
Abstract
Background
Chronic hypersensitivity pneumonitis (CHP) has a variable disease course. Computer analysis of CT features was used to identify a subset of CHP patients with an outcome similar to patients with idiopathic pulmonary fibrosis (IPF).
Methods
Consecutive patients with a multi-disciplinary team diagnosis of CHP (n = 116) had pulmonary function tests (FEV1, FVC, DLco, Kco, and a composite physiologic index [CPI]) and CT variables predictive of mortality evaluated by analysing visual and computer-based (CALIPER) parenchymal features: total interstitial lung disease (ILD) extent, honeycombing, reticular pattern, ground glass opacities, pulmonary vessel volume (PVV), emphysema, and traction bronchiectasis. Mean survival was compared between both CHP and IPF patients (n = 185).
Results
In CHP, visual/CALIPER measures of reticular pattern, honeycombing, visual traction bronchiectasis, and CALIPER ILD extent were predictive of mortality (p < 0 · 05) on univariate analysis. PVV was strongly predictive of mortality on univariate (p < 0 · 0001) and multivariate analysis independent of age, gender and disease severity (represented by the CPI [p < 0 · 01]). CHP patients with a PVV threshold >6 · 5% of the lung had a mean survival (35 · 3 ± 6 · 1 months; n = 20/116 [17%]) and rate of disease progression that closely matched IPF patients (38 · 4 ± 2 · 2 months; n = 185).
Conclusions
Pulmonary vessel volume can identify CHP patients at risk of aggressive disease and a poor IPF-like prognosis.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Background
The majority of patients with hypersensitivity pneumonitis who present to specialist centres have the chronic fibrotic form of the disease [1,2,3]. Within the population of chronic hypersensitivity pneumonitis (CHP) patients, it has been observed that some patients may show an accelerated rate of progression, comparable to the trajectory of idiopathic pulmonary fibrosis (IPF) [2, 4, 5]. The overlap with IPF is also evident in the often-challenging differentiation of CHP from IPF in a multi-disciplinary team (MDT) setting [5,6,7]. The early identification of a rapidly progressive fibrosing lung disease is increasingly relevant in an era of anti-fibrotic therapies, in which treatment guidelines are likely to increasingly broaden the spectrum of diagnoses for which anti-fibrotic treatment is indicated.
Given the varied outcome and unpredictable prognosis in CHP, the American National Heart, Lung and Blood Institute, in collaboration with the Office of Rare Diseases, convened a workshop in 2005 to discuss future research priorities in HP [8]. Amongst the various recommendations was a need to improve the characterisation of different HP phenotypes. The workshop also emphasized the desirability of exploring quantitative CT analysis in longitudinal studies of HP patients [8].
Automated computer-based quantitative imaging has advanced rapidly in recent years, with quantitatively scored CT variables shown to be superior to visual CT scores in predicting mortality in IPF. [9] To date however, quantitative CT tools have not been applied to the study of outcome in patients with CHP. Our study specifically set out to identify whether, in patients without end-stage lung disease, there are visual or quantitative CT features that characterize CHP patients who have a poor IPF-like outcome.
Methods
Study population and clinical information
All new consecutive patients with an MDT diagnosis of CHP and IPF according to published guidelines [10,11,12], over a four and a half year period (January 2007–July 2011) were identified from the Interstital Lung Disease Unit database. Patients with a non-contrast, supine, volumetric thin-section CT were captured. Individuals with no overt CT signs of fibrosis (the absence of honeycombing/reticular pattern/traction bronchiectasis on visual scoring by both radiologist scorers, RE and ALB) were excluded from analysis (Fig. 1).

Flowchart illustrating the selection of patients for the final chronic hypersensitivity pneumonitis study population. ILD = interstitial lung disease, CTD = connective tissue disease, CHP = chronic hypersensitivity pneumonitis, LCH = Langerhans cell histiocytosis, LAM = lymphangioleiomyomatosis, CT = computed tomography
Approval for this retrospective study of clinically indicated CT and pulmonary function test (PFT) data was obtained from the Institutional Ethics Committee and informed patient consent was not required.
CT scanning, echocardiography, PFT protocols were as previously described [9, 13] and are included in the online appendices. CALIPER CT data processing steps [9, 13] are also detailed in the online appendices.
