Stone JB, DeAngelis LM. Cancer-treatment-induced neurotoxicity–focus on newer treatments. Nat Rev Clin Oncol. 2016;13:92–105. https://doi.org/10.1038/nrclinonc.2015.152.
Kessler AT, Bhatt AA. Brain tumour post-treatment imaging and treatment-related complications. Insights Imaging. 2018;9:1057–75. https://doi.org/10.1007/s13244-018-0661-y.
Jablonska PA, et al. Challenges and novel opportunities of radiation therapy for brain metastases in non-small cell lung cancer. Cancers. 2021. https://doi.org/10.3390/cancers13092141.
Soffietti R, Pellerino A, Bruno F, Mauro A, Ruda R. Neurotoxicity from old and new radiation treatments for brain tumors. Int J Mol Sci. 2023. https://doi.org/10.3390/ijms241310669.
Frigault M, et al. Dose fractionation of CAR-T cells A. systematic review of clinical outcomes. J Exp Clin Cancer Res. 2023. https://doi.org/10.1186/s13046-022-02540-w.
Smart D. Radiation toxicity in the central nervous system: mechanisms and strategies for injury reduction. Semin Radiat Oncol. 2017;27:332–9. https://doi.org/10.1016/j.semradonc.2017.04.006.
Baumann M, et al. Radiation oncology in the era of precision medicine. Nat Rev Cancer. 2016;16:234–49. https://doi.org/10.1038/nrc.2016.18.
Fiorino C, Guckemberger M, Schwarz M, van der Heide UA, Heijmen B. Technology-driven research for radiotherapy innovation. Mol Oncol. 2020;14:1500–13. https://doi.org/10.1002/1878-0261.12659.
Beaton L, Bandula S, Gaze MN, Sharma RA. How rapid advances in imaging are defining the future of precision radiation oncology. Br J Cancer. 2019;120:779–90. https://doi.org/10.1038/s41416-019-0412-y.
Ludmir EB, Grosshans DR, Woodhouse KD. Radiotherapy advances in pediatric neuro-oncology. Bioengineering. 2018. https://doi.org/10.3390/bioengineering5040097.
Scaringi C, Agolli L, Minniti G. Technical advances in radiation therapy for brain tumors. Anticancer Res. 2018;38:6041–5. https://doi.org/10.21873/anticanres.12954.
Mangraviti A, Gullotti D, Tyler B, Brem H. Nanobiotechnology-based delivery strategies: New frontiers in brain tumor targeted therapies. J Control Release. 2016;240:443–53. https://doi.org/10.1016/j.jconrel.2016.03.031.
Minniti G, et al. Current status and recent advances in resection cavity irradiation of brain metastases. Radiat Oncol. 2021;16:73. https://doi.org/10.1186/s13014-021-01802-9.
Grunert M, et al. Radiation and brain tumors: an overview. Crit Rev Oncog. 2018;23:119–38. https://doi.org/10.1615/CritRevOncog.2018025927.
Kondziolka D, Shin SM, Brunswick A, Kim I, Silverman JS. The biology of radiosurgery and its clinical applications for brain tumors. Neuro Oncol. 2015;17:29–44. https://doi.org/10.1093/neuonc/nou284.
Hellevik T, Martinez-Zubiaurre I. Radiotherapy and the tumor stroma: the importance of dose and fractionation. Front Oncol. 2014;4:1. https://doi.org/10.3389/fonc.2014.00001.
Moulder JE, Seymour C. Radiation fractionation: the search for isoeffect relationships and mechanisms. Int J Radiat Biol. 2018;94:743–51. https://doi.org/10.1080/09553002.2017.1376764.
Griffin RJ, et al. Understanding high-dose, ultra-high dose rate, and spatially fractionated radiation therapy. Int J Radiat Oncol Biol Phys. 2020;107:766–78. https://doi.org/10.1016/j.ijrobp.2020.03.028.
