
Abstract
Background
Hereditary transthyretin (ATTRv) amyloidosis is a rare, genetically heterogeneous and phenotypically variable systemic disease characterized by deposition of misfolded transthyretin fibrils in various tissues. ATTRv cardiomyopathy and progressive axonal polyneuropathy are the most common manifestations, leading to severe disability and ultimately death within approximately ten years. As disease-modifying treatment options evolve, timely diagnosis and treatment initiation are crucial to prevent rapid disease progression.
Case presentation
Here, we report on a 73-year old patient initially diagnosed with cardiac wild-type ATTR (ATTRwt) amyloidosis by endomyocardial biopsy. Molecular genetic analysis revealed a novel TTR sequence variant (p.Ala65Val) that is highly likely to be amyloidogenic in light of previously reported TTR mutations and the patient’s clinical presentation and family history.
Conclusions
Our findings expand the spectrum of known pathogenic TTR mutations and underline the importance of a thorough diagnostic workup in amyloidosis patients including careful genetic testing to avoid misdiagnosis and missing of treatment opportunities and to enable cascade testing and tracking of carriers.
Similar content being viewed by others


Background
Hereditary transthyretin (ATTRv) amyloidosis is a rare, life-threatening autosomal-dominant systemic disorder characterized by extracellular deposition of misfolded transthyretin fibrils in various tissues. Severe progressive axonal polyneuropathy (ATTRv-PN) and cardiomyopathy (ATTRv-CM) are the most prominent manifestations. Age at symptom onset ranges from 20 to 80 years. Whilst p.Val50Met is the most common underlying genotype globally and in endemic areas such as certain regions of Northern Portugal and Sweden, in non-endemic areas genetic heterogeneity is high with more than 120 amyloidogenic TTR mutations identified to date [1]. There are mutations classically associated with either a predominantly cardiac (p.Val142Ile, p.Ile88Leu) or neuropathic phenotype (p.Val50Met), but both presentations frequently overlap [2, 3]. Genetic heterogeneity and phenotypic variability including varying age at disease onset even in carriers of the same mutation, variable penetrance, insufficient diagnostic workup and low disease awareness frequently lead to a diagnostic delay of several years [1]. As untreated ATTRv amyloidosis leads to death within 10 years on average [4] and highly efficient treatment options for ATTRv-PN emerged in recent years, awareness of typical clinical signs and symptoms is crucial for early diagnosis and treatment initiation. Current treatment options for ATTRv-PN comprise tafamidis, an oral TTR tetramer stabilizer, patisiran, a small interfering RNA, and inotersen, an antisense oligonucleotide, the latter ones both leading to degradation of mutant and wildtype TTR mRNA. Diagnosis of ATTRv amyloidosis relies on sequencing of the whole TTR gene, which is mandatory even in bioptically confirmed cases to distinguish between ATTRv and wildtype ATTR (ATTRwt) amyloidosis because of different treatment approaches. ATTRwt amyloidosis is a frequent cause of heart failure with preserved ejection fraction (HFpEF) occuring almost exclusively in men beyond the age of 60, but usually does not lead to a clinically relevant and progressive peripheral neuropathy. However, the prevalence of ATTRwt amyloidosis, sometimes referred to as senile variant, is far higher than that of ATTRv amyloidosis. In studies of autopsy specimens, cardiac wildtype ATTR deposits can be found in up to 25% of patients aged 80 years or older [5]. Treatment with tafamidis that reduces disease progression is approved for ATTRwt amyloidosis (61 mg daily) as well as ATTRv-CM (61 mg daily) and ATTRv-PN (20 mg daily), while inotersen and patisiran that in some cases even improves functional abilities of affected individuals are only approved for the treatment of patients with ATTRv amyloidosis with peripheral neuropathy [6].
Here, we report on a novel TTR mutation underlying late-onset ATTRv amyloidosis with mixed neuropathic and cardiac phenotype initially misdiagnosed as ATTRwt amyloidosis.
Case presentation
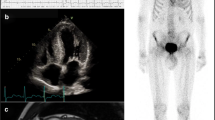
A 73-year old male with progressive impairment of gait, persisting neuropathic pain in his legs, dizziness due to postural changes, and unexplained weight loss of 20 kg (BMI 26.8 kg/m2) during the last 2–3 years presented to our outpatient clinic. Three years ago diagnosis of lumbar spinal stenosis was made and decompression surgery was performed to alleviate neuropathic pain. As symptoms progressed, 1 year before the first presentation a second surgical procedure was scheduled and it was only in the preoperative diagnostic workup that echocardiography revealed left ventricular hypertrophy suspected to represent cardiac amyloidosis (see Fig. 1). Endomyocardial biopsy showed extensive amyloid deposition in Congo red stained tissue with a typcial apple green birefringence and fluorescence signal, which could be classified immunohistochemically as TTR derived amyloid (see Fig. 2). Regarding the cardiac manifestation and the patient’s age and sex ATTRwt amyloidosis was suspected and treatment with tafamidis (61 mg daily) was initiated.