CALIPER CT pattern quantitation
CALIPER evaluation of CT data involved algorithmic identification and volumetric quantification of 15x15x15 voxel volume units into one of six radiological parenchymal features: ground glass opacity (GGO), reticular pattern, honeycombing, emphysema, pulmonary vessel volume (PVV), and normal lung (Figs. 2 and 3).

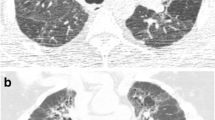
Axial CT image in a 46-year-old female never smoker (a), diagnosed with chronic hypersensitivity pneumonitis following surgical lung biopsy. An upper lobe predominant fibrosing lung disease associated with honeycomb cysts is demonstrated. On the CALIPER colour overlay image (b), ground glass opacities (yellow) are interspaced with reticular pattern (orange) in the lung periphery. In the anterior left upper lobe, there are honeycomb cysts visible (brown). Light and dark green areas represent normal lung parenchyma. Axial CT image in a 55-year-old female 2-pack-year ex-smoker (c), diagnosed with chronic hypersensitivity pneumonitis following surgical lung biopsy with more extensive fibrosis involving the lower lobes. CALIPER colour overlay images show minor misclassification of traction bronchiectasis as honeycomb cysts in the left lower lobe (d), but extensive ground glass opacities (yellow) and interspaced reticular pattern (orange)

Corresponding axial CT images and colour overlay maps of CALIPER quantitation of pulmonary vessel volume (red), a volumetric measure of pulmonary arteries and veins within the lung, excluding vessels at the lung hilum in three chronic hypersensitivity pneumonitis patients with mild (a + b), moderate (c + d) and severe (e + f) extents of fibrosis. Note there is misclassification of small regions of the reticular pattern in the paravertebral regions (b) and right lower lobe (f) as vessels
A fibrosis score represented the sum of reticular pattern and honeycombing whilst the ILD extent additionally summed GGO extent. PVV represented the sum of the volumes of the pulmonary arteries and veins, excluding vessels at the lung hilum. All parenchymal pattern volumes were divided by the total lung volume, also derived by CALIPER, to generate percentages for each pattern.
CT visual quantitation
Each CHP CT was evaluated independently by two radiologists (ALB, RE with 5 and 7 years thoracic imaging experience respectively), blinded to all clinical information. Methods by which discordant CT scores were consensed are given in the Additional file 1.
Additional visually quantified CT features included consolidation, mosaicism (decreased attenuation component), and traction bronchiectasis (no comparable CALIPER score) [details in Additional file 1]. There was no analogous visual score of PVV. A CT index of pulmonary hypertension (main pulmonary artery:ascending aorta ratio) was recorded by a single scorer using electronic caliper measurements of the ascending aorta and pulmonary artery diameters at the level of the pulmonary artery bifurcation [14, 15]. A CT UIP pattern was evaluated by one radiologist (JJ, with 5 years thoracic imaging experience) who was blinded to all clinical information.
Identification of CHP patients with end-stage disease
The study aim was to identify characteristics of the subset of CHP patients who do not have end-stage disease yet progress to death rapidly with an IPF-like trajectory. Two methods were used to identify CHP with end stage disease:
-
1.
Contained within the consecutive CHP cases were individuals with respiratory failure secondary to end-stage fibrosing lung disease, which in itself predicates a poor outcome. Pa02 values were examined to separate out patients who would have an inevitably rapid decline because of end-stage disease at baseline. 102/129 (79%) patients had a measured Pa02. Patients were identified as having end-stage disease if they had a Pa02 < 7 · 5kPa (n = 13). In the 27 patients without a measured Pa02, the use of supplementary oxygen therapy (recommended at a threshold of <7 · 3kPa02 [10]) was used to identify patients with respiratory failure and therefore end-stage disease (n = 3). The first study group consisted of 116 CHP patients.
-
2.
A threshold of FVC ≥ 50% predicted was also used to separate CHP patients into those with and without end-stage disease at baseline. 98/123 (80%) patients had a FVC ≥ 50% predicted. In 6 patients without a measured FVC, only one patient required supplemental home oxygen and was therefore excluded. The second study group consisted of 103 CHP patients. Analysis of this second population of non end-stage CHP patients is provided in Additional files 1, Additional file 2 and 3.