Demaria S, et al. Radiation dose and fraction in immunotherapy: one-size regimen does not fit all settings, so how does one choose? J Immunother Cancer. 2021. https://doi.org/10.1136/jitc-2020-002038.
Sammer M, et al. Normal tissue response of combined temporal and spatial fractionation in proton minibeam radiation therapy. Int J Radiat Oncol Biol Phys. 2021;109:76–83. https://doi.org/10.1016/j.ijrobp.2020.08.027.
Rivers C, et al. Impact of the number of metastatic tumors treated by stereotactic radiosurgery on the dose to normal brain: implications for brain protection. Stereotact Funct Neurosurg. 2017;95:352–8. https://doi.org/10.1159/000480666.
Yang J, et al. Targeting cell death: pyroptosis, ferroptosis, apoptosis and necroptosis in osteoarthritis. Front Cell Dev Biol. 2021;9: 789948. https://doi.org/10.3389/fcell.2021.789948.
Latunde-Dada GO. Ferroptosis: Role of lipid peroxidation, iron and ferritinophagy. Biochim Biophys Acta Gen Subj. 2017. https://doi.org/10.1016/j.bbagen.2017.05.019.
Costa I, et al. Molecular mechanisms of ferroptosis and their involvement in brain diseases. Pharmacol Ther. 2023;244: 108373. https://doi.org/10.1016/j.pharmthera.2023.108373.
Yadav VK, et al. Deeper insight into ferroptosis: association with Alzheimer’s, Parkinson’s disease, and brain tumors and their possible treatment by nanomaterials induced ferroptosis. Redox Rep. 2023;28:2269331. https://doi.org/10.1080/13510002.2023.2269331.
Yu Y, et al. Ferroptosis: a cell death connecting oxidative stress, inflammation and cardiovascular diseases. Cell Death Discov. 2021;7:193. https://doi.org/10.1038/s41420-021-00579-w.
Vitalakumar D, Sharma A, Flora SJS. Ferroptosis: a potential therapeutic target for neurodegenerative diseases. J Biochem Mol Toxicol. 2021;35: e22830. https://doi.org/10.1002/jbt.22830.
Wang HH, Fan SQ, Zhan YT, Peng SP, Wang WY. Suppression of the SLC7A11/glutathione axis causes ferroptosis and apoptosis and alters the mitogen-activated protein kinase pathway in nasopharyngeal carcinoma. Int J Biol Macromol. 2024;254: 127976. https://doi.org/10.1016/j.ijbiomac.2023.127976.
Wang F, He J, Xing R, Sha T, Sun B. Molecular mechanisms of ferroptosis and their role in inflammation. Int Rev Immunol. 2023;42:71–81. https://doi.org/10.1080/08830185.2021.2016739.
Rochette L, et al. Lipid peroxidation and iron metabolism: two corner stones in the homeostasis control of ferroptosis. Int J Mol Sci. 2022. https://doi.org/10.3390/ijms24010449.
Ratan RR. The chemical biology of ferroptosis in the central nervous system. Cell Chem Biol. 2020;27:479–98. https://doi.org/10.1016/j.chembiol.2020.03.007.
Shen L, et al. Ferroptosis in acute central nervous system injuries: the future direction? Front Cell Dev Biol. 2020;8:594. https://doi.org/10.3389/fcell.2020.00594.
Kuang F, Liu J, Tang D, Kang R. Oxidative damage and antioxidant defense in ferroptosis. Front Cell Dev Biol. 2020;8: 586578. https://doi.org/10.3389/fcell.2020.586578.
Liu M, et al. The critical role and molecular mechanisms of ferroptosis in antioxidant systems: a narrative review. Ann Transl Med. 2022;10:368. https://doi.org/10.21037/atm-21-6942.
Stockwell BR. Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell. 2022;185:2401–21. https://doi.org/10.1016/j.cell.2022.06.003.
