
Abstract
Background
Assessing dietary phenylalanine (Phe) tolerance is crucial for managing hyperphenylalaninemia (HPA) in children. However, traditionally, adjusting the diet requires significant time from clinicians and parents. This study aims to investigate the development of a machine-learning model that predicts a range of dietary Phe intake tolerance for children with HPA over 10 years following diagnosis.
Methods
In this multicenter retrospective observational study, we collected the genotypes of phenylalanine hydroxylase (PAH), metabolic profiles at screening and diagnosis, and blood Phe concentrations corresponding to dietary Phe intake from over 10 years of follow-up data for 204 children with HPA. To incorporate genetic information, allelic phenotype value (APV) was input for 2965 missense variants in the PAH gene using a predicted APV (pAPV) model. This model was trained on known pheno-genotype relationships from the BioPKU database, utilizing 31 features. Subsequently, a multiclass classification model was constructed and trained on a dataset featuring metabolic data, genetic data, and follow-up data from 3177 events. The final model was fine-tuned using tenfold validation and validated against three independent datasets.
Results
The pAPV model achieved a good predictive performance with root mean squared error (RMSE) of 1.53 and 2.38 on the training and test datasets, respectively. The variants that cause amino acid changes in the region of 200–300 of PAH tend to exhibit lower pAPV. The final model achieved a sensitivity range of 0.77 to 0.91 and a specificity range of 0.8 to 1 across all validation datasets. Additional assessment metrics including positive predictive value (0.68–1), negative predictive values (0.8–0.98), F1 score (0.71–0.92), and balanced accuracy (0.8–0.92) demonstrated the robust performance of our model.
Conclusions
Our model integrates metabolic and genetic information to accurately predict age-specific Phe tolerance, aiding in the precision management of patients with HPA. This study provides a potential framework that could be applied to other inborn errors of metabolism.
Similar content being viewed by others

Background
Hyperphenylalaninemia (HPA) is characterized by a blood phenylalanine (Phe) concentration exceeding 120 μmol/L. When the blood Phe concentration is between 120 and 360 μmol/L, it is termed mild hyperphenylalaninemia (MHP) and the child only requires regular blood Phe monitoring and general childcare. If the blood Phe concentration persistently exceeds 360 μmol/L, the condition is classified as phenylketonuria (PKU), necessitating treatment with a low Phe diet to reduce blood Phe levels and prevent progressive neurotoxic damage [1]. Approximately 0.45 million individuals with HPA exist worldwide, of whom at least two-thirds require treatment with a low Phe diet [2].
Phe tolerance, representing the dietary Phe intake that maintains a blood Phe concentration within a target range, is a crucial metric for guiding dietary management in patients with HPA [3]. Determining the optimal Phe tolerance range is challenging owing to it changes with age and stabilizes after 5 years, and requires frequent blood Phe monitoring [4, 5].
Variants of the phenylalanine hydroxylase (PAH) gene are the primary cause of HPA. One of the most widely used parameters in the genotype–phenotype relationship is the allelic phenotype value (APV) [6], developed using an algorithm based on pretreatment blood Phe concentration data from 9,336 patients with PAH deficiency, as recorded in the BIOPKU database [7]. However, nearly one-third of PAH variants lack APV data, and the development of APV does not include the actual Phe dietary tolerance of the patients.
Therefore, this study aims to address these limitations by first developing a machine-learning model to predict APV scores for novel variants on PAH. Additionally, a prediction model was developed, incorporating pretreatment Phe concentration, PAH genotypes, predicted APV, and blood Phe levels corresponding to dietary Phe intake during each follow-up from an HPA cohort in northwestern China, where the incidence (1:5230 [8]) is significantly higher than the global average (1:23,930 [2]). Finally, the performance of this prediction model was validated in three independent HPA cohorts.
Methods
Study design
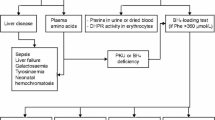
A multicenter retrospective study was conducted to model the follow-up blood Phe test results of patients with HPA under dietary control. The development dataset for this study was collected from clinical records of patients diagnosed at the Neonatal Disease Screening Center of the Children’s Hospital of Xinjiang Uyghur Autonomous Region (Hospital A) between January 2010 and December 2021. The validation datasets were obtained from records of patients diagnosed between December 2021 and March 2023(validation dataset I), other outpatients with HPA at the same hospital (validation dataset II), and patients from the Children’s Hospital of Fudan University (Hospital B) between September 2006 and September 2022 (validation dataset III) (Fig. 1). This study was approved by the Ethics Board of the People’s Hospital of Xinjiang Uyghur Autonomous Region (GZR2017010).

