
Abstract
Background
Idiopathic pulmonary fibrosis (IPF), an interstitial lung disease, is characterized by the exacerbation of progressive pulmonary fibrosis (PF). IPF primarily affects older individuals and can lead to respiratory failure. This study aimed to assess the effects of triiodothyronine (T3) treatment on the lung microbiome of mice with PF.
Methods
Mice were perfused with bleomycin (BLM) to establish a PF model. Using a randomized design, 40 female specific pathogen-free (SPF) C57BL6/N mice were divided into four groups: saline, saline + T3, BLM, and BLM + T3. Histological morphology was assessed through Hematoxylin and Eosin staining as well as Masson’s Trichrome staining. For the identification of lung bacteria, 16S rRNA gene sequencing was employed. An Enzyme-Linked Immunosorbent Assay was used to measure total T3 (TT3), free T3 (FT3, and reverse T3 (rT3) levels in the peripheral serum.
Results
T3 treatment ameliorated BLM-induced lung fibrosis and structural damage. The microbiome experienced a decrease in the abundance of Proteobacteria, Bacteroides, and Actinomycetes and an increase in the abundance of Firmicutes when exposed to BLM; however, T3 treatment reversed this effect. The four groups showed no significant difference in alpha microbiome diversity (P > 0.05). Serum concentrations of TT3 and FT3 were positively correlated with microbiome abundance (P < 0.05). Administration of T3 enhanced the microbiota in PF without affecting the diversity and biological functions of the microbiome (P > 0.05).
Conclusion
The administration of T3 demonstrated a favorable impact on the lung microbiota of mice afflicted with PF, thereby partially substantiating the potential role of T3 as a therapeutic agent in the management of PF.
Similar content being viewed by others


Background
Pulmonary fibrosis (PF) is a devastating lung disease in which fibrotic lesions progressively expand because of the excessive migration and accumulation of multiple mesenchymal cells involved in collagen deposition and irreversible scarring of the lungs [1]. Recently, there has been a reported rise in the incidence of idiopathic pulmonary fibrosis (IPF). In Europe and North America, it is estimated to affect 2.8 to 18 cases per 100,000 individuals annually, whereas in Asia and South America, the reported incidence ranges from 0.5 to 4.2 cases per 100,000 population per year [2]. The pathogenesis of PF are not yet fully elucidated; however, it is commonly recognized as a consequence of genetic abnormalities, autoimmune conditions, or chronic exposure to detrimental gases and particulate matter [3]. IPF is the most common idiopathic form of PF and is frequently pathologically indistinguishable from other forms, especially in the later stages of the disease [4]. The median survival for individuals with IPF without transplantation is approximately three years, rendering it a non-cancerous pulmonary disorder associated with the most ominous prognosis among such diseases [5, 6]. Therefore, there is an urgent need to develop new and effective drugs to prevent or delay disease progression.
The lungs have recently been considered metabolically active organ [7]. Thyroid hormones THs contribute to energy homeostasis by affecting many metabolically relevant tissues [8]. Triiodothyronine (T3) is a thyroid hormone that is secreted by the thyroid gland. T3 regulates PF development through several pathways [9]. T3 regulates the production and activity of matrix metalloproteinases and promotes the degradation of fibrous tissues. In addition, T3 can affect fibroblast activity and remodeling of the extracellular matrix [10, 11]. A previous study found that T3 administration decreased pro-inflammatory cytokine levels in a mouse model of ventilator-induced lung injury [12]. Similarly, another study revealed that in mice models of PF induced by BLM, T3 exhibited superior antifibrotic properties compared to both pirfenidone and nintedanib. This enhanced efficacy was attributed to T3’s capability to safeguard the mitochondria of alveolar type II epithelial cells [13]. Unfortunately, the effects of T3 treatment on lung microbiota in mice with PF remain unclear.
Each lung has its own microbial flora and specialized milieu; when asthma, IPF, or tumors are present, these pulmonary microbiota regulate local immune responses [14]. Health and lung microbiota are not fully understood but the lung microbiome of patients with IPF undergoes changes in composition, diversity, and burden [15]. Previous study has shown an inverse relationship between cystic fibrosis lung function, measured in terms of Forced Expiratory Volume in one second as a percentage of predicted value (FEV1%), and decreased α-diversity [16]. The bacterial community in IPF decreases, while the amounts of potential and pathogenic Haemophilus, Neisseria, Lactobacillus, Enterobacte, and Tetragenococcus increase [17]. A decline in lung function is associated with a notable decrease in microbiome diversity, with an abundance of Bacteroides and Firmicutes identified as factors influencing disease severity [18]. Bronchoalveolar lavage fluid from patients with IPF has a higher bacterial load than that from healthy individuals, and both the total bacterial burden and specific taxa (Streptococcus and Staphylococcus) are associated with more rapid disease progression [19, 20]. Overall, the lung microbiota plays an important role in the regulation of lung health, disease development, and progression.