Statistical analysis
Data are given as medians, means with standard deviations, or numbers of patients with percentages where appropriate. Interobserver variation for visual scores was assessed using the single determination standard deviation. Univariate and multivariate Cox regression analyses of the CHP cohort were used to investigate relationships within and between the three data sets: CALIPER CT evaluation, visual CT evaluation and PFTs. To evaluate group differences, the T-test and Mann Whitney U test were used for mean and median continuous variables and the Chi-squared tests was used for categorical variables.
Cox regression analyses of CALIPER and PFT variables were used to compare the CHP and IPF cohorts. The robustness of the results was confirmed using bootstrapping and resampling of the dataset up to 1000 times. Survival curves were created using Kaplan Meier analyses and statistically significant differences between curves evaluated using the Log-Rank test. In all study analyses, a p-value of <0.05 was considered significant. Assumptions of linearity and proportional hazards were tested by visual inspection of Martingale residuals and scaled Schoenfeld residuals. Statistical analyses were performed with STATA (version 12, StatCorp, College Station, TX, USA).
Results
Demographic data
The initial study population, which has not been previously described, comprised 129 consecutive patients newly presenting with an MDT diagnosis of CHP based on a compatible clinical history and review of the following data: antigen exposure history (positive in 53/129 [41%] patients), precipitating antibodies (positive in 50/129 [39%] patients), bronchoalveolar lavage (BAL) findings (performed in 73/129 [57%] patients), CT imaging (129/129 [100%] of patients), and histopathology (60/129 [46%] of patients).
The median age at presentation was 60 years. Patient status and mean follow up time (58.0 ± 17 · 5 months) were obtained by contacting the patient’s primary care giver on a given date. 50/129 patients (39%) died during the study period. Data on vital status was completed on 98% of cases with 3 patients censored. 41/73 (56%) patients undergoing BAL had a lymphocytosis ≥20% and 25/73 (34%) had a lymphocytosis ≥30%. A right ventricular systolic pressure (RVSP) >50 mmHg measured on transthoracic echocardiography was considered representative of pulmonary hypertension [16], and was found in 10/69 (14%) of the CHP cohort.
When separating the CHP population into those with and without end-stage disease using a PaO2 threshold of 7 · 5kPa02, using Kaplan Meier curves, the 7 · 5kPa02 threshold was shown to clearly distinguish groups with a good and bad outcome (Log rank p < 0.0001) (Fig. 4). The primary study group consisted of 116 CHP patients. Demographic data and average visual score, CALIPER score and PFT data are provided in Table 1. Interobserver variation values for the visual scores are provided in Additional file 1 (Table 1).

Survival curves demonstrating differences in survival between chronic hypersensitivity pneumonitis patients with differing PaO2 thresholds. a Differences between patients with a PaO2 >7 · 5kPa (green)(n = 116; mean survival = 65 · 6 ± 2 · 9 months) and patients with a PaO2 < 7 · 5kPa (blue)(n = 13; mean survival = 14 · 9 ± 2 · 7 months). Log rank test p = <0 · 0001. b Differences in survival between patients with a PaO2 > 8kPa (green)(n = 112; mean survival = 66 · 0 ± 2 · 9 months) and patients with a PaO2 < 8kPa (blue)(n = 17; mean survival = 27 · 4 ± 7 · 6 months). Log rank test p = <0 · 0001
CHP mortality analyses
Strong univariate CALIPER and visually-scored CT predictors of mortality in the CHP cohort included reticular pattern, honeycombing, visual traction bronchiectasis, PVV and the fibrosis score (Table 2). CALIPER and visually scored emphysema were not predictive of survival.
On bivariate analyses, PVV was a stronger predictor of mortality than FVC, DLco, and CPI (Additional file 1 [Table 2]). The results were maintained following adjustment for patient age and gender with no PFTs retaining significance in the model. In a subanalysis of CHP patients with either histopathological confirmation or antibody positivity to precipitating antigens, after adjusting for patient age and gender, PVV (p = 0 · 005) remained a stronger predictor of mortality than FVC (p = 0 · 10), DLco (p = 0 · 009) and CPI (p = 0 · 01).
