
Abstract
Delayed radiation necrosis is a well-known adverse event following radiotherapy for brain diseases and has been studied since the 1930s. The primary pathogenesis is thought to be the direct damage to endothelial and glial cells, particularly oligodendrocytes, which causes vascular hyalinization and demyelination. This primary pathology leads to tissue inflammation and ischemia, inducing various tissue protective responses including angiogenesis. Macrophages and lymphocytes then infiltrate the surrounding areas of necrosis, releasing inflammatory cytokines such as interleukin (IL)-1α, IL-6, and tumor necrosis factor (TNF)-α. Microglia also express these inflammatory cytokines. Reactive astrocytes play an important role in angiogenesis, expressing vascular endothelial growth factor (VEGF). Some chemokine networks, like the CXCL12/CXCR4 axis, are upregulated by tissue inflammation. Hypoxia may mediate the cell–cell interactions among reactive astrocytes, macrophages, and microglial cells around the necrotic core. Recently, bevacizumab, an anti-VEGF antibody, has demonstrated promising results as an alternative treatment for radiation necrosis. The importance of VEGF in the pathophysiology of brain radiation necrosis is being recognized. The discovery of new molecular targets could facilitate novel treatments for radiation necrosis. This literature review will focus on recent work characterizing delayed radiation necrosis in the brain.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Delayed brain radiation necrosis is an adverse event widely known to be associated with therapeutic irradiation to the central nervous system (CNS) and focal injuries of the late radiation effects with focal neurologic abnormalities [1]. Late radiation injuries can occur from a few months to 10 or more years after radiotherapy [2]. Histological changes in delayed radiation effects were initially described by Fischer and Holfelder [3] in 1930. Although CNS was first thought to be highly radio-resistant, an animal study showed that CNS, particularly the white matter in the deep part of the brain was more radio-responsive than previously supposed [4]. Different conditions of data acquisition mean that the reported incidence of radiation necrosis varies from 3 to 24 % [5]. In long-term survivors with metastatic brain tumors treated with gamma knife radiosurgery, 64 % of patients developed radiation necrosis, with median survival of 32 months [6]. The predictive factors for developing radiation necrosis in patients with gliomas were radiation dose, use of chemotherapy, and length of survival [5]. Therapeutic irradiation is an important treatment modality to locally control disease in malignant CNS tumors, as there are limited effective chemotherapeutic agents that are able to pass through blood–brain barrier (BBB). Recently, high-dose radiotherapies such as intensity-modulated radiation therapy, proton beam radiotherapy, and boron neutron capture therapy have been used for treatment of malignant gliomas, with better outcomes expected than conventional radiotherapy. However, there is a high incidence of radiation necrosis or symptomatic change requiring surgical management in these high-dose radiotherapies [7–9]. To conquer CNS malignancies, it is necessary to manage radiation necrosis in addition to developing high-dose radiotherapies.
However, diagnosis and treatment of radiation necrosis remain challenging as it is usually difficult to distinguish radiation necrosis from tumor recurrence. In malignant gliomas, pseudoprogression has also become a problem, and is difficult to differentiate from early disease progression following chemoradiotherapy [10]. Pseudoprogression is treatment-related necrosis occurring within 2 months of treatment [11]. The therapeutic strategies for tumor progression and treatment-related necrosis are completely different. Radiation necrosis initially responds to corticosteroids, but progressive radiation necrosis is refractory to medical treatment. However, the vascular endothelial growth factor (VEGF) has recently been reported to play an important role in the development of radiation necrosis [12–16]. In addition, bevacizumab, a monoclonal anti-VEGF antibody, has been reported to be effective in treating radiation necrosis and reducing perilesional edema [17, 18]. We recently published a review of radiation necrosis [19], providing an overview of radiation necrosis from pathophysiology to treatment for clinicians with the aim of helping the management of patients with radiation necrosis. However, further clarifying the molecular biological pathology of radiation necrosis may lead to the discovery and development of a novel, effective therapy for radiation necrosis such as anti-VEGF therapy. Therefore, in the present review, we focus on reviewing the literature on the pathology of radiation necrosis, and highlight recent molecular pathological findings with regard to radiation necrosis.