Previous studies have demonstrated an association between IPF progression and lung microbiome, assessing the lung microbiome as a therapeutic target for IPF treatment [21]. In this study, we established a PF model by bleomycin (BLM) induction to investigate the effect of T3 treatment on the lung microbiota in PF mice, providing a more comprehensive theoretical basis for T3 as a potential treatment for IPF.
Methods
Experimental materials
Weitong Lihua Laboratory Animal Technology Co. Ltd. provided specific pathogen-free (SPF) C57BL6/N mice with permit number SCXK(Jing) 2006-006. A pathogen-free environment and regular laboratory diet was provided to all mice. Five animals were kept in a cage at a time with 12 h of light and 12 h of darkness daily. Modeling was started after a week of adaptation, and there were no confounding factors to control. The experimental protocols were approved by the Institutional Committee for Use and Care of Henan Normal University (approval no. 2019HNU-043).
Forty female SPF C57BL/6 N mice, each weighing between 18 and 25 g, were randomly assigned into four groups utilizing the random number table method. Inhalation anesthesia was induced by diluting isoflurane with 1, 2-propylene glycol to 40% concentration. Anesthesia was terminated when the mice exhibited prolonged inspiratory breathing and brief exhalation. PF was initiated by the intratracheal instillation of 50 µL of BLM (1.5 U/kg, Hanhui Pharmaceutical, China) to anesthetized mice, while the control group was given the same volume of saline. Briefly, the mice were anesthetized and bleomycin or saline was instilled into the trachea during the inspiratory phase, followed by inhalation into the alveoli for even distribution. On the 10th to 20th day of Saline + T3 and BLM + T3 groups, a small household atomizer was used to treat mice with T3 (the dose was 40 µg/kg every other day, diluted to 4 ml with PBS); the Saline group and BLM group were given 4 ml PBS atomization. As BLM perfusion established fibrosis after ten days, fibrosis peaked on day 14. T3 atomization treatment on the 10th day, and nebulization treatment was performed every other day for ten days. On day 21, mice were euthanized after an intraperitoneal injection of 20% Urethan 0.1 ml. Each group required three whole lungs of mice to extract lung microorganisms for DNA sequencing; two whole lungs were fixed with 4% paraformaldehyde for successful histological and pathological verification, and blood was collected for Enzyme-Linked Immunosorbent Assay (ELISA) testing (Fig. 1).

Flow chart of the experimental scheme. All experimental animals were included in the statistics because it was necessary to kill the mice on the 21st day and HE staining of pathological sections to determine whether the mice were successfully induced PF by pulmonary infusion of BLM. The laboratory animal modeling experience is mature and stable, and the design of the number of experimental animals considers the individual differences of animals
Hematoxylin and eosin (HE) staining
HE staining of paraffin-embedded lungs fixed with 4% paraformaldehyde was performed to observe histological morphology. Briefly, after deparaffinization and rehydration, the tissue sections were stained with hematoxylin solution (Solarbio, China) for 5 min, followed by five dips in 1% acid ethanol (1% HCl in 75% ethanol), and then rinsed in distilled water. Sections were stained with an eosin solution (Solarbio, China) for 3 min, dehydrated with graded alcohol, and cleared in xylene. Mounted slides were examined and photographed using a Nikon E200 microscope (Nikon, Tokyo, Japan).
Masson’s tricolor straining
The sections were stained with Masson’s trichrome following the manufacturer’s protocol (G1340, Solarbio, China). A Nikon microscope (E200, Japan) was used to observe illumination using bright light, and photographs were acquired using a Discovery C30 camera (Tucsen Imaging Technology Co., Ltd. Fujian, China).
16S rRNA gene sequencing
Lungs were obtained from each group and DNA was extracted for 16S rRNA microbial sequencing. A DNA extraction kit (DP712, TianGen, China), MiSeq Reagent Kit v3 (MS-102-3001, Illumina, USA), and NEXTFLEX Rapid DNA-Seq Kit (NOVA-5188-01, Bio-Scientific, USA) were used. The corresponding region of the primer used: 16S V3-V4 region, the upstream primer 338 F: ACTCCTACGGGAGGCAGCAG; and the downstream primer 806 R: GGACTACHVGGGTWTCTAAT. DNA extraction, PCR amplification, library preparation, and standard protocols were performed using an Illumina MiSeq sequencing platform (Illumina, San Diego, CA, USA) (Fig. 2A).

16S rRNA sequencing flow chart. (A) Experimental flow chart. (B) Information analysis flow chart
By the “Atacama soil microbiome tutorial” in the QIIME2 (2019.1) docs and customized scripts, the analysis was conducted (https://docs.qiime2.org/2019.1/) [22]. Briefly, this was performed according to the barcode information (sample unique) with the QIIME2 demux plug-in; the sequences of different samples were split from the original sequence (FASTQ format) [23]. Plugging in the QIIME2 dada2, a final feature fragment form was generated, the chimeras were removed, and quality control, trimming, denoising, and splicing were performed [24]. A QIIME2 feature classifier plug-in removed contaminating mitochondrial and chloroplast sequences after aligning the ASV sequences with the GreenGenes database [25]. The DADA2 (1.22.0) plug-in in the QIIME2 software controls the quality of all the original sequences of the samples (input) by filtering, denoising, merging, and non-chimeric to form an OTU (Table 1; Fig. 2B).