Benchmarking of an IPF outcome in patients with CHP
A consecutive population of IPF patients (n = 283), presenting to our institution over the same period as the CHP study population was analyzed to establish a mortality profile of IPF. The IPF population has been previously evaluated in two studies [9, 13]. To identify and exclude patients with respiratory failure and end-stage disease, in a similar way to the CHP cohort, all IPF patients without a measured Pa02 (n = 70), as well as those patients with a Pa02 < 7 · 5kPa (n = 28) were excluded. 185 IPF patients were subsequently evaluated with Kaplan Meier analysis which identified the mean survival to be 38 · 4 ± 2 · 2 months (Fig. 5). Of the final population of 185 IPF patients, 74/185 (40%) patients demonstrated a definite UIP pattern on CT and 102/185 (55%) patients demonstrated a possible UIP pattern on CT. 9/185 (5%) patients had a CT pattern that was inconsistent with a UIP pattern, but which was characterised as UIP on open lung biopsy, and confirmed as IPF following MDT discussion with no support for a diagnosis of CHP or connective tissue disease related-ILD (alternative explanations of UIP).

Survival curves for patients with idiopathic pulmonary fibrosis (IPF) (yellow) and chronic hypersensitivity pneumonitis (CHP) (green and blue). CHP cases with end stage disease (denoted by a PaO2 < 7 · 5kPa) were excluded. IPF cases without a measured PaO2 or with end stage disease (denoted by a PaO2 < 7 · 5kPa) were excluded. IPF patients (n = 185) were compared to CHP cases at pulmonary vessel volume (PVV) thresholds above (green) and below (blue) 5%, 5 · 5%, 6 · 5% and 7% (a–d). The IPF and poor-outcome CHP curves were most comparable at a CHP PVV threshold of 6 · 5% where the mean survival in the IPF population (38 · 4 ± 2 · 2 months; n = 185) was found to be similar to the poor-outcome CHP population (35 · 3 ± 6 · 1 months; n = 20) and distinct from the good-outcome CHP population (71 · 3 ± 2 · 8 months; n = 96). The PVV > 6% threshold contained only two patients more than the PVV > 6 · 5% threshold which did not significantly alter the results and accordingly was not been formally plotted
Derivation of poor-outcome using PVV thresholds
CHP patients with an IPF-like outcome were identified by separating the CHP population into two age and gender-matched cohorts comprising 58 patients (Additional file 1 [Table 3]). No significant differences were identified for PFTs or CT parenchymal pattern scores between groups. Various PVV thresholds predictive of mortality were investigated. A PVV threshold >6 · 5% of the lung showed a mean survival most comparable to IPF patients in both CHP groups: Group 1 mean survival = 39 · 7 ± 7 · 8 months (n = 11; 19%); Group 2 mean survival = 28 · 2 ± 8 · 1 months (n = 9; 16%).
The two CHP groups were combined and PVV thresholds were reanalyzed and compared to outcomes in the IPF population (Fig. 5). No statistically significant difference between Kaplan Meier survival curves was identified on comparison of the IPF and poor-outcome CHP groups when using a PVV threshold of 6 · 5% (Log rank test p = 0 · 89).
Percent-predicted DLco and CPI thresholds indicative of an IPF-equivalent outcome were examined in the CHP population (n = 116) and compared to the 6 · 5% PVV threshold. A DLco of <25% predicted was associated with a mean survival of 41 · 8 ± 8 · 1 months and a CPI threshold >65 was associated with a mean survival of 38 · 1 ± 6 · 1 months. The PVV threshold was a stronger predictor of mortality than either PFT threshold.
Combined analysis of IPF and poor-outcome CHP cases
The 20 CHP cases with an IPF-like outcome were combined with the 185 IPF cases and evaluated together using Cox proportional hazards analyses. An MDT diagnosis of CHP was not associated with an improved outcome over patients diagnosed with IPF on univariate analysis. Results were confirmed following correction for age, gender and disease severity (as estimated by the CPI) and bootstrapping of 1000 samples at a confidence interval level of 95%. Results did not change when the 70 IPF patients without a measured Pa02.were included in the IPF cohort.
When patients were categorized as non end-stage disease using an FVC ≥ 50% predicted threshold the results were essentially unchanged (see Additional files 1, Additional file 2 and 3).