Individual inclusion and exclusion process for training and validation datasets. HPA, hyperphenylalaninemia; Phe, phenylalanine; PKU, phenylketonuria; PAH, phenylalanine hydroxylase
Participants and data collection
Patients were diagnosed with HPA according to the Chinese national consensus statement and European guidelines [9, 10]. Blood Phe concentration > 360 μmol/L was defined as phenylketonuria (PKU), requiring treatment with a low-Phe diet or tetrahydrobiopterin, such as Sapropterin dihydrochloride, to lower blood Phe concentration. Without treatment, progressive neurotoxic damage may occur. Patients were further filtered based on the following inclusion criteria: (1) individuals with one homozygous or two heterozygous variants in the PAH gene, and (2) complete clinical information, including pretreatment blood Phe concentration, diagnosis time, blood Phe concentration, and corresponding dietary Phe intake measured at each follow-up. Patients were excluded if they underwent any treatment other than diets such as Sapropterin or Pegvaliase. Included individuals were further reviewed for the following parameters: (1) demographic information, sex, value, and age at the re-screening test of blood Phe concentration via fluorometric assay; value and age at the diagnostic test of blood Phe and tyrosine concentration using TMS; family history; and consanguinity. The Phe concentration from re-screening test was defined as screening Phe, while the Phe concentration from the diagnostic test was defined as diagnostic Phe.
Outcomes of model
The outcome of the model was defined as the blood Phe testing results from each subsequent follow-up visit. Normal: The target ranges for blood Phe concentrations are 120–240 μmol/L for ages 0–12 months, 120–360 μmol/L for ages 1–12 years, and 120–600 μmol/L for individual > 12 years [9].
High: The Phe levels exceeding 240 μmol/L for ages 0–12 months, 360 μmol/L for ages 1–12 years, and 600 μmol/L for individuals > 12 years. Low: The Phe levels below 120 μmol/L for all age groups.
Feature collection
Metabolic features
The screening Phe levels (Phe1) from dried blood spots and each Phe concentration from fresh peripheral blood during the follow-up were measured using a fluorometric assay with the Neonatal Phenylalanine kit (Fenghua, China). According to the protocol of the manufacturer, calibrators, high control, low control, and samples were simultaneously incubated with the extraction solution. After incubating for 120 min at 37 °C, 200 μL of copper reagent was added into each well and incubated for 60 min at 26 °C without shaking. Fluorescence was measured using an Auto Fluoroimmunoassay Analyzer (Auto TRFIA-2, Fenghua, China).
The diagnostic Phe levels (Phe2) were measured using TMS with the NeoBase Non-derivatized TMS S kit (PerkinElmer, USA). The assay was performed according to the protocolof the manufacturer. The concentrations of Phe and Tyr in samples were measured with an ACQUITY TQD mass spectrometer (Waters, Milford, MA, USA), and the data were analyzed using the MassLynx 4.1 version software.
Genetic features
PAH genotyping and variant annotations
PAH genotyping was performed using Sanger sequencing of all PAH exons before 2016 and next-generation sequencing subsequently. Details on sample collection, DNA isolation, PCR amplification, product sequencing, and data analysis were conducted as described previously [8]. The pathogenicity of each PAH variant was reassessed according to the ClinVar annotations (https://www.ncbi.nlm.nih.gov/clinvar/). Novel variants and variants of uncertain significance in the ClinVar were reassigned based on the guidelines of the ACMG and Phenylketonuria Variant Curation Expert Panel of ClinGen (https://www.clinicalgenome.org/affiliation/50015). RefSeq NM_000277.1 was used the reference transcript for PAH. Variants were annotated using the variant effect predictor (VEP) [11] to integrate information such as variant types, population frequencies, and computational predicting scores from dbNSFP [12]. Since HPA is an autosomal recessive disorder, biallelic effects of the PAH gene should be considered. Therefore, our model included variants from both alleles.
Allelic phenotype value
APV is a model used for predicting phenotypes based on genotypes in PKU, utilizing the frequencies of the metabolic phenotypes associated with genotypes in a functionally hemizygous state, as identified from the bioPKU databases. For novel variants lacking a known APV, a predicted APV (pAPV) was imputed using a regression machine-learning model, which involved the following four steps. (1) Preparing training and testing datasets: 219 missense variants with known APV scores from Garbade et al. [6] were randomly divided into training (80%, n = 176) and testing (20%, n = 43) datasets. (2) Processing feature: Initially, 41 features related to conservation, protein structure, pathogenicity, population allele frequency, and residue interactions in (Additional file 1: Table S1) were considered for modeling. Feature selection included evaluating the contributions and correlations of these features. Features with average Shapley Additive exPlanations (SHAP) values [13] > 0.01 were retained, while highly correlated features (Spearman’s correlation > 0.70) were removed to reduce redundancy. This process resulted in a final set of 31 features for modeling. (3) Model development: Using the selected features, a gradient boosting machine algorithm was trained. Hyperparameters were optimized through tenfold validation and grid search to select the model with the lowest root-mean-square error (RMSE). (4) Precomputed scores for all missense variants: To compute the pAPV for missense variants, all possible single nucleotide variants were simulated in the PAH coding sequence and selected with those missense consequences according to VEP annotation. The pAPV model was then applied to the annotated features to calculate the pAPV for each variant. Nonsense variants and variants with a SpliceAI score > 0.5 were assigned a pAPV of zero. Additionally, if a missense variant was predicted to impact splicing (SpliceAI > 0.5), its pAPV was assigned as zero.