Shi W, et al. Reprimo (RPRM) mediates neuronal ferroptosis via CREB-Nrf2/SCD1 pathways in radiation-induced brain injury. Free Radic Biol Med. 2024;213:343–58. https://doi.org/10.1016/j.freeradbiomed.2024.01.021.
Ye LF, et al. Radiation-induced lipid peroxidation triggers ferroptosis and synergizes with ferroptosis inducers. ACS Chem Biol. 2020;15:469–84. https://doi.org/10.1021/acschembio.9b00939.
Li J, et al. Ferroptosis: past, present and future. Cell Death Dis. 2020;11:88. https://doi.org/10.1038/s41419-020-2298-2.
Chen L, et al. Enhanced defense against ferroptosis ameliorates cognitive impairment and reduces neurodegeneration in 5xFAD mice. Free Radic Biol Med. 2022;180:1–12. https://doi.org/10.1016/j.freeradbiomed.2022.01.002.
Wang X, Wang Z, Cao J, Dong Y, Chen Y. Ferroptosis mechanisms involved in hippocampal-related diseases. Int J Mol Sci. 2021. https://doi.org/10.3390/ijms22189902.
Tang S, et al. The role of iron, its metabolism and ferroptosis in traumatic brain injury. Front Cell Neurosci. 2020;14: 590789. https://doi.org/10.3389/fncel.2020.590789.
Gao M, et al. Ferroptosis is an autophagic cell death process. Cell Res. 2016;26:1021–32. https://doi.org/10.1038/cr.2016.95.
Valko M, Jomova K, Rhodes CJ, Kuca K, Musilek K. Redox- and non-redox-metal-induced formation of free radicals and their role in human disease. Arch Toxicol. 2016;90:1–37. https://doi.org/10.1007/s00204-015-1579-5.
Wang Y, et al. The iron chaperone poly C binding protein 1 regulates iron efflux through intestinal ferroportin in mice. Blood. 2023. https://doi.org/10.1182/blood.2023020504.
Gaschler MM, et al. FINO(2) initiates ferroptosis through GPX4 inactivation and iron oxidation. Nat Chem Biol. 2018;14:507–15. https://doi.org/10.1038/s41589-018-0031-6.
Meng P, et al. Arsenite induces testicular oxidative stress in vivo and in vitro leading to ferroptosis. Ecotoxicol Environ Saf. 2020;194: 110360. https://doi.org/10.1016/j.ecoenv.2020.110360.
Mao H, Zhao Y, Li H, Lei L. Ferroptosis as an emerging target in inflammatory diseases. Prog Biophys Mol Biol. 2020;155:20–8. https://doi.org/10.1016/j.pbiomolbio.2020.04.001.
Hall ED, Bosken JM. Measurement of oxygen radicals and lipid peroxidation in neural tissues. Curr Protoc Neurosci. 2009. https://doi.org/10.1002/0471142301.ns0717s48.
Winterbourn CC. Toxicity of iron and hydrogen peroxide: the Fenton reaction. Toxicol Lett. 1995;82–83:969–74. https://doi.org/10.1016/0378-4274(95)03532-x.
Xu YY, Wan WP, Zhao S, Ma ZG. L-type calcium channels are involved in iron-induced neurotoxicity in primary cultured ventral mesencephalon neurons of rats. Neurosci Bull. 2020;36:165–73. https://doi.org/10.1007/s12264-019-00424-2.
Yu H, et al. Sulfasalazine-induced ferroptosis in breast cancer cells is reduced by the inhibitory effect of estrogen receptor on the transferrin receptor. Oncol Rep. 2019;42:826–38. https://doi.org/10.3892/or.2019.7189.
Xie J, et al. Graphdiyne nanoparticles with high free radical scavenging activity for radiation protection. ACS Appl Mater Interfaces. 2019;11:2579–90. https://doi.org/10.1021/acsami.8b00949.
Rodencal J, Dixon SJ. A tale of two lipids: Lipid unsaturation commands ferroptosis sensitivity. Proteomics. 2023;23: e2100308. https://doi.org/10.1002/pmic.202100308.