Subanalysis of the poor outcome CHP population
No difference in patient age, gender or smoking history was identified between good and bad outcome CHP patients. 13/20 (65%) of poor outcome CHP patients had histopathological or antibody-positive confirmation of a CHP diagnosis and more ILD measured visually (p = 0 · 008) and by CALIPER (p < 0 · 0001) than good outcome patients (Table 3). Overall, CALIPER variables were better able to identify differences between CHP outcome groups than visual CT scores. The mean FVC, DLco and CPI between groups were significantly different (p < 0 · 0001). However no difference was identified in either the mean RVSP (p = 0 · 96), or an RVSP value indicative of pulmonary hypertension [>50 mm Hg] [16] between outcome groups. The lack of a vasculopathic signal was reinforced by the lack of an appreciable difference in Kco values between groups (p = 0 · 88).
Discussion
Our study is the first to our knowledge to evaluate the prognostic value of quantitatively derived CT variables in patients with chronic hypersensitivity pneumonitis. Importantly, we have shown that the pulmonary vessel volume (PVV) provides a robust prognostic signal, independent of PFTs and thresholding of the PVV allowed clear prognostic distinctions to be made in CHP patients. 17% of the CHP study population had a PVV above 6 · 5% of the total lung volume, and these patients demonstrated a rate of disease progression, nearly identical to that of IPF.
Previous studies have reported that a proportion of CHP patients exhibit rapid decline and progression to death, similar to patients with IPF [4, 5]. A study of 47 histologically-confirmed UIP patients performed by Pérez-Padilla et al [4] showed that after correcting for CT disease extent, patients exposed to avian antigens had similar outcomes to patients unexposed to avian antigens [4]. A study by Mooney et al [17] demonstrated that the quartile of CHP patients with the most extensive fibrosis (the sum of visual CT reticulation and honeycomb extent scores), had an outcome comparable to IPF at 4 years. However the study by Mooney et al included CHP patients with respiratory failure and end-stage disease who would be expected to have a poor, IPF-like outcome. In our study our specific aim was to demonstrate that even with the exclusion of patients with end-stage disease, a proportion of CHP patients progress to death with an IPF-like disease course.
A prior study in CHP patients with interspaced CT imaging demonstrated that computer-derived CT variables were strongly predictive of mortality [18]. However the interspaced nature of the dataset precluded certain variables such as the pulmonary vessel volume form being adequately examined as a predictor of mortality. Given that a previous report in patients with IPF demonstrated the PVV to be the strongest CT variable for predicting mortality [9], it was a logical step to examine this key variable in a separate CHP population with volumetric CT imaging. The similar severity-adjusted survival between a subgroup of CHP patients and IPF patients identified in the current study corroborates previous observations that a subset of CHP patients have an outcome nearly identical to IPF. [2, 4, 5] Whilst our findings do not identify the mechanism for this difference, three potential contributing factors could be considered.
Firstly, the PVV threshold may identify CHP patients that have more aggressive lung disease. Alternatively PVV may be providing added information regarding pulmonary vasculopathy, though the similarities in RVSP and Kco between good and bad outcome CHP groups makes this unlikely. Lastly vessel size may be a better marker of interstitial severity than functional indices. The high negative intrathoracic pressures generated during respiration in patients with fibrosing lung disease may increase as fibrosis worsens, drawing extra blood into the lungs as well as concomitantly exerting a pull on pulmonary vessel walls. The mortality signal associated with a high PVV may therefore reflect worsening pulmonary fibrosis.
Intriguingly, a study evaluating pulmonary angiogenesis in patients with CHP has identified elevated levels of vascular endothelial growth factor in the lungs of CHP patients when compared to control subjects [19]. Angiogenesis represents a physiological, hypoxia-induced response within the tissues to facilitate improvements in gas exchange. Therefore an increase in the PVV may represent a response to the progressive impairment in gas exchange that results as the interstitial damage progresses.
The PVV threshold of >6 · 5% of the lung demonstrated a stronger distinction between groups than any individual PFT and has the additional benefit of avoiding the issue of observer disagreement associated with subjective visual CT scoring. Furthermore, given recent evidence suggesting variability between MDTs in making a CHP diagnosis [7], we were able to confirm the PVV signal strength in patients with either a histopathological CHP diagnosis or antibody positivity.