Phe intake estimation
Based on the consensus statement [9], children with PKU were treated with free-Phe amino acid formula (Periflex® and Periflex Ad®) and low-Phe rice and flour (Wei Si Duo®) at different ages. The recommended Phe intake ranges from 130 ~ 430 mg/day for infants aged 0 to 3 months. For other age groups, the Phe intake followed the guidelines outlined in the consensus statement.
The dietary Phe intake was assessed by a clinical follow-up expert during each visit using sophisticated dietary calculation software called Rainbow Diet Calculator, developed by Nutricia and food composition reference [14]. Dietary Phe intake was managed according to the guidelines [9, 15] and calculated based on information from 24-h dietary recall for 3 to 4 days. Nutrition professionals reviewed all Phe intake in this study.
Model development
After feature collection, 10 features were used including the blood Phe level from the initial screening (Phe1), the age at screening (Age1), Phe levels from the TMS test (Phe2), and age at TMS test (Age2), APVs for both allele (APV1 and APV2), alleles frequencies for both allele (AF1 and AF2), and Phe intake and age at each follow-up event. Missing values of APV were imputed using the method described earlier (see the section of APVs). The values of the 10 features were normalized. They were then transformed using the Yeo-Johnson method. Multiclass classification models were trained on the training dataset using three different supervised machine-learning algorithms: gradient boosting machine, random forest, and supporting vector machine. Hyperparameters were through tenfold validation and grid search to select the model with the highest area under the curve (AUC). Additional file 1: Table S2 shows the hyperparameters selected for the final models.
Model validation
To evaluate model robustness, validation was performed on three independent datasets (datasets I–III) using the overall accuracy and kappa metric. Furthermore, the one-versus-rest strategy was employed to assess the performance of the multiclass model using the receiver operating characteristic curve (ROC) and precision-recall (PR) Curve for high and low classes. Sensitivity, specificity, positive prediction value, negative prediction value, F1 score, and balanced accuracy were also utilized to assess model performance across the three independent datasets.
Personalized optimal Phe intake estimation
To determine the upper and lower thresholds for Phe intake for each patient, the class boundary in the Phe intake versus age plot is estimated. Simulated datasets were generated by fixing the patient-specific features (Phe1, Age1, Phe2, Age2, APV1, APV2, AF1, AF2) and iterating through event-specific features. This process involved varying Phe intake from 0 to 1000 mg across ages ranging from 0 to 180 months. The model was then used to predict outcomes for each Phe intake and age pair. The curve connecting the lowest Phe intake that resulted in an H-class outcome at each age was considered the upper boundary for optimal Phe intake. Similarly, the curve connecting the highest Phe intake, which resulted in an L-class outcome, was considered the lower boundary for optimal Phe intake.
Statistical analysis
The performance of pAPV models was measured using mean absolute error (MAE), RMSE, and R2. All statistical analyses were performed in R (version 4.2) [R Core Team (2023). R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria. < https://www.R-project.org/ > .]. Machine learning models were built using the caret package (v6.0–88) [16]. Feature significance was calculated with SHAP (Shapley Additive exPlanations) [17] using the xgboost package [18]. Plots were generated by using the ggplot2 package v3.3.5 [19].
Results
Characteristics of training and validation cohorts
Overall, 275 patients were diagnosed with HPA between January 2010 and December 2021. Of these 139 patients (68 male and 71 female) underwent PAH genotyping and their 3177 following-up blood Phe test results were used as developmental datasets to train the prediction model. To assess the prediction model, three independent validation datasets were collected from 28 outpatient patients (383 follow-up events) who missed newborn screening tests, 19 patients (168 follow-up events) diagnosed after 2022, and 18 patients (55 follow-up events) from another health center (Fig. 1 and Table 1), respectively. Table 1 shows the characteristics of the training and testing cohorts. The follow-up Phe test results were categorized as high, normal, and low if the testing results were higher, within, or lower than the target ranges, respectively. Additional file 2: Fig. S1 shows the distribution of blood Phe levels for all patients at each follow-up. There were 1951 (61.4%) high, 831 (26.2%) normal, and 395 (12.4%) low events in the training cohorts. There were 266 (69.5%), 75 (44.6%), and 22 (40%) high events, for the validation I, II, and III cohorts, and 26 (6.8%), 36 (21.4%), and 5 (9.1%) low events for the corresponding validation cohorts.