Pan Q, Luo Y, Xia Q, He K. Ferroptosis and liver fibrosis. Int J Med Sci. 2021;18:3361–6. https://doi.org/10.7150/ijms.62903.
Yang WS. Ferroptosis: whERe is the critical site of lipid peroxidation? Front Cell Dev Biol. 2023;11:1179245. https://doi.org/10.3389/fcell.2023.1179245.
He L, et al. Antioxidants maintain cellular redox homeostasis by elimination of reactive oxygen species. Cell Physiol Biochem. 2017;44:532–53. https://doi.org/10.1159/000485089.
Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. https://doi.org/10.1152/physrev.00018.2001.
Lei L, Zhang J, Decker EA, Zhang G. Roles of lipid peroxidation-derived electrophiles in pathogenesis of colonic inflammation and colon cancer. Front Cell Dev Biol. 2021;9: 665591. https://doi.org/10.3389/fcell.2021.665591.
Sha W, Hu F, Xi Y, Chu Y, Bu S. Mechanism of ferroptosis and its role in type 2 diabetes mellitus. J Diabetes Res. 2021;2021:9999612. https://doi.org/10.1155/2021/9999612.
Feng H, Stockwell BR. Unsolved mysteries: How does lipid peroxidation cause ferroptosis? PLoS Biol. 2018;16: e2006203. https://doi.org/10.1371/journal.pbio.2006203.
Zhang X, et al. Ferroptosis, a new form of cell death defined after radiation exposure. Int J Radiat Biol. 2022;98:1201–9. https://doi.org/10.1080/09553002.2022.2020358.
Mizugaki A, et al. Cystine reduces mitochondrial dysfunction in C2C12 myotubes under moderate oxidative stress induced by H(2)O(2). Amino Acids. 2022;54:1203–13. https://doi.org/10.1007/s00726-022-03176-y.
Chen M, et al. Prospects for anti-tumor mechanism and potential clinical application based on glutathione peroxidase 4 mediated ferroptosis. Int J Mol Sci. 2023. https://doi.org/10.3390/ijms24021607.
Wu L, et al. Effects of curcumin on oxidative stress and ferroptosis in acute ammonia stress-induced liver injury in gibel carp (carassius gibelio). Int J Mol Sci. 2023. https://doi.org/10.3390/ijms24076441.
Dixon SJ, et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife. 2014;3: e02523. https://doi.org/10.7554/eLife.02523.
Magtanong L, Dixon SJ. Ferroptosis and Brain Injury. Dev Neurosci. 2018;40:382–95. https://doi.org/10.1159/000496922.
Adeniyi PA, et al. Ferroptosis of microglia in aging human white matter injury. Ann Neurol. 2023. https://doi.org/10.1002/ana.26770.
Tarangelo A, Dixon S. The p53–p21 pathway inhibits ferroptosis during metabolic stress. Oncotarget. 2018;9:24572–3. https://doi.org/10.18632/oncotarget.25362.
Ji Q, et al. ACSL4 is essential for radiation-induced intestinal injury by initiating ferroptosis. Cell Death Discov. 2022;8:332. https://doi.org/10.1038/s41420-022-01127-w.
Pruzincova L, et al. MR imaging of late radiation therapy- and chemotherapy-induced injury: a pictorial essay. Eur Radiol. 2009;19:2716–27. https://doi.org/10.1007/s00330-009-1449-8.
Ivanov SD, Semenov AL, Kovan’ko EG, Yamshanov VA. Effects of iron ions and iron chelation on the efficiency of experimental radiotherapy of animals with gliomas. Bull Exp Biol Med. 2015;158:800–3. https://doi.org/10.1007/s10517-015-2865-1.
Sandoval-Acuna C, et al. Targeting mitochondrial iron metabolism suppresses tumor growth and metastasis by inducing mitochondrial dysfunction and mitophagy. Cancer Res. 2021;81:2289–303. https://doi.org/10.1158/0008-5472.CAN-20-1628.