Significant univariate predictors of mortality in the current study are in line with previous reports that have identified CT markers of fibrosis including reticulation [17], honeycombing [20, 21], traction bronchiectasis [3] and a fibrosis score (sum of reticulation and honeycombing) [1, 17, 22] as predictors of a poor prognosis in CHP. A mosaic attenuation pattern (scored as the decreased attenuation component), however, was not predictive of mortality in contrast to the findings in histopathologically proven CHP cases of Lima et al. [1] Emphysema extent scored on CT had no bearing on prognosis, a result which could arguably be reconciled with the relatively limited extent of emphysema in our CHP cohort.
In the CHP group with an IPF-like outcome, the majority (79%) of patients were never smokers and unlike previous reports in CHP, smoking was not associated with a worse outcome [23]. Similarly, the presence of pulmonary hypertension was not associated with a worsened prognosis in contrast to the study by Koschel et al. [24] However the study by Koschel et al [24] did not correct for baseline disease extent when analyzing outcome in CHP patients with pulmonary hypertension which could have negated the study findings.
Of the PFTs, the total lung capacity (TLC) was the strongest predictor of mortality on univariate analysis, echoing the findings of Mooney et al [17]. However, the same association of TLC with mortality was not replicated in a study by Vourlekis et al [2]. On multivariate analysis, CT variables, particularly CALIPER PVV, were stronger predictors of mortality than FVC, DLco, and CPI. The findings mirror the results of the study by Walsh et al which reported the superiority of visual CT variables over any PFTs in predicting mortality in CHP [3].
In our analysis, we deliberately excluded patients with end-stage fibrosing lung disease to allow the identification of patients at risk of a poor outcome, but in whom, targeted intervention might reasonably be expected to modify disease progression. Patients with end-stage disease, characterized by the requirement of oxygen therapy at rest, more usually enter palliative care pathways rather than aggressive medical treatment. The poor outcome of such end-stage CHP cases was highlighted in the study by Mooney et al in which, unsurprisingly, oxygen therapy was shown to be predictive of mortality on multivariate analysis [17]. In our study, with the exclusion of end-stage patients, a significant proportion of CHP patients (17%) had an outcome comparable to IPF.
There are limitations to the current study. In an era in which the MDT diagnosis is the accepted diagnostic standard, the number of patients undergoing surgical biopsy for tissue diagnosis is inevitably reduced. Though only 46% of patients in the final study had a surgical biopsy, the diagnosis of CHP on a histopathological examination alone may not be definitive, as studies have shown that the majority of biopsy samples do not show all of the classical pathological features [1, 6]. The PVV variable was found to be liable to minor contamination as areas of reticular pattern, predominantly in the lung periphery, were captured and classified as vessels. The noise associated with the PVV signal when improved in future iterations of similar software would likely strengthen our observations. Though the median age of our CHP population was slightly higher than several prior CHP studies [2, 25, 26], it was similar to the study by Mooney et al [17] whilst the median age of the poor-outcome CHP group was similar to the HP population in the study by Lacasse et al [25]. We do not believe that these limitations diminish the applicability of our results to patients with an MDT diagnosis of CHP.
Conclusions
In conclusion, we have demonstrated that a significant proportion of CHP patients have an IPF-like outcome and that this can be predicted from the PVV derived from CALIPER software. This novel finding may have a role in identifying the subgroup of CHP patients who may benefit from close monitoring and intervention to control disease progression.
Abbreviations
- Ao:
-
Aorta
- BAL:
-
Broncho-alveolar lavage
- CALIPER:
-
Computer-aided lung informatics for pathology evaluation and rating
- CHP:
-
Chronic hypersensitivity pneumonitis
- CI:
-
Confidence interval
- CPI:
-
Composite physiologic index
- CT:
-
Computed tomography
- DA:
-
Decreased attenuation
- DLco:
-
Diffusing capacity for carbon monoxide
- FEV1:
-
Forced expiratory volume in one second
- FVC:
-
Forced vital capacity
- GGO:
-
Ground glass opacity
- HR:
-
Hazard ratio
- ILD:
-
Interstitial lung disease
- IPF:
-
Idiopathic pulmonary fibrosis
- Kco:
-
Carbon monoxide transfer coefficient
- MDT:
-
Multi-disciplinary team
- PA:
-
Pulmonary artery
- Pa02 :
-
Partial pressure of oxygen
- PFT:
-
Pulmonary function test
- PVV:
-
Pulmonary vessel volume
- RVSP:
-
Right ventricular systolic pressure
- TLC:
-
Total lung capacity
- TxBx:
-
Traction bronchiectasis
- UIP:
-
Usual interstitial pneumonia
References
Lima MS, Coletta ENAM, Ferreira RG, et al. Subacute and chronic hypersensitivity pneumonitis: Histopathological patterns and survival. Respir Med. 2009;103(4):508–15.