Vascular damage as the primary cause of radiation necrosis
In the 1930s, seminal case reports demonstrated radiation necrosis as the late radiation injury to the brain, presenting as neurological deterioration after cranial irradiation with a long latent interval. Pennybacker and Russell [20] reported case series including nine patients who encountered harmful effects to the brain after radiation therapy. The authors described a pathological observation of a resected lesion in a patient without brain tumor (irradiated for a rodent ulcer of the scalp). They demonstrated architectural changes of the gyrus and vasculature. Further, loss of neurons and gliosis were observed in the cortex. Hemorrhage, thrombosis, and collagenous thickening and fibrinoid necrosis of vessels were also observed in the white matter. From their clinical and experimental studies, the authors concluded that vascular damage played an important role in the development of radiation necrosis [21]. The authors also speculated that an increase in capillary permeability preceded the lesion, but could not show direct pathological evidence. A second pathological study using electron microscope showed capillary basal lamina thickening, swelling of endothelial cells, and morphological changes in glial cells [22]. These were suggested to be secondary changes following capillary dysfunction. In a human study, Martins et al. [23] tested the hypothesis that the primary site of radiation injury was endothelial cells. Their cases showed that white matter was damaged with thickened and hyalinized small vessels. Many of the fibrinoid, necrotic, and fragmented vessels were surrounded by hemorrhage and edema. Necrosis was found in not only the cerebral parenchyma but also the white matter with gliosis. Radiation injuries to blood vessels are most likely to be initiated by breaks in double-strand deoxyribonucleic acid, leading to phenotypic changes in endothelium [24].
Angiogenesis and VEGF
Tsao et al. [12] proposed that BBB dysfunction caused by endothelial cell damage played a major role in the pathogenesis of late radiation injury. The authors demonstrated that VEGF protein and mRNA were expressed in astrocytes around areas of white matter necrosis and were temporally and spatially associated with albumin extravasation as a surrogate of blood–spinal cord barrier (BSCB) breakdown in an irradiated rat spinal cord [12]. Using this animal model, the authors showed that a hypoxic marker of the nitroimidazole group and VEGF were colocalized in cells surrounding the necrotic core, and that hypoxia-inducible factor-1α (HIF-1α) and VEGF were expressed in astrocytes around areas of necrosis [13, 14]. HIF-1α and VEGF expression were colocalized following BSCB disruption. The authors postulated that endothelial cells were initially damaged after irradiation, and subsequently, the barrier was disrupted. In a study of experimental radiosurgery, intraparenchymal edema was identified as the initial histological change after radiosurgery [25]. An increase in vascular permeability may also result in hypoxia. Astrocytes responded to HIF-1α upregulated VEGF, which further increased vascular permeability and exacerbated tissue hypoxia. Approximately 74 % of VEGF-expressing cells were astrocytes, whereas only 43 % of HIF-1α-positive cells were identified as astrocytes [13, 14].
We reported a pathological examination of radiation necrosis in specimens from 18 patients who underwent surgical removal of necrotic tissue [15]. Hematoxylin and eosin staining showed enlarged vessels with thin walls, as observed with telangiectasis, at the border zone between the normal brain and necrotic core (perinecrotic area) (Fig. 1). These telangiectatic vessels were often accompanied by microbleeding and interstitial edema and could be leaky and fragile. Similar findings occurred in radiation necrosis irrespective of the original tumor pathology and modality of radiation therapy. These telangiectatic vessels could be caused by angiogenesis and affected by VEGF. In an immunohistological analysis, reactive astrocytes in the perinecrotic area exhibited strong positive staining for VEGF expression (Fig. 2). Further, glial fibrillary acidic protein (GFAP)-positive cells expressed VEGF as assessed by double immunofluorescence staining. HIF-1α was also expressed in cells that resembled glial cells around the necrotic core [26]. Thus, reactive astrocytes are likely to play an important role in angiogenesis in radiation necrosis.