ELISA
Whole blood was refrigerated overnight at 4 ℃, coagulated naturally, centrifuged, and separated into the serum. ELISA for total T3 (TT3), free T3 (FT3), and reverse T3 (rT3) levels in peripheral serum (n ≥ 4) was performed as per the following steps (JL13028, JL20674, JL143442, J&L Biological). First, 50 µl sample serum was added to each well. Biotin-labeled antibody (50 µl; standard, except blank well) was added to each well and bathed at 37 ℃ for 30 min. Discard liquid, clapper, and 1× washing solution for 1 min×5 times; Horseradish peroxidase-labeled antibody 100 µl (except blank hole), 37 ℃ water bath for 30 min; Abandon the liquid, clapping board, and 1× washing liquid 1 min×5 times; Adding chromogenic agents A and B were 50 µl at 37 ℃ for 15 min; Then 50 µl of stopping solution was added to each well to immediately terminate the reaction when the blue turned yellow. The measurement was carried out within 15 min after the addition of the stopping solution, the blank well was used to adjust to zero, The absorbance (OD value) of each well was measured at the wavelength of 450 nm by a microplate reader (Biotek ElX800, Biotek, Vermont, USA).
Experimental data processing
The identification of bacteria exhibiting differential abundance was conducted using various statistical methods, including ANCOM, ANOVA, Kruskal Wallis, LEfSe, and DEseq2 [26, 27]. The R language ggtree package (v1.4.11) was used to visualize the evolutionary tree. ELISA data were presented as x ± SEM, and statistical analysis was performed using GraphPad Prism 7.0 (San Diego, USA) with a significance level set at P < 0.05. In cases where the data exhibited heterogeneous variance, the nonparametric Kruskal-Wallis test was utilized alongside the homogeneity of variance test (ANOVA). When there are differences among groups, Tukey’s Honestly Significant Difference is used for pairwise comparisons between groups. However, applying an effect size with a confidence interval was not possible in this experiment. Using the 16S rRNA sequence of the bacterial genome under investigation, PICRUSt (v2.4.2) employed a computational approach to estimate the gene function spectra of their shared ancestors. Additionally, it constructs gene function prediction spectra for Archaea and bacteria by inferring the gene function spectra of other untested species from the Greengenes database [28]. By mapping the sequencing-derived composition of the bacterial community in the aforementioned database, it is possible to predict the metabolic functionality of the bacteria [29].
Results
T3 could affect mice’s lung microbiome and organization structure with BLM -induced PF
BLM-induced thickening of the alveolar wall, deformation of the alveolar structure, and proliferation of the lung interstitium were observed, and T3 treatment reduced alveolar structure damage and pulmonary interstitial hyperplasia (Fig. 3A). Masson’s trichrome staining showed that T3 treatment reduced the tissue structure of BLM-induced blue collagen deposition in the lungs (Fig. 3B). Four-group Venn diagrams showed that the bacterial composition comprised 687, 1322, 541, and 1492 genera in Saline, T3, BLM and BLM + T3 groups, respectively. The bacterial population in the BLM-treated group also decreased. T3 treatment increased the lung flora of mice (Fig. 3C). BLM group Firmicutes (n = 3, average 84.81%) compared to the Saline group (n = 3, average 67.39%) are increased; BLM group Proteobacteria (n = 3, average 10.74%), Actinomycetes (n = 3, average 1.96%), unknown bacteria (n = 3, average 0.89%, no bacteria identified when the sequence is compared with the database ) and Bacteroides (n = 3, average 0.78%) is lower than Saline group (n = 3, average 24.41%, 2.52%, 2.51%, and 1.23%). BLM + T3 group Firmicutes (n = 3, average 43.63%) were down-regulated compared to that in the BLM group (n = 3, average 84.81). In the BLM + T3 group, Proteobacteria (n = 3, average 38.16%), Actinomycetes (n = 3, average 5.58%), unknown bacteria (n = 3, average 6.40%), and Bacteroides (n = 3, average 3.16%) were upregulated compared to the BLM group (n = 3, average 10.74%, 1.96%, 0.89%, and 0.78%, respectively) (Fig. 3D).

T3 Treatment can reverse the microbiome changes caused by Bleomycin damage. (A) A representative section of mouse lung stained with HE (n = 2). (B) A representative mouse lung section stained with Masson’s trichrome (n = 2). (C) Common or unique species Venn diagram display (An ellipse represents a group and an OTU’s standard or unique number among them, n = 3). (D) Phylum-level annotations to all annotated sequences ratio (n = 3). Sequences that are not annotated as unclassified
Results of information analysis of synergistic or reciprocal inhibition of microbial populations
An interconnected network was established utilizing the igraph package (version 1.2.6) within the R programming environment. Among the lung microbiota, Firmicutes exhibited the highest abundance. Notably, Firmicutes demonstrated negative correlations with Proteobacteria, Actinobacteria, and Bacteroidetes. Conversely, a robust positive correlation was identified among Proteobacteria, Actinobacteria, and Bacteroidetes, aligning with the observed trends in the relative abundance fluctuations of these phyla following T3 intervention (Fig. 4).