Alborzinia H, et al. Golgi stress mediates redox imbalance and ferroptosis in human cells. Commun Biol. 2018;1:210. https://doi.org/10.1038/s42003-018-0212-6.
Gao X, et al. Ibuprofen induces ferroptosis of glioblastoma cells via downregulation of nuclear factor erythroid 2-related factor 2 signaling pathway. Anticancer Drugs. 2020;31:27–34. https://doi.org/10.1097/CAD.0000000000000825.
Yokoi I, Toma J, Liu J, Kabuto H, Mori A. Adenosines scavenged hydroxyl radicals and prevented posttraumatic epilepsy. Free Radic Biol Med. 1995;19:473–9. https://doi.org/10.1016/0891-5849(95)00050-8.
Mori A, Yokoi I, Noda Y, Willmore LJ. Natural antioxidants may prevent posttraumatic epilepsy: a proposal based on experimental animal studies. Acta Med Okayama. 2004;58:111–8. https://doi.org/10.18926/AMO/32111.
Ponka P. Rare causes of hereditary iron overload. Semin Hematol. 2002;39:249–62. https://doi.org/10.1053/shem.2002.35638.
David S, Jhelum P, Ryan F, Jeong SY, Kroner A. Dysregulation of iron homeostasis in the central nervous system and the role of ferroptosis in neurodegenerative disorders. Antioxid Redox Signal. 2022;37:150–70. https://doi.org/10.1089/ars.2021.0218.
Keep RF, Hua Y, Xi G. Intracerebral haemorrhage: mechanisms of injury and therapeutic targets. Lancet Neurol. 2012;11:720–31. https://doi.org/10.1016/S1474-4422(12)70104-7.
Liu H, et al. Bakuchiol attenuates oxidative stress and neuron damage by regulating Trx1/TXNIP and the phosphorylation of AMPK after subarachnoid hemorrhage in mice. Front Pharmacol. 2020;11:712. https://doi.org/10.3389/fphar.2020.00712.
Duan X, Wen Z, Shen H, Shen M, Chen G. Intracerebral hemorrhage, oxidative stress, and antioxidant therapy. Oxid Med Cell Longev. 2016;2016:1203285. https://doi.org/10.1155/2016/1203285.
Qu J, Chen W, Hu R, Feng H. The injury and therapy of reactive oxygen species in intracerebral hemorrhage looking at mitochondria. Oxid Med Cell Longev. 2016;2016:2592935. https://doi.org/10.1155/2016/2592935.
Zille M, et al. Neuronal death after hemorrhagic stroke in vitro and in vivo shares features of ferroptosis and necroptosis. Stroke. 2017;48:1033–43. https://doi.org/10.1161/STROKEAHA.116.015609.
Li Q, et al. Inhibition of neuronal ferroptosis protects hemorrhagic brain. JCI Insight. 2017;2: e90777. https://doi.org/10.1172/jci.insight.90777.
Chen J, Li M, Liu Z, Wang Y, Xiong K. Molecular mechanisms of neuronal death in brain injury after subarachnoid hemorrhage. Front Cell Neurosci. 2022;16:1025708. https://doi.org/10.3389/fncel.2022.1025708.
Chen S, et al. Targeting oxidative stress and inflammatory response for blood-brain barrier protection in intracerebral hemorrhage. Antioxid Redox Signal. 2022;37:115–34. https://doi.org/10.1089/ars.2021.0072.
Hong Y, et al. High-frequency repetitive transcranial magnetic stimulation improves functional recovery by inhibiting neurotoxic polarization of astrocytes in ischemic rats. J Neuroinflammation. 2020;17:150. https://doi.org/10.1186/s12974-020-01747-y.
Cui C, Yang F, Li Q. Post-translational modification of GPX4 is a promising target for treating ferroptosis-related diseases. Front Mol Biosci. 2022;9: 901565. https://doi.org/10.3389/fmolb.2022.901565.