Histological findings. Micrographs showing enlarged thin-walled vessels (a black arrows), hyalinized vessels (a white arrows), and reactive astrocytes (b black arrows) in the surrounding area of the necrotic core. (a hematoxylin and eosin stain, magnification 100×; b Glial fibrillary acidic protein immunohistochemistry, magnification 200×)

Immunohistochemical findings. Immunohistochemistry for vascular endothelial growth factor-A (VEGFA) revealed that VEGFA-expressing cells were distributed around the necrotic core (a magnification 100×). Reactive astrocytes (b, c black arrows), reactive microglia (ameboid microglia) (b white arrows), and endothelial cells (c white arrowheads) showed positive staining for VEGFA in the surrounding area of the necrotic core (b, c magnification 400×)
Inflammation, cytokines, and angiogenesis
It was reported that the myelin sheath and oligodendrocytes were directly damaged by radiation in the irradiated injury model of the spinal cord [27]. In the brain radiation injury model, the demyelinating process preceded white matter necrosis before delayed radiation necrosis, which appeared before the onset of any vascular changes [28]. In radiosurgery using a rat glioma model, cellular edema and hypocellular architectural change without alteration in blood vessel morphology were suggested to be related to the direct cytotoxic effect of radiation [29]. Degeneration of white matter associated with variable degrees of astrogliosis and demyelination were frequently observed in a postmortem study of patients with intracranial gliomas [30]. Inflammatory reactions were seen with myelin damage, hemorrhage, and edema. Kureshi et al. [31] investigated lymphocyte and macrophage infiltration as well as cytokine expression in human specimens of radiation necrosis. T cell lymphocytes and macrophages were diffusely infiltrated in the perivascular and parenchymal spaces. Macrophages heavily infiltrated the surrounding areas of necrosis and perivascular proliferation, and strongly expressed Interleukin (IL) and tumor necrosis factor (TNF)-α. Although IL-6 and TNF-α were also identified in reactive astrocytes, the staining of IL-6 and TNF-α was weaker in reactive astrocytes than in infiltrating macrophages. IL-1α was expressed in activated macrophages or microglia. The authors suggested that these inflammatory cytokines may be released for cellular protection [32], but may also be involved in the gliosis, endothelial cell proliferation, and tissue necrosis at the late phase after irradiation. A second human specimen study revealed that many inflammatory cells, including macrophages and lymphocytes, were observed with telangiectatic vascularization in the area around the necrosis [33]. The author concluded that overexpression of inflammatory cytokines played a key role in the progression of radiation necrosis. In addition, the author suggested that coagulation necrosis was accompanied by the disorder of endothelial cells.
We presumed that inflammation and angiogenesis were major contributors to the progression of radiation necrosis and investigated the relationship between these two pathophysiologies. Although HIF-1α is a key regulator for inducing VEGF expression, VEGF is also upregulated through a HIF-1α-independent pathway [34–36]. Interestingly, chemokines stimulate not only inflammation, but also angiogenesis [37–39]. In human glioblastomas, it was reported that expression of CXCL12 and CXCR4 were colocalized in the regions of necrosis and angiogenesis [40], and furthermore, function of the CXCL12/CXCR4 axis promoted VEGFA expression in glioma cells [41, 42].
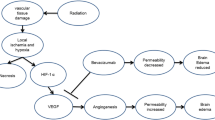
In our study, CXCL12 was detected in GFAP-positive cells, and CXCR4 was expressed in CD68-positive cells in human specimens of radiation necrosis [26]. Most of the CD68-positive cells were identical to the human glucose transporter 5-positive cells. This finding indicated that microglia expressed CXCR4. Microglia also expressed IL-1α, IL-6, and TNF-α. Therefore, chemokines could stimulate microglial cells to produce these cytokines, exacerbating inflammation. However, VEGF was mostly expressed in reactive astrocytes, not microglia. The relationship between CXCL12/CXCR4 signaling and VEGF production is currently unclear. Lymphocytes expressing CD45 also expressed CXCR4, but did not express any cytokines. We concluded that microglia played a key role in inflammation in radiation necrosis. Our hypothetical mechanism of radiation necrosis is shown in Fig. 3.