The interaction network diagram is in the phylum classification level. (Circle’s size indicates its relative abundance, and the line represents a significant correlation between these two species, P < 0.05, positively related in red and negatively related in blue lines)
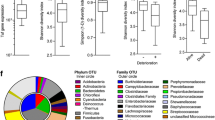
Lung microbiome alpha diversity
Our Alpha diversity analysis results of the 12 samples in the four groups showed no significant differences (P > 0.05, Fig. 5A-D). The saline group and saline + T3 group had a large sample gap, while the BLM group and BLM + T3 group had a small gap between samples (Fig. 5E). The lung microbiome of each mouse varied. However, the differences in the microbiome after BLM-induced PF were reduced, indicating that the model had a relatively stable microbiome. T3 treatment did not increase the differences in the microbiomes of PF mice (Fig. 5F).

The Alpha diversity index. (A) The chao1 index (an index use-d to measure the abundance of species) is not significant. (B) Faith’s Phylogenetic Diversity (is an index based on phylogenetic trees that measure diversity) is not a significant difference. (C) Shannon index is not a significant difference. (D) Simpson index is not significantly different. (E) No significant difference in Weighted Unifrac distance between groups. (F) NMDS analysis of differences in microbiome between the four groups (stress < 0.05, the sample is representative)
The increase of serum-free T3 was more beneficial to the growth of Proteobacteria
Serum TT3 (n = 3, P < 0.05) and FT3 (n = 3, P < 0.001) levels significantly affected the lung microbiome at the blood plasma level, whereas rT3 (n = 3, P > 0.05) had little effect. Linking Firmicutes and Proteobacteria to their origins and examining the cosine of the angle between them in relation to environmental variables revealed that the correlation coefficient between Proteobacteria and FT3 exceeded that of Firmicutes and FT3. This finding supported the notion that an elevated serum free T3 concentration more favorably impacted the proliferation of Proteobacteria (Fig. 6A). We used enzyme-linked immunosorbent assay (ELISA) to detect peripheral blood TT3, FT3, and rT3 levels in all mice in the same batch. Compared with the saline group, the peripheral serum TT3 of the BLM group was significantly lower (n = 5, P < 0.01), FT3 was also significantly reduced (n = 5, P < 0.01), and the rT3 level was significantly increased (n = 5, P < 0.01) (Fig. 6B-D), which also proves that the Firmicutes phyla are more accessible for growth than Proteobacteria in the low T3 state. In contrast, when T3 treatment increased peripheral blood FT3 levels, Proteobacteria grew faster than Firmicutes.

The increase of serum-free T3 was more beneficial to the growth of Proteobacteria. Levels. (A) Within the RDA species sorting diagram, serum TT3 and FT3 considerably influenced the abundance of Firmicutes, Proteobacteria, and rT3 had little effect. (B)-(D) ELISA method to detect TT3, FT3, and rT3 in peripheral blood of mice. *P < 0.05, **P < 0.01
T3 as an environmental factor is closely related to the lung microbiome
Bacteroidetes, Actinobacteria, Firmicutes, Proteobacteria, Planctomycetes and Chloroflexi were found to be enriched in the lung microflora of mice. BLM increased bacterial abundance in PF mice compared with controls; and bacterial abundance was further enhanced in PF mice after T3 intervention (Fig. 7).

T3 can regulate bacterial abundance in mice with pulmonary fibrosis. A phylogenetic tree and a heat map of the distribution of abundances between groups. (A phylogenetic tree is shown on the left, each branch representing an OTU. Branches with different colors represent different phyla, and the end has annotated the genus. Bacterial quantity is represented by the heat map on the right, with higher values representing the higher relative abundance of the phylum)
T3 treatment does not affect the function of the microbiome community
We performed the following analysis to determine whether T3 affected the function of microorganisms in the body’s metabolism. According to the prediction of the participation of the microbial community in the metabolism of the body, the first 20 metabolic functions mainly involve the metabolism of carbohydrates, amino acids, energy, nucleotides, vitamins, and lipids (Fig. 8). Treatment with either BLM or T3 resulted in no significant differences between the groups.