Yang WS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317–31. https://doi.org/10.1016/j.cell.2013.12.010.
Hambright WS, Fonseca RS, Chen L, Na R, Ran Q. Ablation of ferroptosis regulator glutathione peroxidase 4 in forebrain neurons promotes cognitive impairment and neurodegeneration. Redox Biol. 2017;12:8–17. https://doi.org/10.1016/j.redox.2017.01.021.
Abdulla S, Saada J, Johnson G, Jefferies S, Ajithkumar T. Tumour progression or pseudoprogression? a review of post-treatment radiological appearances of glioblastoma. Clin Radiol. 2015;70:1299–312. https://doi.org/10.1016/j.crad.2015.06.096.
Greene-Schloesser D, et al. Radiation-induced brain injury: a review. Front Oncol. 2012;2:73. https://doi.org/10.3389/fonc.2012.00073.
Ruan Y, Jiang S, Musayeva A, Gericke A. Oxidative stress and vascular dysfunction in the retina: therapeutic strategies. Antioxidants. 2020. https://doi.org/10.3390/antiox9080761.
Ali FS, et al. Cerebral radiation necrosis: incidence, pathogenesis, diagnostic challenges, and future opportunities. Curr Oncol Rep. 2019;21:66. https://doi.org/10.1007/s11912-019-0818-y.
McCullough LD. Neurovascular disease: 2022 update. Free Neuropathol. 2022. https://doi.org/10.17879/freeneuropathology-2022-3910.
Rakhshandeh A, de Lange CFM, Htoo JK, Gheisari A, Rakhshandeh AR. Immune system stimulation increases the plasma cysteine flux and whole-body glutathione synthesis rate in starter pigs1. J Anim Sci. 2019;97:3871–81. https://doi.org/10.1093/jas/skz211.
Li X, et al. Abnormal neuronal damage and inflammation in the hippocampus of kainic acid-induced epilepsy mice. Cell Biochem Funct. 2021;39:791–801. https://doi.org/10.1002/cbf.3651.
Piao J, et al. Human embryonic stem cell-derived oligodendrocyte progenitors remyelinate the brain and rescue behavioral deficits following radiation. Cell Stem Cell. 2015;16:198–210. https://doi.org/10.1016/j.stem.2015.01.004.
Hassanein SM, Ibrahim YA. Posttraumatic acute disseminated encephalomyelitis in a child resolved by steroid therapy: case report. J Clin Neurol. 2016;12:245–7. https://doi.org/10.3988/jcn.2016.12.2.245.
Klaus C, Liao H, Allendorf DH, Brown GC, Neumann H. Sialylation acts as a checkpoint for innate immune responses in the central nervous system. Glia. 2021;69:1619–36. https://doi.org/10.1002/glia.23945.
Liu Y, et al. Mesenchymal stem cells inhibit lipopolysaccharide-induced inflammatory responses of BV2 microglial cells through TSG-6. J Neuroinflammation. 2014;11:135. https://doi.org/10.1186/1742-2094-11-135.
Akeret K, et al. Cerebrospinal fluid hemoglobin drives subarachnoid hemorrhage-related secondary brain injury. J Cereb Blood Flow Metab. 2021;41:3000–15. https://doi.org/10.1177/0271678X211020629.
Wagner KR, Sharp FR, Ardizzone TD, Lu A, Clark JF. Heme and iron metabolism: role in cerebral hemorrhage. J Cereb Blood Flow Metab. 2003;23:629–52. https://doi.org/10.1097/01.WCB.0000073905.87928.6D.
Wu J, et al. Iron and iron-handling proteins in the brain after intracerebral hemorrhage. Stroke. 2003;34:2964–9. https://doi.org/10.1161/01.STR.0000103140.52838.45.
Wan S, Zhan R, Zheng S, Hua Y, Xi G. Activation of c-Jun-N-terminal kinase in a rat model of intracerebral hemorrhage: the role of iron. Neurosci Res. 2009;63:100–5. https://doi.org/10.1016/j.neures.2008.10.013.