pAPV of novel variants of PAH
Thirteen variants were detected in our cases without APVs, including one splicing, two stop-gained, and 10 missense variants. Considering most null variants are severe, an APV of 0 was assigned to the stop-gained variants. A model incorporating 31 features was trained using 174 missense variants with known APV from the BIOPKU database, to predict the pAPV for the ten missense variants without APV (Additional file 1: Table S3). The performance of the prediction model, based on the test datasets yielded an RMSE of 2.38 (Fig. 2B). SHAP values revealed the contributions of various features to the pAPV model (Fig. 2A). Among all the features, the EVE score, an unsupervised deep learning model that captured evolutionary patterns of protein sequences from 140,000 species, contributed the most to the pAPV, followed by the EVM epistatic score, which considers sequence co-variation through evolution, allele frequencies in the gnomAD database, changes in amino acid isoelectric point and the GERP score.

pAPV model of missense variants in PAH. A Feature contributions of the pAPV model. B Comparison of predicted pAPV score and known APV score of variants from the test dataset. C Red dots indicate the variants with known APV in the BIOPKU database, while green dots represent variants with pAPV predicted in this study. The blue line illustrates the trend of pAPVs along the protein sequence. The iron/copper binding sites (IPR018301) within residues 281–292 residues are highlighted in yellow
The pAPV was computed for 2965 missense variants to interpret the severity of all possible missense variants in PAH. The landscape of pAPV along the protein sequence was shown in Fig. 2. The variants that cause amino acid changes in the region between residues 200 and 300 are more likely to have lower pAPV, indicating that this is a functionally important region of PAH. This region includes residues 281–292, which are annotated as iron/copper binding sites (IPR018301) in the InterPro database (Fig. 2C).
Phe model prediction and validation
To predict age-specific Phe tolerance, machine-learning models were developed using data from 3177 follow-up events with 10 features (Fig. 3A), including the initial blood Phe level age at screening, Phe levels from the TMS test, age at TMS test, APVs and allele frequencies for both alleles, Phe intake, and age at each follow-up event. These data were collected from screening, genetic tests, and follow-up processes. Three commonly used machine learning algorithms, including gradient boost machine (GBM), random forest, and support vector machine (SVM) were employed. The GBM model was selected as the final model based on validation results from three independent datasets (Additional file 1: Table S2). In the validation dataset I (Fig. 3B), the model achieved an area under the receiver operating characteristic curve (ROC-AUC) of 0.965 and an area under the precision-recall curve (PR-AUC) of 0.965 for predicting high events versus normal and low events. It also showed high performance with a ROC-AUC (0.955) and PR-AUC (0.834) for predicting low events versus normal and high events, indicating strong predictive ability for abnormal blood Phe test results in outpatient cases. The model performed well when used in predicting high (ROC-AUC = 0.943, PR-AUC = 0.976) and low (ROC-AUC = 0.974, PR-AUC = 0.868) categories in the validation dataset II (Fig. 3C). The model showed robust performance in the validation dataset III (Fig. 3D) which consists of patients from a different health center (high: ROC-AUC = 0.909, PR-AUC = 0.909; low: ROC-AUC = 0.912, PR-AUC = 0.835). With a probability cutoff of 0.5, the model achieved sensitivity between 0.77 and 0.91 and specificity between 0.8 and 1 across all validation sets. Additional metrics, including positive predictive value (0.68–1), negative predictive values (0.8–0.98), F1 score (0.71–0.92), and balanced accuracy (0.8–0.92), further confirmed the strong performance of the model (Table 2).

Model development and evaluation. A Study design: Data collected during clinical procedures for each sample, spanning from birth to the last follow-up, were used to construct the model. B–D ROC and precision-recall curves: These curves demonstrate the efficacy of the model in predicting events above (H) and below (L) the target region in validation datasets I, II, and III, respectively. Phe1: Blood phenylalanine concentration from re-screening, measured by a fluorometric assay. Age 1: Age of the individual at the time of the re-screening test. Phe2: Blood phenylalanine concentration from diagnosis, measured by tandem mass spectrometry. Age 2: Age of the individual at the time of the diagnostic test. APV1: Allelic phenotype value score for the first allele. APV2: Allelic phenotype value score for the second allele. AF1: Variant allelic frequency from the gnomAD database for the first allele. AF2: Variant allelic frequency from the gnomAD database for the second allele
Sensitivity analyses
To evaluate the influence of metabolic and genetic factors on the prediction model, alternative models were constructed using subsets of the full feature set. The baseline model comprised only Phe intake and age at each follow-up event as features. The metabolic model expands on this, by incorporating additional features such as Phe levels from the initial screening, age at screening, Phe levels from TMS tests, and age at TMS tests. The genetic model has augmented the baseline model by integrating APVs and allele frequencies for both alleles. Validation results showed that the full model is generally more accurate and robust than models using only metabolic or genetic features. However, the metabolic model had slightly higher ROC-AUC (0.947) and PR-AUC (0.978) values compared to that of the full model (ROC-AUC = 0.943, PR-AUC = 0.976) for predicting events above the target range in validation dataset I. For predicting events below the target range, in validation dataset II, the full model performed best on all other metrics across the three validation datasets.