T3 treatment does not change the function of the microbiome. From top to bottom, the results of KEGG are displayed in sequence. The vertical axis represents an annotation’s proportion to a specific function; the horizontal axis represents its group name; each color block corresponds to a function category in the right leg
Discussion
The lung microbiome is closely associated with the development and progression of PF, and some pathogenic microorganisms may promote PF development by inducing inflammation and fibrosis [30]. T3 has shown potential in inhibiting tissue fibrosis, encompassing even PF. Consequently, we conjectured that T3 might exert an indirect influence on the composition of the lung microbiota community, potentially mediated through its anti-inflammatory properties and metabolic regulatory effects. The findings revealed that T3 administration ameliorated both the BLM-induced PF and associated structural impairments. Secondly, in the presence of BLM, the microbial ecosystem underwent a reduction in the prevalence of Proteobacteria, Bacteroidetes, and Actinobacteria, coupled with an elevation in Firmicutes; however, T3 intervention reversed these shifts. Notably, no substantial variations in α-diversity of the microbiomes across the groups were observed. Furthermore, there existed a positive correlation between the serum levels of TT3 and FT3 and the abundance of microbiota populations. Lastly, T3 supplementation augmented the microbiome profile in PF without altering its overall diversity or compromising its biological functionalities.
The Human Microbiome Project was launched in 2007 to study the microbial inhabitants of the human body and the microbes that live on human mucosal surfaces [31]. The human lung was originally thought to be a sterile organ and was not included in the original Human Microbiome Project [32]. However, a growing number of studies have revealed the presence of microbial communities in the lungs [33, 34]. Through the analysis of bacterial abundance alterations in IPF using multiple bacterial quantification methodologies, it is evident that while a connection persists between decreased microbial diversity and patient survival, there also exists a signaling cascade within the airways that fosters inflammation and fibrotic processes. These findings collectively suggest that individuals diagnosed with IPF harbor a distinct lung microbiome composition, which is intimately linked to the severity of the disease manifestation [35]. Yang et al. found that alterations in lung microbiota promote the development of lung inflammation and fibrosis in mouse models, suggesting that dysbiosis of the lung flora is a key causative factor in the development of IPF [36]. Microbiota structure, bacterial burden, and diversity of bacterial species are the three main factors that affect the pathogenesis and prognosis of IPF [37]. Individuals with reduced lung microbiome diversity are worse health off; therefore, a less diverse lung bacterial community in IPF may be a significant factor in its pathogenesis [38]. Furthermore, a decrease in lung bacterial diversity is associated with increased alveolar cytokines and growth factors associated with inflammation and fibrosis. The lung microbiome may be related to IPF [39].
Evidence shows that changes in the gut microbiome and metabolites are related to PF through the “gut-lung axis” [40]. Based on the intestinal-pulmonary axis theory, the immune response caused by the spread of gastrointestinal bacteria can worsen lung condition [41]. In a PF mouse model, there was a negative correlation between the intestinal flora Alloprevotella, Helicobacter, Rikenella, and PF severity, whereas a positive correlation was found between Dubosiella, Parasutterella, and PF outcome [42]. Tong X et al. found that the β diversity of microbiome was reduced in the IPF group. The signatures of the IPF microbes were enriched for Streptococcus, Pseudobutyrivibrio, and Anaerorhabdus. After the analysis of gene function, ABC transporter systems, biofilm formation, and two-component regulatory systems were enriched in the microbiome of patients with IPF [43]. Morris et al. also showed an increased bacterial load and decreased microbial diversity in BAL samples from patients with IPF [44]. Molyneaux et al. prospectively explored the lung microbiomes of patients with IPF and found increased reads of potentially pathogenic Haemophilus, Streptococcus, Neisseria and Veillonella spp. in patients with IPF compared to healthy controls. They were unable to identify any bacterial community structure or composition that differed between patients with stable or progressive IPF [45]. In this study, we sequenced the resultant flora to be mainly Firmicutes, Proteobacteria, Actinomycetes, and Bacteroidetes; Firmicutes were more abundant in pulmonary fibrosis, while Proteobacteria were less abundant, which was also validated in our model, and T3 treatment reversed these changes. Although the statistical analysis of alpha diversity and the prediction of microbial community function did not show significant differences, it cannot be proven whether the therapeutic effect of T3 on PF is based on the upregulation of lung microbiome alpha diversity and the influence of microbial community function. In addition, our study found that BLM-treated mice with PF had decreased TT3 and FT3 levels in the peripheral blood and increased rT3. The model was preliminarily judged to have low T3 syndrome, a symptom widely associated with acute and chronic diseases, including acute diseases such as Covid-19 [46]. Therefore, more experimental data are needed to determine whether T3 therapy is a potential therapeutic method for the clinical treatment of IPF.
In mouse models, we verified that PF increased the risk of an abundance of Streptococcus, Staphylococcus, Pseudomonas aeruginosa, Pseudomonas, and Escherichia [47]. T3 treatment downregulated their abundance. However, the reported association between IPF and the upregulation of Prevotella, Veillonella, Haemophilus, and Veillonella cocci is unverified [36, 48]. In view of this, further research is necessary, and human experiments may be considered in the future to verify whether the results observed in mouse models can be replicated in humans. In addition, a deeper understanding of these bacterial species and their roles in PF is important.