Perez de la Ossa N, et al. Iron-related brain damage in patients with intracerebral hemorrhage. Stroke. 2010;41:810–3. https://doi.org/10.1161/STROKEAHA.109.570168.
Neubert J, Wagner S, Kiwit J, Brauer AU, Glumm J. New findings about iron oxide nanoparticles and their different effects on murine primary brain cells. Int J Nanomedicine. 2015;10:2033–49. https://doi.org/10.2147/IJN.S74404.
Donley DW, Realing M, Gigley JP, Fox JH. Iron activates microglia and directly stimulates indoleamine-2,3-dioxygenase activity in the N171–82Q mouse model of Huntington’s disease. PLoS ONE. 2021;16: e0250606. https://doi.org/10.1371/journal.pone.0250606.
Saleppico S, et al. Iron regulates microglial cell-mediated secretory and effector functions. Cell Immunol. 1996;170:251–9. https://doi.org/10.1006/cimm.1996.0159.
Chang YQ, et al. Treatment of radiation-induced brain injury with bisdemethoxycurcumin. Neural Regen Res. 2023;18:416–21. https://doi.org/10.4103/1673-5374.346549.
Zhou D, et al. Astrocytes-derived VEGF exacerbates the microvascular damage of late delayed RBI. Neuroscience. 2019;408:14–21. https://doi.org/10.1016/j.neuroscience.2019.03.039.
Li M, Hamilton R, Salapa HE, Levin MC. Pro-inflammatory cytokines and antibodies induce hnRNP A1 dysfunction in mouse primary cortical neurons. Brain Sci. 2021. https://doi.org/10.3390/brainsci11101282.
Yang P, et al. Ionizing radiation upregulates glutamine metabolism and induces cell death via accumulation of reactive oxygen species. Oxid Med Cell Longev. 2021;2021:5826932. https://doi.org/10.1155/2021/5826932.
Hayashi T, et al. Radiation-induced apoptosis of stem/progenitor cells in human umbilical cord blood is associated with alterations in reactive oxygen and intracellular pH. Mutat Res. 2004;556:83–91. https://doi.org/10.1016/j.mrfmmm.2004.07.002.
Phaniendra A, Jestadi DB, Periyasamy L. Free radicals: properties, sources, targets, and their implication in various diseases. Indian J Clin Biochem. 2015;30:11–26. https://doi.org/10.1007/s12291-014-0446-0.
Eid R, Arab NT, Greenwood MT. Iron mediated toxicity and programmed cell death: a review and a re-examination of existing paradigms. Biochim Biophys Acta Mol Cell Res. 1864;399–430:2017. https://doi.org/10.1016/j.bbamcr.2016.12.002.
Abe C, Miyazawa T, Miyazawa T. Current use of fenton reaction in drugs and food. Molecules. 2022. https://doi.org/10.3390/molecules27175451.
Long H, Zhu W, Wei L, Zhao J. Iron homeostasis imbalance and ferroptosis in brain diseases. MedComm. 2023. https://doi.org/10.1002/mco2.298.
Pham-Huy LA, He H, Pham-Huy C. Free radicals, antioxidants in disease and health. Int J Biomed Sci. 2008;4:89–96.
Logan MP, Parker S, Shi R. Glutathione and ascorbic acid enhance recovery of Guinea pig spinal cord white matter following ischemia and acrolein exposure. Pathobiology. 2005;72:171–8. https://doi.org/10.1159/000086786.
Vaziri ND, Lee YS, Lin CY, Lin VW, Sindhu RK. NAD(P)H oxidase, superoxide dismutase, catalase, glutathione peroxidase and nitric oxide synthase expression in subacute spinal cord injury. Brain Res. 2004;995:76–83. https://doi.org/10.1016/j.brainres.2003.09.056.