Validation and explanation for model performance on three independent datasets
We showed the prediction results of individual cases under three different conditions from the training cohorts. For patients requiring low-protein diet therapy during long-term follow-up, the model predicts the upper and lower bounds for the ideal Phe intake over time, using inputs such as pretreatment blood Phe concentration and PAH genotypes. Figure 4A illustrates the bounded curves that successfully distinguish between historical high and low events. For a patient who did not require a restricted diet, the model generated only a lower bound, as the blood Phe concentration was below 360 μmol/L during follow-up (Fig. 4B). The patient in Fig. 4C received low-protein diet therapy for a period and was able to maintain blood Phe concentration between 120 and 360 μmol/L after discontinuing the treatment. In this case, two curves are shown during the diet therapy period, while only the lower limit curve is displayed after the diet treatment is stopped. Figure 4D–F presents cases from the validation datasets. For example, the predicted upper and lower bounds accurately reflect the actual diet tolerance for case p167, who required dietary Phe intake control of 200–250 mg/day at the age of 1 year, between 220 and 400 mg/day at the age of 2 years and less than 450 mg/day after 7 years. The analyzed data was provided in (Additional file 1: Table S4).

Upper and lower bounds for optimal Phe intake. A–C Model results for three different conditions of training sets. D–F Model results for three validation datasets of patients undergoing dietary therapy. The X-axis represents age, and the Y-axis represents dietary Phe intake. Dashed lines illustrate the upper (red) and lower (green) bounds for dietary Phe intake calculated from the full model. Phe intake within these bounds indicates a safe intake amount to maintain blood Phe levels within the target range. Dots represent actual Phe intake records from follow-up visits, categorized high (red), low (green), or normal (blue) based on Phe levels relative to age-specific reference ranges. MHP = 0: the patient required long-term low-protein diet therapy. Mild hyperphenylalaninemia (MHP). MHP = 1: the patient did not require low-protein diet therapy. MHP = 2: the patient initially accepted low-protein diet therapy, however, eventually discontinued it
Discussion
Managing HPA is a major challenge owing to the time required to determine appropriate dietary Phe intake. High Phe levels can damage the brain through several mechanisms: Phe causes disturbances of neuronal dendritic outgrowth, leading to white matter disruption [20], Phe-mediated inhibition of pyruvate kinase or other enzymes of glycolysis, leading to cerebral metabolism disorder [21], and Phe-mediated competition for binding to LAT1 and impair the flux of the other large neutral amino acids into the brain, leading to their deficiency in the brain [22]. Alternatively, low Phe levels can affect skin and growth [23]. Our machine learning model addresses this challenge by providing personalized recommendations for the dietary Phe tolerance ranges in children with HPA over 10 years following diagnosis. With accuracies of 0.81, 0.85, and 0.78 across three external validation datasets, our model demonstrated robust performance. It achieved a mean AUC of 0.94 for distinguishing between high and low blood Phe concentrations, highlighting its effectiveness in facilitating dietary management and its potential to enhance patient outcomes. By offering predictive results, our model can alleviate the burden on clinicians, who would otherwise need to calculate each dietary Phe intake, monitor blood Phe concentrations, and adjust diets iteratively during long-term follow-up.
With the widespread application of artificial intelligence (AI) and big data in the medical field, machine-learning models play important roles in disease management and prognosis. Some machine learning approaches can be used to predict glucose trends in hospitals, helping to mitigate and prevent suboptimal hypoglycemic and hyperglycemic outcomes [24]. Additionally, AI-based nutritionist programs that utilize advanced language and image recognition models have been developed. These programs can identify ingredients from images of meals of patients and provide nutritional guidance and dietary recommendations [25]. For some rare diseases, prediction models using clinical and genetic biomarkers can aid in the early diagnosis of SCN1A-related epilepsies, such as Dravet syndrome, which is characterized by drug-resistant seizures and intellectual disability, or milder genetic epilepsy with febrile seizures plus, which is characterized by normal cognition [26]. In summary, for diseases that require long-term control through diet or medication, machine-learning models offer significant advantages.
Our results showed that the full model performs best in most scenarios, while alternative models remain useful in certain situations where metabolic or genetic data are inaccessible (Fig. 3). The strong performance of the full model may be related to the automatic inclusion of patients with potential interallelic complementation of PAH genes and responsiveness to BH4. The widespread implementation of metabolic-based newborn screening for PKU highlights the broader applicability of the metabolic model [10, 27]. The metabolic model achieves ROC-AUC scores exceeding 0.883 and PR-AUC scores above 0.88 for predicting events above the target range across all validation sets. Similarly, for predicting events below the target range, the model achieves ROC-AUC scores above 0.94 and PR-AUC scores exceeding 0.80 across all validation datasets.