Hypothetical mechanisms of radiation necrosis. Endothelial cells of cerebral vessels are initially damaged by irradiation, resulting in tissue hypoxia. Reactive astrocytes, microglia, and macrophages respond to hypoxia by endothelial dysfunction after radiotherapy, and express hypoxia-inducible factor (HIF)-1α. HIF-1α strongly mediates up-regulation of vascular endothelial growth factor (VEGF). VEGF is a vascular permeability factor as well as an angiogenic factor. Overexpression of VEGF causes leaky and fragile angiogenesis accompanied by microscopic bleeding, despite protection from hypoxic injury. Reactive astrocytes express CXCL12, and recruiting microglia and macrophages express CXCR4 via CXCL12/CXCR4 chemotaxis. These monocytes contribute to inflammation by releasing inflammatory cytokines including NF-κB, interleukin (IL)-1α, IL-6, and tumor necrosis factor (TNF)-α. These processes of angiogenesis and inflammation exacerbate tissue hypoxia and vasogenic edema, resulting in the progression of radiation necrosis
Platelet-derived growth factors (PDGFs) are currently of interest as an angiogenic factor [43, 44]. PDGF was identified as a constituent of whole blood serum and was subsequently purified from human platelets [45]. PDGFs consist of disulphide-linked dimers and members of the PDGF/VEGF family of growth factors. PDGFs were overexpressed in pathological conditions including malignancies, atherosclerosis, and fibroproliferative diseases.
In our study using human specimens, PDGF-C and -D were expressed at a significantly higher level than PDGF-A and -B around the necrotic core [46]. PDGF-C and -D were mainly expressed by macrophages, microglial cells, reactive astrocytes, lymphocytes, and endothelial cells. Platelet-derived growth factor receptor (PDGFR)-α was also expressed by these cells. PDGF-C is potentially an angiogenic factor, similar to VEGF and PDGF-A and -B. PDGF-D has been shown to stimulate angiogenesis [47]. However, in radiation necrosis, PDGFR-β, which PDGF-D binds to with high affinity, was only expressed in endothelial cells. Therefore, PDGF-C is the critical regulator in PDGFs. Macrophages, microglia, and reactive astrocytes significantly contribute to inflammation and angiogenesis through PDGF-C/PDGF-α autocrine and paracrine signaling in radiation necrosis. The expression of inflammatory and angiogenic molecules in our study of radiation necrosis is summarized in Table 1.
Treatment of radiation necrosis
Although existing treatments of radiation necrosis are theoretically effective, based on pathophysiology, these treatments are not always effective. Corticosteroids are commonly used and are mildly effective on radiation necrosis [48]. Corticosteroids suppress inflammation and stabilize BBB, resulting in a reduction in vasogenic edema. Anticoagulants could improve microcirculation and are expected to prevent radiation necrosis, by improving ischemic and hypoxic conditions. Surprisingly, anticoagulants showed only mild effectiveness on symptomatic radiation necrosis [49]. Hyperbaric oxygen therapy (HBO) could raise the partial pressure of oxygen in the irradiated tissue. However, it was difficult to resolve neurological symptoms by HBO [50]. Further, prophylactic HBO did not reduce the incidence of radiation necrosis after stereotactic radiosurgery (SRS) for brain metastasis, although white matter injury was less frequent in patients with HBO after SRS than in patients without HBO [51]. In the surgical approach, resection of necrotic tissue is a useful option to improve perilesional edema. This treatment can remove not only necrotic tissue, but also cells responsible for producing VEGF and/or inflammatory cytokines in tissue surrounding the necrosis. However, radiation therapy is typically applied to unresectable residual tumors adjacent to the eloquent area after surgical removal of tumors. Therefore, surgical removal of necrotic tissue has a high morbidity of neurological deterioration. In a review of surgically treatments, 35 of 44 patients (79.5 %) showed a good outcome; however, two patients (4.5 %) experienced deterioration after surgery [31]. Furthermore, seven patients (15.9 %) died because of surgical complications. Although this review was published in 1980s, the mortality rate was far from low. In a recent report on surgical management for radiation necrosis using neuronavigation and intraoperative magnetic resonance imaging, there was no mortality [35]. However, surgical complications and neurological deterioration were still observed in 54 % of patients [52]. As previously mentioned, bevacizumab, a monoclonal anti-VEGF antibody, has been highlighted as a new alternative treatment for radiation necrosis. Bevacizumab can efficiently reduce perilesional edema through stabilizing BBB and preventing angiogenesis via trapping of VEGF. Gonzalez et al. [53] first reported the efficacy of bevacizumab on radiation necrosis of the brain. They retrospectively reviewed 15 cases who underwent bevacizumab treatment. Of the 15 patients, eight patients were retrospectively diagnosed with radiation necrosis and responded better to bevacizumab than other patients with recurrent tumors. Subsequently, several case studies and one small, randomized controlled trial have reported clinical and radiological improvements in all patients who received bevacizumab [17, 54–60]. However, there are no further clinical trials designed to acquire on-label approval for use in the United States and Europe. Therefore, we conducted a prospective multicenter clinical trial in Japan.