Limitations
Owing to the reading length limitation of the second-generation sequencing platform, only a single v region, double v region, or triple v region can be selected as the target fragment region for 16S sequencing. Primers 341 F and 806R are often selected in the V3V4 region during second-generation sequencing, which has a high coverage rate in bacteria and archaea and can be used to detect the diversity distribution of bacteria and archaea simultaneously; this is the most significant limitation compared to the low alignment rate and identification accuracy of third-generation sequencing (full-length 16S rRNA).
Conclusion
This study proves that T3 therapy can benefit microecological diversity and equilibrium, and further verifies it as an effective drug for IPF treatment. T3 treatment has a relatively low cost and better therapeutic effects have been observed in animal models, introducing new hope for IPF treatment. Future study may necessitate additional mouse experiments to ascertain whether T3 can alter the microbiome of colonized mice and its impact on pulmonary fibrosis, as well as to elucidate the mechanisms of the lung-brain axis in IPF by which the lung microbiome signals central neurons and glial cells.
Data availability
The datasets generated and/or analysed during the current study are available in NCBI. BioProject, accession number: PRJNA890142. (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA890142).
References
Wei P, Xie Y, Abel PW, Huang Y, Ma Q, Li L, Hao J, Wolff DW, Wei T, Tu Y. Transforming growth factor (TGF)-β1-induced miR-133a inhibits myofibroblast differentiation and pulmonary fibrosis. Cell Death Dis. 2019;10(9):670.
Hutchinson J, Fogarty A, Hubbard R, McKeever T. Global incidence and mortality of idiopathic pulmonary fibrosis: a systematic review. Eur Respir J. 2015;46(3):795–806.
Wang J, Xiang Y, Yang SX, Zhang HM, Li H, Zong QB, Li LW, Zhao LL, Xia RH, Li C, et al. MIR99AHG inhibits EMT in pulmonary fibrosis via the miR-136-5p/USP4/ACE2 axis. J Transl Med. 2022;20(1):426.
Jeon S, Jin H, Kim JM, Hur Y, Song EJ, Lee YJ, Na Y, Cho J, Lee YS. The miR-15b-Smurf2-HSP27 axis promotes pulmonary fibrosis. J Biomed Sci. 2023;30(1):2.
Sgalla G, Biffi A, Richeldi L. Idiopathic pulmonary fibrosis: diagnosis, epidemiology and natural history. Respirology. 2016;21(3):427–37.
Shen YH, Cheng MH, Liu XY, Zhu DW, Gao J. Sodium Houttuyfonate inhibits Bleomycin Induced Pulmonary Fibrosis in mice. Front Pharmacol. 2021;12:596492.
Breitzig MT, Alleyn MD, Lockey RF, Kolliputi N. Thyroid hormone: a resurgent treatment for an emergent concern. Am J Physiol Lung Cell Mol Physiol. 2018;315(6):L945–50.
Mullur R, Liu YY, Brent GA. Thyroid hormone regulation of metabolism. Physiol Rev. 2014;94(2):355–82.
Kohon MY, Zaaroor Levy M, Hornik-Lurie T, Shalom A, Berl A, Drucker L, Levy Y, Tartakover Matalon S. αvβ3 integrin as a link between the development of fibrosis and thyroid hormones in systemic sclerosis. Int J Mol Sci 2023, 24(10).
Wang L, Li Z, Wan R, Pan X, Li B, Zhao H, Yang J, Zhao W, Wang S, Wang Q, et al. Single-cell RNA sequencing provides New insights into therapeutic roles of thyroid hormone in idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol. 2023;69(4):456–69.
Vamesu BM, Nicola T, Li R, Hazra S, Matalon S, Kaminski N, Ambalavanan N, Kandasamy J. Thyroid hormone modulates hyperoxic neonatal lung injury and mitochondrial function. JCI Insight 2023, 8(8).
Barca-Mayo O, Liao XH, DiCosmo C, Dumitrescu A, Moreno-Vinasco L, Wade MS, Sammani S, Mirzapoiazova T, Garcia JG, Refetoff S, et al. Role of type 2 deiodinase in response to acute lung injury (ALI) in mice. Proc Natl Acad Sci U S A. 2011;108(49):E1321–1329.
Yu G, Tzouvelekis A, Wang R, Herazo-Maya JD, Ibarra GH, Srivastava A, de Castro JPW, DeIuliis G, Ahangari F, Woolard T, et al. Thyroid hormone inhibits lung fibrosis in mice by improving epithelial mitochondrial function. Nat Med. 2018;24(1):39–49.
Hosang L, Canals RC, van der Flier FJ, Hollensteiner J, Daniel R, Flugel A, Odoardi F. The lung microbiome regulates brain autoimmunity. Nature. 2022;603(7899):138–44.
Invernizzi R, Wu BG, Barnett J, Ghai P, Kingston S, Hewitt RJ, Feary J, Li Y, Chua F, Wu Z, et al. The respiratory microbiome in chronic hypersensitivity pneumonitis is distinct from that of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2021;203(3):339–47.