Davis CK, Vemuganti R. Antioxidant therapies in traumatic brain injury. Neurochem Int. 2022;152: 105255. https://doi.org/10.1016/j.neuint.2021.105255.
Fesharaki-Zadeh A. Oxidative stress in traumatic brain injury. Int J Mol Sci. 2022. https://doi.org/10.3390/ijms232113000.
Kale A, et al. Neuroprotective effects of Quercetin on radiation-induced brain injury in rats. J Radiat Res. 2018;59:404–10. https://doi.org/10.1093/jrr/rry032.
Zhang Y, et al. Neuroprotective effects of kukoamine a against radiation-induced rat brain injury through inhibition of oxidative stress and neuronal apoptosis. Neurochem Res. 2016;41:2549–58. https://doi.org/10.1007/s11064-016-1967-0.
Abrams RP, Carroll WL, Woerpel KA. Five-membered ring peroxide selectively initiates ferroptosis in cancer cells. ACS Chem Biol. 2016;11:1305–12. https://doi.org/10.1021/acschembio.5b00900.
Lei G, Zhuang L, Gan B. Targeting ferroptosis as a vulnerability in cancer. Nat Rev Cancer. 2022;22:381–96. https://doi.org/10.1038/s41568-022-00459-0.
Tang D, Chen X, Kang R, Kroemer G. Ferroptosis: molecular mechanisms and health implications. Cell Res. 2021;31:107–25. https://doi.org/10.1038/s41422-020-00441-1.
Ji Y, et al. Insight into the potential role of ferroptosis in neurodegenerative diseases. Front Cell Neurosci. 2022;16:1005182. https://doi.org/10.3389/fncel.2022.1005182.
Guo S, et al. Radiation-induced tumor immune microenvironments and potential targets for combination therapy. Signal Transduct Target Ther. 2023;8:205. https://doi.org/10.1038/s41392-023-01462-z.
Lei G, Mao C, Yan Y, Zhuang L, Gan B. Ferroptosis, radiotherapy, and combination therapeutic strategies. Protein Cell. 2021;12:836–57. https://doi.org/10.1007/s13238-021-00841-y.
Porras CA, Rouault TA. Iron homeostasis in the CNS: an overview of the pathological consequences of iron metabolism disruption. Int J Mol Sci. 2022. https://doi.org/10.3390/ijms23094490.
Beretta GL, Zaffaroni N. Radiotherapy-induced ferroptosis for cancer treatment. Front Mol Biosci. 2023;10:1216733. https://doi.org/10.3389/fmolb.2023.1216733.
Zhang X, et al. Hematopoietic protection and mechanisms of ferrostatin-1 on hematopoietic acute radiation syndrome of mice. Int J Radiat Biol. 2021;97:464–73. https://doi.org/10.1080/09553002.2021.1876956.
Funding
This research received support from the 2023 Project Foundation of Tianjin Baodi Hospital (Grant No. BDYYYB03).
Author information
Authors and Affiliations
Contributions
The authors declare no competing interests. Lifang Li (L.L.), Xia Liu (X.L.), and Chunfeng Han (C.H.) have contributed equally to this work. Their primary responsibilities included manuscript writing, drafting the text, and preparing figures. Licheng Tian (L.T.) and Yongzhi Wang (Y.W.) provided critical revisions for important intellectual content and offered valuable suggestions for improvement. Baolin Han (B.H.), the corresponding author, played a pivotal role in conceptualizing and designing the study, overseeing the entire project to ensure achievement of its objectives, and ensuring the accuracy and integrity of the work. All authors have read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Given that this manuscript is a review article and does not involve any direct research on human or animal subjects, ethical approval and consent to participate are not applicable.
Consent for publication
Not applicable, as this manuscript does not contain any personal data from individuals in any form.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, L., Liu, X., Han, C. et al. Ferroptosis in radiation-induced brain injury: roles and clinical implications. BioMed Eng OnLine 23, 93 (2024). https://doi.org/10.1186/s12938-024-01288-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12938-024-01288-y