Conclusions
Initial injury by irradiation is prominent in the myelin and the endothelial cells. Inflammation and angiogenesis are related to these cell injuries and are responsible for delayed brain radiation necrosis. These two processes interact in a complex manner, and it is unknown which occurs first. Chronological changes in molecular mechanisms should be elucidated to resolve this question. On the other hand, the observation that bevacizumab can improve outcomes in patients with radiation necrosis demonstrated that VEGF is one of key molecules controlling the pathology of delayed radiation injury in the brain. Bevacizumab is now expected to be utilized as an alternative therapy for intractable delayed radiation necrosis. We showed that PDGF/PDGFR contributed to radiation necrosis and predict that targeted therapy for these molecules could be a novel treatment for radiation necrosis. In the future, delayed radiation necrosis may be prevented by a novel therapy that blockades certain molecules, if the molecular mechanism preceding the progression of delayed radiation necrosis is elucidated.
References
Schultheiss TE, Kun LE, Ang KK, Stephens LC (1995) Radiation response of the central nervous system. Int J Radiat Oncol Biol Phys 31(5):1093–1112
Valk PE, Dillon WP (1991) Radiation injury of the brain. AJNR Am J Neuroradiol 12(1):45–62
Fischer AW, Holfelder H (1930) Lokales amyloid in gehirn. Dtsch Z Chir 227:475–483
Arnold A, Bailey P, Laughlin JS (1954) Effects of beatron radiations on the brain of primates. Neurology 4(3):165–178
Ruben JD, Dally M, Bailey M, Smith R, McLean CA, Fedele P (2006) Cerebral radiation necrosis: incidence, outcomes, and risk factors with emphasis on radiation parameters and chemotherapy. Int J Radiat Oncol Biol Phys 65(2):499–508
Jagannathan J, Petit JH, Balsara K, Hudes R, Chin LS (2004) Long-term survival after gamma knife radiosurgery for primary and metastatic brain tumors. Am J Clin Oncol 27(5):441–444
Iuchi T, Hatano K, Narita Y, Kodama T, Yamaki T, Osato K (2006) Hypofractionated high-dose irradiation for the treatment of malignant astrocytomas using simultaneous integrated boost technique by IMRT. Int J Radiat Oncol Biol Phys 64(5):1317–1324
Kawabata S, Miyatake S, Kuroiwa T, Yokoyama K, Doi A, Iida K, Miyata S, Nonoguchi N, Michiue H, Takahashi M, Inomata T, Imahori Y, Kirihata M, Sakurai Y, Maruhashi A, Kumada H, Ono K (2009) Boron neutron capture therapy for newly diagnosed glioblastoma. J Radiat Res 50(1):51–60
Mizumoto M, Tsuboi K, Igaki H, Yamamoto T, Takano S, Oshiro Y, Hayashi Y, Hashii H, Kanemoto A, Nakayama H, Sugahara S, Sakurai H, Matsumura A, Tokuuye K (2010) Phase I/II trial of hyperfractionated concomitant boost proton radiotherapy for supratentorial glioblastoma multiforme. Int J Radiat Oncol Biol Phys 77(1):98–105
Brandes AA, Franceschi E, Tosoni A, Blatt V, Pession A, Tallini G, Bertorelle R, Bartolini S, Calbucci F, Andreoli A, Frezza G, Leonardi M, Spagnolli F, Ermani M (2008) MGMT promoter methylation status can predict the incidence and outcome of pseudoprogression after concomitant radiochemotherapy in newly diagnosed glioblastoma patients. J Clin Oncol 26(13):2192–2197
Brandsma D, Stalpers L, Taal W, Sminia P, van den Bent MJ (2008) Clinical features, mechanisms, and management of pseudoprogression in malignant gliomas. Lancet Oncol 9(5):453–461