Avalos-Fernandez M, Alin T, Metayer C, Thiebaut R, Enaud R, Delhaes L. The respiratory microbiota alpha-diversity in chronic lung diseases: first systematic review and meta-analysis. Respir Res. 2022;23(1):214.
Yoon HY, Moon SJ. Lung tissue microbiome is Associated with Clinical outcomes of Idiopathic Pulmonary Fibrosis. Front Med (Lausanne). 2021;8:744523.
Metzger MI, Graeber SY, Stahl M, Sommerburg O, Mall MA, Dalpke AH. A volatile and dynamic longitudinal microbiome is Associated with Less reduction in lung function in adolescents with cystic fibrosis. Front Cell Infect Microbiol. 2021;11:763121.
Invernizzi R, Barnett J, Rawal B, Nair A, Ghai P, Kingston S, Chua F, Wu Z, Wells AU, Renzoni ER et al. Bacterial burden in the lower airways predicts disease progression in idiopathic pulmonary fibrosis and is independent of radiological disease extent. Eur Respir J 2020, 55(4).
Han MK, Zhou Y, Murray S, Tayob N, Noth I, Lama VN, Moore BB, White ES, Flaherty KR, Huffnagle GB, et al. Lung microbiome and disease progression in idiopathic pulmonary fibrosis: an analysis of the COMET study. Lancet Respir Med. 2014;2(7):548–56.
Varone F, Gibiino G, Gasbarrini A, Richeldi L. Evaluation of the lung microbiome as a therapeutic target in the management of idiopathic pulmonary fibrosis: role of antioxidant/antibiotic combination therapy. Eur Rev Med Pharmacol Sci. 2019;23(14):6379–86.
Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. 2019;37(8):852–7.
A T: Qiime Artifact eXtractor (qax): a fast and versatile Tool to interact with Qiime2 archives. BioTech (Basel) 2021, 10(1):5.
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, Holmes SP. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13(7):581–3.
Dania MI, Faraji B. Micronutrient Biosynthesis potential of spontaneous grain fermentation microbiomes. Int J Environ Res Public Health. 2022;19(24):16621.
Jiang H, Li J, Zhang B, Huang R, Zhang J, Chen Z, Shang X, Li X. Intestinal Flora disruption and novel biomarkers Associated with nasopharyngeal carcinoma. Front Oncol. 2019;9:1346.
Xue Z, Li R, Liu J, Zhou J, Zhang X, Zhang T, Zhang M, Yang Y. Preventive and synbiotic effects of the soluble dietary fiber obtained from Lentinula edodes byproducts and Lactobacillus plantarum LP90 against dextran sulfate sodium-induced colitis in mice. J Sci Food Agric. 2022;103(2):616–26.
Xu Y, Wang Q, Wu H. Evaluating the contingency treatment performance of advanced electro-catalysis oxidation processes for marine bacteria in ballast water. Water Sci Technol. 2021;84(8):1885–95.
Bharti R, Grimm DG. Current challenges and best-practice protocols for microbiome analysis. Brief Bioinform. 2021;22(1):178–93.
Quan Y, Yin Z, Chen S, Lang J, Han L, Yi J, Zhang L, Yue Q, Tian W, Chen P, et al. The gut-lung axis: gut microbiota changes associated with pulmonary fibrosis in mouse models induced by bleomycin. Front Pharmacol. 2022;13:985223.
Peterson J, Garges S, Giovanni M, McInnes P, Wang L, Schloss JA, Bonazzi V, McEwen JE, Wetterstrand KA, Deal C, et al. The NIH Human Microbiome Project. Genome Res. 2009;19(12):2317–23.
Zhang J, Wu Y, Liu J, Yang Y, Li H, Wu X, Zheng X, Liang Y, Tu C, Chen M, et al. Differential oral microbial input determines two microbiota pneumo-types Associated with Health Status. Adv Sci (Weinh). 2022;9(32):e2203115.
Sodhi CP, Wohlford-Lenane C, Yamaguchi Y, Prindle T, Fulton WB, Wang S, McCray PB Jr., Chappell M, Hackam DJ, Jia H. Attenuation of pulmonary ACE2 activity impairs inactivation of des-Arg(9) bradykinin/BKB1R axis and facilitates LPS-induced neutrophil infiltration. Am J Physiol Lung Cell Mol Physiol. 2018;314(1):L17–31.
Qi C, Hountras P, Pickens CO, Walter JM, Kruser JM, Singer BD, Seed P, Green SJ, Wunderink RG. Detection of respiratory pathogens in clinical samples using metagenomic shotgun sequencing. J Med Microbiol. 2019;68(7):996–1002.
Wilson AM, Clark AB, Cahn T, Chilvers ER, Fraser W, Hammond M, Livermore DM, Maher TM, Parfrey H, Swart AM, et al. Effect of co-trimoxazole (Trimethoprim-Sulfamethoxazole) vs placebo on death, lung transplant, or hospital admission in patients with moderate and severe idiopathic pulmonary fibrosis: the EME-TIPAC Randomized Clinical Trial. JAMA. 2020;324(22):2282–91.