Vourlekis JS, Schwarz MI, Cherniack RM, et al. The effect of pulmonary fibrosis on survival in patients with hypersensitivity pneumonitis. Am J Med. 2004;116:662–8.
Walsh SL, Sverzellati N, Devaraj A, et al. Chronic hypersensitivity pneumonitis: high resolution computed tomography patterns and pulmonary function indices as prognostic determinants. Eur Radiol. 2012;22(8):1672–9.
Perez-Padilla R, Salas J, Chapel R, et al. Mortality in Mexican patients with chronic pigeon breeder's lung compared to those with usual interstitial pneumonia. Am Rev Respir Dis. 1993;148:49–53.
Churg A, Sin DD, Everett D, et al. Pathologic patterns and survival in chronic hypersensitivity pneumonitis. Am J Surg Pathol. 2009;33(12):1765–70.
Morell F, Villar A, Montero M-I, et al. Chronic hypersensitivity pneumonitis in patients diagnosed with idiopathic pulmonary fibrosis: a prospective case-cohort study. Lancet Respir Med. 2013;1(9):685–94.
Walsh SLF, Wells AU, Desai SR, et al. Multicentre evaluation of multidisciplinary team meeting agreement on diagnosis in diffuse parenchymal lung disease: a case-cohort study. Lancet Respir Med. 2016;4(7):557–65.
Fink JN, Ortega HG, Reynolds HY, et al. Needs and opportunities for research in hypersensitivity pneumonitis. Am J Respir Crit Care Med. 2005;171(7):792–8.
Jacob J, Bartholmai B, Rajagopalan S, et al. Mortality prediction in idiopathic pulmonary fibrosis: evaluation of automated computer tomographic analysis with conventional severity measures. Eur Respir J. 2016; doi: 10.1183/13993003.01011-2016.
Wells AU, Hirani N. Interstitial lung disease guideline. Thorax. 2008;63 Suppl 5:v1–v58.
Travis WD, Costabel U, Hansell DM, et al. An official American thoracic society/European respiratory society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013;188(6):733–48.
Raghu G, Collard HR, Egan JJ, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183(6):788–824.
Jacob J, Bartholmai B, Rajagopalan S, et al. Automated quantitative CT versus visual CT scoring in idiopathic pulmonary fibrosis: validation against pulmonary function. J Thorac Imaging. 2016;31:304–11.
Ng CS, Wells AU, Padley SP. A CT sign of chronic pulmonary arterial hypertension: the ratio of main pulmonary artery to aortic diameter. J Thorac Imaging. 1999;14(4):270–8.
Edwards PD, Bull RK, Coulden R. CT measurement of main pulmonary artery diameter. Br J Radiol. 1998;71:1018–20.
Bossone E, D’Andrea A, D’Alto M, et al. Echocardiography in pulmonary arterial hypertension: from diagnosis to prognosis. J Am Soc Echocardiogr. 2013;26(1):1–14.
Mooney JJ, Elicker BM, Urbania TH, et al. Radiographic fibrosis score predicts survival in hypersensitivity pneumonitis. Chest. 2013;144(2):586–92.
Jacob J, Bartholmai B, Rajagopalan S, et al. Automated computer-based CT stratification as a predictor of outcome in hypersensitivity pneumonitis. Eur Radiol. 2016;Accepted for publication.
Navarro C, Ruiz V, Gaxiola M, et al. Angiogenesis in hypersensitivity pneumonitis. Arch Physiol Biochem. 2003;111(4):365–8.
Gaxiola M, Buendía-Roldán I, Mejía M, et al. Morphologic diversity of chronic pigeon breeders disease: clinical features and survival. Respir Med. 2011;105(4):608–14.
Tateishi T, Ohtani Y, Takemura T, et al. Serial high-resolution computed tomography findings of acute and chronic hypersensitivity pneumonitis induced by avian antigen. J Comput Assist Tomogr. 2011;35(2):272–9.