Tsao MN, Li YQ, Lu G, Xu Y, Wong CS (1999) Upregulation of vascular endothelial growth factor is associated with radiation-induced blood-spinal cord barrier breakdown. J Neuropathol Exp Neurol 58(10):1051–1060
Li YQ, Ballinger JR, Nordal RA, Su ZF, Wong CS (2001) Hypoxia in radiation-induced blood-spinal cord barrier breakdown. Cancer Res 61(8):3348–3354
Nordal RA, Nagy A, Pintilie M, Wong CS (2004) Hypoxia and hypoxia-inducible factor-1 target genes in central nervous system radiation injury: a role for vascular endothelial growth factor. Clin Cancer Res 10(10):3342–3353
Nonoguchi N, Miyatake S, Fukumoto M, Furuse M, Hiramatsu R, Kawabata S, Kuroiwa T, Tsuji M, Ono K (2011) The distribution of vascular endothelial growth factor-producing cells in clinical radiation necrosis of the brain: pathological consideration of their potential roles. J Neurooncol 105(2):423–431
Furuse M, Nonoguchi N, Kawabata S, Miyata T, Toho T, Kuroiwa T, Miyatake S (2015) Intratumoral and peritumoral post-irradiation changes, but not viable tumor tissue, may respond to bevacizumab in previously irradiated meningiomas. Radiat Oncol 10:156
Furuse M, Kawabata S, Kuroiwa T, Miyatake S (2011) Repeated treatments with bevacizumab for recurrent radiation necrosis in patients with malignant brain tumors: a report of 2 cases. J Neurooncol 102(3):471–475
Furuse M, Nonoguchi N, Kawabata S, Yoritsune E, Takahashi M, Inomata T, Kuroiwa T, Miyatake S (2013) Bevacizumab treatment for symptomatic radiation necrosis diagnosed by amino acid PET. Jpn J Clin Oncol 43(3):337–341
Miyatake S, Nonoguchi N, Furuse M, Yoritsune E, Miyata T, Kawabata S, Kuroiwa T (2015) Pathophysiology, diagnosis, and treatment of radiation necrosis in the brain. Neurol Med Chir 55(Suppl 1):50–59
Pennybacker J, Russell DS (1948) Necrosis of the brain due to radiation therapy; clinical and pathological observations. J Neurol Neurosurg Psychiatry 11(3):183–198
Russell DS, Wilson CW, Tansley K (1949) Experimental radio-necrosis of the brain in rabbits. J Neurol Neurosurg Psychiatry 12(3):187–195
McDonald LW, Hayes TL (1967) The role of capillaries in the pathogenesis of delayed radionecrosis of brain. Am J Pathol 50(5):745–764
Martins AN, Johnston JS, Henry JM, Stoffel TJ, Di Chiro G (1977) Delayed radiation necrosis of the brain. J Neurosurg 47(3):336–345
O’Connor MM, Mayberg MR (2000) Effects of radiation on cerebral vasculature: a review. Neurosurgery 46(1):138–149
Kondziolka D, Lunsford LD, Claassen D, Maitz AH, Flickinger JC (1992) Radiobiology of radiosurgery: part I. The normal rat brain model. Neurosurgery 31(2):271–279
Yoritsune E, Furuse M, Kuwabara H, Miyata T, Nonoguchi N, Kawabata S, Hayasaki H, Kuroiwa T, Ono K, Shibayama Y, Miyatake S (2014) Inflammation as well as angiogenesis may participate in the pathophysiology of brain radiation necrosis. J Radiat Res 55(4):803–811
Mastaglia FL, McDonald WI, Watson JV, Yogendran K (1976) Effects of x-radiation on the spinal cord: an experimental study of the morphological changes in central nerve fibres. Brain 99(1):101–122
Arnold A, Bailey P, Harvey RA, Haas LL, Laughlin JS (1954) Changes in the central nervous system following irradiation with 23-mev X-rays from the betatron. Radiology 62(1):37–46
Kondziolka D, Lunsford LD, Claassen D, Pandalai S, Maitz AH, Flickinger JC (1992) Radiobiology of radiosurgery: part II. The rat C6 glioma model. Neurosurgery 31(2):280–287