Yang D, Chen X, Wang J, Lou Q, Lou Y, Li L, Wang H, Chen J, Wu M, Song X, et al. Dysregulated lung commensal Bacteria drive Interleukin-17B production to promote pulmonary fibrosis through their outer membrane vesicles. Immunity. 2019;50(3):692–e706697.
Takahashi Y, Saito A, Chiba H, Kuronuma K, Ikeda K, Kobayashi T, Ariki S, Takahashi M, Sasaki Y, H T. Impaired diversity of the lung microbiome predicts progression of idiopathic pulmonary fibrosis. Respir Res. 2018;19(1):34.
Schuliga M, Read J, DA K. Ageing mechanisms that contribute to tissue remodeling in lung disease. Ageing Res Rev. 2021;70:101405.
Spence CD, Vanaudenaerde B, Einarsson GG, Mcdonough J, Lee AJ, Johnston E, Verleden GM, Elborn JS, Dupont LJ, Van Herck A, et al. Influence of azithromycin and allograft rejection on the post-lung transplant microbiota. J Heart Lung Transpl. 2020;39(2):176–83.
Wu Y, Li Y, Luo Y, Zhou Y, Wen J, Chen L, Liang X, Wu T, Tan C, Liu Y. Gut microbiome and metabolites: the potential key roles in pulmonary fibrosis. Front Microbiol. 2022;13:943791.
Donati Zeppa S, Agostini D, Piccoli G, Stocchi V. Gut microbiota status in COVID-19: an unrecognized player? Front Cell Infect Microbiol. 2020;10:576551.
Hu H, Wang F, Han P, Li P, Wang K, Song H, Zhao G, Li Y, Lu X, Tao W, et al. Bu-Fei-Huo-Xue capsule alleviates bleomycin-induced pulmonary fibrosis in mice through modulating gut microbiota. Front Pharmacol. 2023;14:1084617.
Tong X, Su F, Xu X, Xu H, Yang T, Xu Q, Dai H, Huang K, Zou L, Zhang W, et al. Alterations to the lung Microbiome in Idiopathic Pulmonary Fibrosis patients. Front Cell Infect Microbiol. 2019;9:149.
Morris A, Gibson K, Collman RG. The lung microbiome in idiopathic pulmonary fibrosis. What does it mean and what should we do about it? Am J Respir Crit Care Med. 2014;190(8):850–2.
Molyneaux PL, Cox MJ, Willis-Owen SA, Mallia P, Russell KE, Russell AM, Murphy E, Johnston SL, Schwartz DA, Wells AU, et al. The role of bacteria in the pathogenesis and progression of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2014;190(8):906–13.
Geslot A, Chanson P, Caron P. Covid-19, the thyroid and the pituitary - the real state of play. Ann Endocrinol (Paris). 2022;83(2):103–8.
Oda K, Yatera K, Fujino Y, Kido T, Hanaka T, Sennari K, Fushimi K, Matsuda S. Respiratory comorbidities and risk of mortality in hospitalized patients with idiopathic pulmonary fibrosis. Respir Investig. 2018;56(1):64–71.
Han MK, Zhou Y, Murray S, Tayob N, Noth I, Lama VN, Moore BB, White ES, Flaherty KR, Huffnagle GB, et al. Lung microbiome and disease progression in idiopathic pulmonary fibrosis: an analysis of the COMET study. Lancet Respiratory Med. 2014;2(7):548–56.
Acknowledgements
The authors thank Professor Xinsheng Zhang for assistance with the experimental techniques used in this study.
Funding
Ministry of Science and Technology, PR China grant 2019YFE0119500; Key R&D Program of Henan province grant 231111310400; Zhongyuan scholar 244000510009; Henan Project of Science and Technology grants 232102521025, and GZS2023008. The funder of the study had no role in the study design, data collection, data analysis, data interpretation, or writing of the report.
Author information
Authors and Affiliations
Contributions
Conceptualization, investigation, GY.Y., XS.G., Formal analysis, XS.G., KX, Animal experiment, XS.G., KX, QW.W., ZY.H., Supervision, Validation, Funding acquisition, and GY.Y.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study protocol was approved by the Ethics Committee of the Henan Normal University (approval no. 2019HNU-043). This study does not involve human samples. We confirmed that all experimental protocols were approved by the Ethics Committee of the Henan Normal University (approval no. 2019HNU-043). We confirmed that all methods were carried out in accordance with relevant guidelines and regulations. We confirmed that all methods are reported in accordance with ARRIVE guidelines (https://arriveguidelines.org) for the reporting of animal experiments.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Guo, X., Xu, K., Wang, Q. et al. Assessing the impact of triiodothyronine treatment on the lung microbiome of mice with pulmonary fibrosis. BMC Pulm Med 24, 405 (2024). https://doi.org/10.1186/s12890-024-03214-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12890-024-03214-3