Various factors such as feeding, preterm birth, and uterine environments can influence screening Phe levels [28, 29]. In cases heavily influenced by these factors, the genetic model could be useful [30]. The genetic model demonstrated predictive efficacy, with ROC-AUC values of 0.928, 0.937, and 0.707 for events above the target range, and PR-AUC values of 0.970, 0.936, and 0.689 across validation sets I, II, and III, respectively. It also predicted events below the target range with ROC-AUC values of 0.959, 0.932, and 0.922 and PR-AUC values of 0.85, 0.677, and 0.837 for validation sets I, II, and III respectively. Moreover, the advancements in sequencing technology have made it possible to resolve genotypes in prenatal cases through genetic testing [31]. However, metabolic phenotypes may not be observable in such cases. Our genetic model can predict Phe tolerance, which reflects disease severity, based on genotypic information. By obtaining the genotype of the fetus from a prenatal genetic test, our model can forecast Phe metabolism. Therefore the genetic model holds a potential utility for prenatal screening [32] for HPA.
This study utilized an APV as a key genetic index, derived from metabolic phenotype frequencies observed in functionally hemizygous states. Despite the significant contributions from the BioPKU database, many variants, including 13 from the cohort, remain uncharacterized. To address this gap, we developed a novel machine-learning model was developed, leveraging information from known BioPKU variants to assign APV scores to novel variants. Alterations in protein sequences are hypothesized to directly reflect phenotypic variations in hemizygous states. Consequently, protein sequence and structural data were used to impute APV scores for novel variants. This approach enhances the predictive utility of in silico scores for assessing phenylalanine tolerance, particularly in large-scale genetic testing scenarios where novel PAH variants are identified.
However, since PAH is a recessive genetic disorder, metabolic phenotypes should ideally be considered in their biallelic states. Quantifying the combined effects of two alleles is a well-recognized challenge in genetic studies. Garbade et al. introduced the genotypic phenotype value (GPV) as APVmax, which represents the higher APV of the two alleles [6]. Fang et al.incorporate allelic interactions using features derived from allelic mutation linkage graphs [33]. The reliance on weighted contributions from known variant combinations in this method raises questions about its applicability to novel variants and uncharacterized genotype combinations. In developing our phenylalanine tolerance model, potential complex interactions between alleles were considered. Instead of, using integrated GPV, separate APVs were selected for each allele. This approach avoids assuming allele dominance and enables the model to learn the potential additive contributions of individual alleles to phenotypic variation.
Studies have reported the phenomenon of interallelic complementation in PAH [34, 35], however, this study did not comprehensively explore its influence. Our models may capture patterns of allelic interaction by treating the APVs of the two alleles as separate features. Furthermore, integrating metabolic phenotype data could partially account for the phenotypic variability resulting from genetic interactions. However, the current data are insufficient for a thorough investigation and reliable conclusions on allelic interactions. More data are needed for future research in this area.
This study had two other limitations. Firstly, the model did not account for geographical factors and variations in dietary patterns. However, testing the model on datasets from two hospitals in distinct geographical regions yielded robust results, indicating its applicability to patients from diverse backgrounds. Secondly, the model was developed exclusively using PAH pathogenic variants and did not account for the varied genetic backgrounds of patients or the potential impact of undiscovered modifier genes related to the PAH gene [34, 35].
Conclusions
In this cohort study, we employed 10-year follow-up data from 264 children with PAH deficiency. The model integrates metabolic and genetic information to predict age-specific Phe tolerance, aiding in the precise management of patients. This approach serves as a model for developing similar strategies to improve dietary management in other inherited metabolic diseases, including maple syrup urine disease (leucine restriction) [36] or glutaric aciduria type 1 (lysine restriction) [37].
Availability of data and materials
The datasets used or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- APV:
-
Allelic phenotype value
- AUC:
-
Area under the curve
- HPA:
-
Hyperphenylalaninemia
- MAE:
-
Mean absolute error
- MHP:
-
Mild hyperphenylalaninemia
- PAH:
-
Phenylalanine hydroxylase
- PAH :
-
Phenylalanine hydroxylase
- pAPV:
-
Predicted APV
- Phe:
-
Phenylalanine
- PKU:
-
Phenylketonuria
- PR:
-
Precision-recall
- RMSE:
-
Root mean squared error
- ROC:
-
Receiver operating characteristic curve
- SHAP:
-
Shapley Additive exPlanations
- VEP:
-
Variant effect predictor
References
Rajabi F, Rohr F, Wessel A, Martell L, Dobrowolski SF, Guldberg P, et al. Phenylalanine hydroxylase genotype-phenotype associations in the United States: a single center study. Mol Genet Metab. 2019;128(4):415–21.