Burger PC, Mahley MS Jr, Dudka L, Vogel FS (1979) The morphologic effects of radiation administered therapeutically for intracranial gliomas: a postmortem study of 25 cases. Cancer 44(4):1256–1272
Kureshi SA, Hofman FM, Schneider JH, Chin LS, Apuzzo ML, Hinton DR (1994) Cytokine expression in radiation-induced delayed cerebral injury. Neurosurgery 35(5):822–829
Pizzi M, Sarnico I, Boroni F, Benarese M, Dreano M, Garotta G, Valerio A, Spano P (2004) Prevention of neuron and oligodendrocyte degeneration by interleukin-6 (IL-6) and IL-6 receptor/IL-6 fusion protein in organotypic hippocampal slices. Mol Cell Neurosci 25(2):301–311
Yoshii Y (2008) Pathological review of late cerebral radionecrosis. Brain Tumor Pathol 25(2):51–58
Mizukami Y, Li J, Zhang X, Zimmer MA, Iliopoulos O, Chung DC (2004) Hypoxia-inducible factor-1-independent regulation of vascular endothelial growth factor by hypoxia in colon cancer. Cancer Res 64(5):1765–1772
Pore N, Jiang Z, Gupta A, Cerniglia G, Kao GD, Maity A (2006) EGFR tyrosine kinase inhibitors decrease VEGF expression by both hypoxia-inducible factor (HIF)-1-independent and HIF-1-dependent mechanisms. Cancer Res 66(6):3197–3204
Pore N, Liu S, Shu HK, Li B, Haas-Kogan D, Stokoe D, Milanini-Mongiat J, Pages G, O’Rourke DM, Bernhard E, Maity A (2004) Sp1 is involved in Akt-mediated induction of VEGF expression through an HIF-1-independent mechanism. Mol Biol Cell 15(11):4841–4853
Koch AE, Polverini PJ, Kunkel SL, Harlow LA, DiPietro LA, Elner VM, Elner SG, Strieter RM (1992) Interleukin-8 as a macrophage-derived mediator of angiogenesis. Science 258(5089):1798–1801
Strieter RM, Kunkel SL, Elner VM, Martonyi CL, Koch AE, Polverini PJ, Elner SG (1992) Interleukin-8. A corneal factor that induces neovascularization. Am J Pathol 141(6):1279–1284
Maione TE, Gray GS, Petro J, Hunt AJ, Donner AL, Bauer SI, Carson HF, Sharpe RJ (1990) Inhibition of angiogenesis by recombinant human platelet factor-4 and related peptides. Science 247(4938):77–79
Rempel SA, Dudas S, Ge S, Gutierrez JA (2000) Identification and localization of the cytokine SDF1 and its receptor, CXC chemokine receptor 4, to regions of necrosis and angiogenesis in human glioblastoma. Clin Cancer Res 6(1):102–111
Yang SX, Chen JH, Jiang XF, Wang QL, Chen ZQ, Zhao W, Feng YH, Xin R, Shi JQ, Bian XW (2005) Activation of chemokine receptor CXCR4 in malignant glioma cells promotes the production of vascular endothelial growth factor. Biochem Biophys Res Commun 335(2):523–528
Ping YF, Yao XH, Jiang JY, Zhao LT, Yu SC, Jiang T, Lin MC, Chen JH, Wang B, Zhang R, Cui YH, Qian C, Wang J, Bian XW (2011) The chemokine CXCL12 and its receptor CXCR4 promote glioma stem cell-mediated VEGF production and tumour angiogenesis via PI3K/AKT signalling. J Pathol 224(3):344–354
Li X, Kumar A, Zhang F, Lee C, Li Y, Tang Z, Arjuna P (2010) VEGF-independent angiogenic pathways induced by PDGF-C. Oncotarget 1(4):309–314
Wang D, Huang HJ, Kazlauskas A, Cavenee WK (1999) Induction of vascular endothelial growth factor expression in endothelial cells by platelet-derived growth factor through the activation of phosphatidylinositol 3-kinase. Cancer Res 59(7):1464–1472
Heldin CH, Westermark B (1999) Mechanism of action and in vivo role of platelet-derived growth factor. Physiol Rev 79(4):1283–1316
Miyata T, Toho T, Nonoguchi N, Furuse M, Kuwabara H, Yoritsune E, Kawabata S, Kuroiwa T, Miyatake S (2014) The roles of platelet-derived growth factors and their receptors in brain radiation necrosis. Radiat Oncol 9:51