Hanak V, Golbin JM, Hartman TE, et al. High-resolution CT findings of parenchymal fibrosis correlate with prognosis in hypersensitivity pneumonitis. Chest. 2008;134(1):133–8.
Ohtsuka Y, Munakata M, Tanimura K, et al. Smoking promotes insidious and chronic farmer's lung disease, and deteriorates the clinical outcome. Intern Med. 1995;34(10):966–71.
Koschel DS, Cardoso C, Weidemann B, Hoffken G, Halank M. Pulmonary hypertension in chronic hypersensitivity pneumonitis. Lung. 2012;190:295–302.
Lacasse Y, Selman M, Costabel U, et al. Clinical diagnosis of hypersensitivity pneumonitis. Am J Respir Crit Care Med. 2003;168(8):952–8.
Hanak V, Golbin JM, Ryu JH. Causes and presenting features in 85 consecutive patients with hypersensitivity pneumonitis. Mayo Clin Proc. 2007;82(7):812–6.
Acknowledgements
The manuscript was supported by the National Institute of Health Research Respiratory Disease Biomedical Research Unit at the Royal Brompton and Harefield NHS Foundation Trust and Imperial College London.
We would also like to thank Professor David Hansell without whom the bulk of this work would not have been possible.
Funding
There is no funding source for the current study. Joseph Jacob had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Availability of data and materials
The datasets analysed during the current study are available from the corresponding author on reasonable request. Permission for use of the database was obtained from the Institutional Ethics Committee of the Royal Brompton Hospital.
Authors’ contributions
JJ, MK, RE, ALB, AUW, DMH were involved in either the acquisition, or analysis or interpretation of data for the study. JJ, AUW and DMH were also involved in the conception and design of the study. BJB, RK and SR invented and developed CALIPER. They were involved in processing the raw CT scans and in generation of figures but were not involved with the analysis or interpretation of the data in the study. All authors revised the work for important intellectual content and gave final approval for the version to be published. All authors agree to be accountable for the all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Competing interests
JJ reports personal fees from Boehringer Ingelheim unrelated to the work in the current manuscript. BJB, RK, SR report a grant from the Royal Brompton Hospital during the conduct of the study; another from Imbio, LLC, was outside the submitted work; and all have a patent: SYSTEMS AND METHODS FOR ANALYZING IN VIVO TISSUE VOLUMES USING MEDICAL IMAGING DATA licensed to Imbio, LLC. AUW receives personal fees for participating in advisory boards and speaking at symposia from Boehringer Ingelheim, Intermune, Roche and Bayer, and for participating in advisory boards from Gilead, MSD and speaker fees from Chiesi. DMH has received a grant from Intermune for creating an educational website and consultancy and receives personal consultancy fees from AstraZeneca, Boehringer Ingelheim, Intermune, Roche, Sanofi, Glaxo Smith Kline. DMH is the recipient of a National Institute of Health Research Senior Investigator Award.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Approval for this study of clinically indicated CT and PFT data was obtained from the Institutional Ethics Committee of the Royal Brompton Hospital and the Institutional Review Board of the Mayo Clinic Rochester.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1:
Brief description of data: Methodological information regarding: pulmonary function test, echocardiography and CT scanning protocols and details regarding CALIPER CT scoring and visual CT scoring and consensus formulation of visual CT scores. A further analysis of end-stage patients defined using an FVC threshold <50% predicted is also included. Two tables highlighting the single determination standard deviation values for the two radiologist scores and a comparison of age and gender matched hypersensitivity pneumonitis groups are also included. (DOCX 125 kb)
Additional file 2: Figure S1.
Brief description of data: Survival curves demonstrating outcomes in non end-stage (defined using an FVC threshold >50% predicted) hypersensitivity pneumonitis patients. (PNG 70 kb)
Additional file 3: Figure S2.
Brief description of data: Survival curves comparing poor-outcome (defined using an FVC threshold >50% predicted) chronic hypersensitivity pneumonitis patients and idiopathic pulmonary fibrosis patients using Kaplan Meier curve analysis. (PNG 80 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

{kind=link}
{kind=link}
Cite this article
Jacob, J., Bartholmai, B.J., Egashira, R. et al. Chronic hypersensitivity pneumonitis: identification of key prognostic determinants using automated CT analysis. BMC Pulm Med 17, 81 (2017). https://doi.org/10.1186/s12890-017-0418-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12890-017-0418-2