Hillert A, Anikster Y, Belanger-Quintana A, Burlina A, Burton BK, Carducci C, et al. The Genetic Landscape and Epidemiology of Phenylketonuria. Am J Hum Genet. 2020;107(2):234–50.
Camp KM, Parisi MA, Acosta PB, Berry GT, Bilder DA, Blau N, et al. Phenylketonuria Scientific Review Conference: state of the science and future research needs. Mol Genet Metab. 2014;112(2):87–122.
van Spronsen FJ, van Rijn M, Dorgelo B, Hoeksma M, Bosch AM, Mulder MF, et al. Phenylalanine tolerance can already reliably be assessed at the age of 2 years in patients with PKU. J Inherit Metab Dis. 2009;32(1):27–31.
Darling PB, Dunn M, Gilani GS, Ball RO, Pencharz PB. Phenylalanine kinetics differ between formula-fed and human milk-fed preterm infants. J Nutr. 2004;134(10):2540–5.
Garbade SF, Shen N, Himmelreich N, Haas D, Trefz FK, Hoffmann GF, et al. Allelic phenotype values: a model for genotype-based phenotype prediction in phenylketonuria. Genet Med. 2019;21(3):580–90.
Available from: http://www.biopku.org/home/pah.asp.
Su Y, Wang H, Rejiafu N, Wu B, Jiang H, Chen H, et al. The molecular epidemiology of hyperphenylalaninemia in Uygur population: incidence from newborn screening and mutational spectra. Ann Transl Med. 2019;7(12):258.
Subspecialty Group of Newborn Screening, Society of Birth Defects Prevention and Control, Chinese Preventive Medicine Association; Subspecialty Group of Clinical Nutrition, the Society of Pediatrics, Chinese Medical Association; Subspecialty Committee of Clinical Biochemistry and Genetics, the Society of Medical Genetics, Chinese Medical Doctor Association; Subspecialty Group of Clinical Genetics, The Society of Adolescent Medicine, Chinese Medical Doctor Association. Consensus statement on dietary treatment and nutritional management for phenylalanine hydroxylase deficiency. Zhonghua Er Ke Za Zhi. 2019,57(6):405-9. Chinese. https://doi.org/10.3760/cma.j.issn.0578-1310.2019.06.002.
van Wegberg AMJ, MacDonald A, Ahring K, Belanger-Quintana A, Blau N, Bosch AM, et al. The complete European guidelines on phenylketonuria: diagnosis and treatment. Orphanet J Rare Dis. 2017;12(1):162.
McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, et al. The Ensembl Variant Effect Predictor. Genome Biol. 2016;17(1):122.
Liu X, Li C, Mou C, Dong Y, Tu Y. dbNSFP v4: a comprehensive database of transcript-specific functional predictions and annotations for human nonsynonymous and splice-site SNVs. Genome medicine. 2020;12(1):103.
Lundberg S, Lee S-I. A Unified Approach to Interpreting Model Predictions. 31st Conference on Neural Information Processing Systems (NIPS 2017); Long Beach, CA, USA.2017. p. 4768–77. https://dl.acm.org/doi/10.5555/3295222.3295230.
Yuexin Y, Guangya W, Xingchang P. China Food Composition. 2nd ed. Peking University Medical Press; 2009. ISBN:9787811167276.
MacDonald A, van Wegberg AMJ, Ahring K, Beblo S, Bélanger-Quintana A, Burlina A, et al. PKU dietary handbook to accompany PKU guidelines. Orphanet J Rare Dis. 2020;15(1):171.
Kuhn M. Building Predictive Models in R Using the caret Package. J Stat Softw. 2008;28(6):1–26.
Singh R, Lanchantin J, Sekhon A, Qi Y. Attend and Predict: Understanding Gene Regulation by Selective Attention on Chromatin. Adv Neural Inf Process Syst. 2017;30:6785–95.
Chen T, Guestrin C. XGBoost: A Scalable Tree Boosting System. Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining. 2016. p. 785–94. https://dl.acm.org/doi/proceedings/10.1145/2939672.
Wickham H. Ggplot2: Elegant graphics for data analysis. Second ed. Switzerland AG: Springer Nature; 2016.
Hörster F, Schwab MA, Sauer SW, Pietz J, Hoffmann GF, Okun JG, et al. Phenylalanine reduces synaptic density in mixed cortical cultures from mice. Pediatr Res. 2006;59(4 Pt 1):544–8.
Miller AL, Hawkins RA, Veech RL. Phenylketonuria: phenylalanine inhibits brain pyruvate kinase in vivo. Science (New York, NY). 1973;179(4076):904–6.
de Groot MJ, Hoeksma M, Blau N, Reijngoud DJ, van Spronsen FJ. Pathogenesis of cognitive dysfunction in phenylketonuria: review of hypotheses. Mol Genet Metab. 2010;99(Suppl 1):S86–9.
van Spronsen FJ, Blau N, Harding C, Burlina A, Longo N, Bosch AM. Phenylketonuria. Nat Rev Dis Prime. 2021;7(1):36.