Reigstad LJ, Varhaug JE, Lillehaug JR (2005) Structural and functional specificities of PDGF-C and PDGF-D, the novel members of the platelet-derived growth factors family. FEBS J 272(22):5723–5741
Glass JP, Hwang TL, Leavens ME, Libshitz HI (1984) Cerebral radiation necrosis following treatment of extracranial malignancies. Cancer 54(9):1966–1972
Glantz MJ, Burger PC, Friedman AH, Radtke RA, Massey EW, Schold SC Jr (1994) Treatment of radiation-induced nervous system injury with heparin and warfarin. Neurology 44(11):2020–2027
Bui QC, Lieber M, Withers HR, Corson K, van Rijnsoever M, Elsaleh H (2004) The efficacy of hyperbaric oxygen therapy in the treatment of radiation-induced late side effects. Int J Radiat Oncol Biol Phys 60(3):871–878
Ohguri T, Imada H, Kohshi K, Kakeda S, Ohnari N, Morioka T, Nakano K, Konda N, Korogi Y (2007) Effect of prophylactic hyperbaric oxygen treatment for radiation-induced brain injury after stereotactic radiosurgery of brain metastases. Int J Radiat Oncol Biol Phys 67(1):248–255
McPherson CM, Warnick RE (2004) Results of contemporary surgical management of radiation necrosis using frameless stereotaxis and intraoperative magnetic resonance imaging. J Neurooncol 68(1):41–47
Gonzalez J, Kumar AJ, Conrad CA, Levin VA (2007) Effect of bevacizumab on radiation necrosis of the brain. Int J Radiat Oncol Biol Phys 67(2):323–326
Torcuator R, Zuniga R, Mohan YS, Rock J, Doyle T, Anderson J, Gutierrez J, Ryu S, Jain R, Rosenblum M, Mikkelsen T (2009) Initial experience with bevacizumab treatment for biopsy confirmed cerebral radiation necrosis. J Neurooncol 94(1):63–68
Wong ET, Huberman M, Lu XQ, Mahadevan A (2008) Bevacizumab reverses cerebral radiation necrosis. J Clin Oncol 26(34):5649–5650
Liu AK, Macy ME, Foreman NK (2009) Bevacizumab as therapy for radiation necrosis in four children with pontine gliomas. Int J Radiat Oncol Biol Phys 75(4):1148–1154
Jeyaretna DS, Curry WT Jr, Batchelor TT, Stemmer-Rachamimov A, Plotkin SR (2011) Exacerbation of cerebral radiation necrosis by bevacizumab. J Clin Oncol 29(7):e159–162
Matuschek C, Bolke E, Nawatny J, Hoffmann TK, Peiper M, Orth K, Gerber PA, Rusnak E, Lammering G, Budach W (2011) Bevacizumab as a treatment option for radiation-induced cerebral necrosis. Strahlenther und Onkol 187(2):135–139
Benoit A, Ducray F, Cartalat-Carel S, Psimaras D, Ricard D, Honnorat J (2011) Favorable outcome with bevacizumab after poor outcome with steroids in a patient with temporal lobe and brainstem radiation necrosis. J Neurol 258(2):328–329
Levin VA, Bidaut L, Hou P, Kumar AJ, Wefel JS, Bekele BN, Grewal J, Prabhu S, Loghin M, Gilbert MR, Jackson EF (2011) Randomized double-blind placebo-controlled trial of bevacizumab therapy for radiation necrosis of the central nervous system. Int J Radiat Oncol Biol Phys 79(5):1487–1495
Acknowledgments
This work was partly supported by a Grant-in-Aid for Scientific Research (C) (26462222) given to M.F. from the Japanese Ministry of Education, Culture, Sports, Science and Technology.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Furuse, M., Nonoguchi, N., Kawabata, S. et al. Delayed brain radiation necrosis: pathological review and new molecular targets for treatment. Med Mol Morphol 48, 183–190 (2015). https://doi.org/10.1007/s00795-015-0123-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00795-015-0123-2