Zale A, Mathioudakis N. Machine Learning Models for Inpatient Glucose Prediction. Curr DiabRep. 2022;22(8):353–64.
Sun H, Zhang K, Lan W, Gu Q, Jiang G, Yang X, et al. An AI Dietitian for type 2 diabetes mellitus management based on large language and image recognition models: preclinical concept validation study. J Med Internet Res. 2023;25:e51300.
Brunklaus A, Pérez-Palma E, Ghanty I, Xinge J, Brilstra E, Ceulemans B, et al. Development and validation of a prediction model for early diagnosis of SCN1A-related epilepsies. Neurology. 2022;98(11):e1163–74.
Singh RH, Cunningham AC, Mofidi S, Douglas TD, Frazier DM, Hook DG, et al. Updated, web-based nutrition management guideline for PKU: An evidence and consensus based approach. Mol Genet Metab. 2016;118(2):72–83.
Ponzone A, Spada M, Roasio L, Porta F, Mussa A, Ferraris S. Impact of neonatal protein metabolism and nutrition on screening for phenylketonuria. J Pediatr Gastroenterol Nutr. 2008;46(5):561–9.
Porta F, Ponzone A, Spada M. Neonatal phenylalanine wash-out in phenylketonuria. Metab Brain Dis. 2020;35(7):1225–9.
Blau N. Genetics of Phenylketonuria: Then and Now. Hum Mutat. 2016;37(6):508–15.
Roman TS, Crowley SB, Roche MI, Foreman AKM, O’Daniel JM, Seifert BA, et al. Genomic sequencing for newborn screening: results of the NC NEXUS Project. Am J Hum Genet. 2020;107(4):596–611.
Jelin AC, Sagaser KG, Wilkins-Haug L. Prenatal Genetic Testing Options. Pediatr Clin North Am. 2019;66(2):281–93.
Shen N, Heintz C, Thiel C, Okun JG, Hoffmann GF, Blau N. Co-expression of phenylalanine hydroxylase variants and effects of interallelic complementation on in vitro enzyme activity and genotype-phenotype correlation. Mol Genet Metab. 2016;117(3):328–35.
Klaassen K, Djordjevic M, Skakic A, Kecman B, Drmanac R, Pavlovic S, et al. Untreated PKU patients without intellectual disability: SHANK gene family as a candidate modifier. Mol Genet Metab Rep. 2021;29:100822.
Li Y, Tan Z, Zhang Y, Zhang Z, Hu Q, Liang K, et al. A noncoding RNA modulator potentiates phenylalanine metabolism in mice. Science (New York, NY). 2021;373(6555):662–73.
Mengler K, Garbade SF, Gleich F, Thimm E, May P, Lindner M, et al. Treatment outcomes for maple syrup urine disease detected by newborn screening. Pediatr. 2024;154(2):e2023064370.
Boy N, Mühlhausen C, Maier EM, Ballhausen D, Baumgartner MR, Beblo S, et al. Recommendations for diagnosing and managing individuals with glutaric aciduria type 1: Third revision. J Inherit Metab Dis. 2023;46(3):482–519.
Acknowledgements
We are sincerely grateful to all the parents who allowed their children to participate in our study. We want to thank all maternity and child healthcare providers in southern Xinjiang. They help us to recall the patients and collect peripheral blood regularly. We also want to thank Xingran Dong and Wenbo Sheng for providing constructive suggestions.
Funding
This study was supported by the Natural Science Foundation of Xinjiang Uygur Autonomous Region (2021D01A137), the National Natural Science Foundation of China (81741102), and the Science and Technology Introduction Innovation Project of People’s Hospital in Xinjiang Uygur Autonomous Region (20170312).
Author information
Authors and Affiliations
Contributions
YS, XA and HJ collected and curated the data. YS checked the data of Phe intake. YS and YW performed the statistical analyses and wrote the manuscript. JH, WL and LL followed up the patients. HW, WZ, LL,YS and YW designed the study and revised the manuscript. WZ and LL supervised the project. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Ethics Board of the People’s Hospital of Xinjiang Uyghur Autonomous Region (GZR2017010).
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
12916_2024_3602_MOESM1_ESM.xlsx
Additional file 1: Table S1-S4. Table S1-Features for developing the pAPV model. Table S2-The hyperparameters selected for the final models. Table S3-Features and pAPV for 10 missense variants in this study. Table S4-The analyzed data of each patient in the model
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Su, Y., Wang, Y., He, J. et al. Development and validation of machine-learning models of diet management for hyperphenylalaninemia: a multicenter retrospective study. BMC Med 22, 377 (2024). https://doi.org/10.1186/s12916-024-03602-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12916-024-03602-w