The patient’s echocardiography. Four-chamber view with prominent left-ventricular hypertrophy and granular sparkling typical for amyloidosis

Results of the myocardial biopsy. Hematoxylin and eosin stain (panel A, magnification 300x), Congo red fluorescence showing amyloid deposition (panel B, magnification 20x, and C, magnification 300x), immunohistochemical staining using antibodies directed against immunoglobulin light chains without relevant reaction (panel D, magnification 300x), and immunohistochemical staining positive for transthyretin (panel E, magnification 20x, and F, magnification 300x)
At the time of referral, the patient was not able to walk unaided for almost 1 year due to weakness of his legs and impairment of balance. Neuropathic pain and dizziness due to orthostatic dysregulation had progressed and the patient reported no obvious improvement after approximately 1 year of treatment with tafamidis. The patient’s family history revealed frequent deaths due to cardiac events in his first-degree relatives (see Fig. 3). His father died at the age of 67 from a cardiac event not further specified, as well as two of the patient’s brothers. One of them reportedly suffered cardiomyopathy and polyneuropathy. The only living brother, aged 68, was considerably impaired by a cardiomyopathy and peripheral neuropathy of unknown etiology.

The patient’s pedigree. The reported patient and his only living brother both show cardiomyopathy (hatched) and polyneuropathy (black shading), and the p.Ala65Val variant. One of the three other brothers suffered cardiomyopathy and polyneuropathy. The patient’s father and two of his brothers died from cardiac events. *own documented evaluation, E (Evaluation) = molecular genetic analysis of the TTR gene
The clinical examination showed a severe impairment of gait due to sensory ataxia, bilateral distal paralyses of the lower extremities (ankle dorsiflexion MRC grade 2/5, plantar flexion MRC grade 2–3/5 on the right side, MRC grade 3–4/5 on the left side). A slight weakness of both hands (MRC grade 4/5) was evident, as well as moderate atrophy of distal limb muscles, distal symmetric hypesthesia, and lost tendon reflexes. Nerve conduction studies (NCS) confirmed a severe axonal sensorimotor neuropathy with bilaterally unobtainable sural and tibial nerve amplitudes. Corneal confocal microscopy (CCM) showed a highly reduced corneal nerve fiber density (7 fibers/mm2, reference: > 24 fibers/mm2) indicating prominent small fiber involvement. Extensive laboratory testing for potential causes of the neuropathy including HbA1c, vitamin B1, B6, B12, folic acid, TSH, ANA, ANCA, rheumatid factor, hepatitis serology, serum electrophoresis and immunofixation did not yield any relevant abnormalities. There was no history of alcohol abuse or any other exposure to neurotoxic agents. However, molecular genetic analysis (Sanger sequencing, bioinformatic analysis of collected data by means of Mutation Surveyor Version 3.10 and Alamut Visual Version 2.6.1) revealed a heterozygous sequence variant in exon 2 of the TTR gene (NM_000371.3 (TTR): c.194C > T, p.Ala65Val, s. Table 1 and Fig. 4), classified as likely pathogenic (class 4) according to the American College of Medical Genetics (ACMG) classification system, which had been reported in ClinVar twice (ClinVar Accession: VCV000448841.4, ClinVar Variation ID: 448841). In ClinVar, the variant is classified as a variant of uncertain significance (two submissions) and once with the condition amyloidogenic transthyretin amyloidosis, but there is no literature on individuals with TTR-related conditions with this genotype. ACMG criteria PM1 (variant located in a mutational hot spot), PM2 (variant absent from general population in databases gnomAD/ExAC), PM5 (novel missense change at an amino acid residue where a different missense change determined to be pathogenic has been seen before), and PP3 (multiple lines of computational evidence supported a deleterious effect on the gene product) were fulfilled [7]. Presuming this genotype to be amyloidogenic, the patient’s brother underwent targeted genetic testing by Sanger sequencing that revealed the same heterozygous sequence variant.

DNA sequencing electropherogram. Reference trace (above) and sample trace (below) with heterozygous C > T point mutation (yellow shaded)
Discussion and conclusions
Here, we report on a novel point mutation (p.Ala65Val) underlying late-onset ATTRv amyloidosis with a mixed cardiac and neuropathic phenotype, thereby extending the spectrum of known amyloidogenic TTR mutations. The amyloidogenic nature of the p.Ala65Val mutation is obvious for several reasons. Firstly, our patient and his brother both show a classical ATTRv amyloidosis phenotype with HFpEF and rapidly progressive peripheral neuropathy with prominent small fiber involvement and autonomic dysfunction leading to orthostatic hypotension, weight loss and neuropathic pain. Lumbar spinal stenosis is common among patients with ATTRv amyloidosis because of amyloid deposition in the ligamentum flavum. Secondly, different amino acid replacements at the same locus have been reported to cause isolated ATTRv-CM (p.Ala65Ser, p.Ala65Thr, p.Ala65Gly) or ATTRv-CM and ATTRv-PN (p.Ala65Asp) previously [8] and in silico analyses via different algorithms relying on evolutionary conservation of sequences (FATHMM) and protein structure/function relationship (SIFT, Sorting Invariant from Tolerated; PolyPhen-2; MutationTaster; Align-GVGD; PROVEAN) predicted a pathogenic effect on amino acid level. Thirdly, the family history of frequent cardiac events without a different common risk factor suggests a genetic background. Although the exact cause of death remained undetermined in the patient’s relatives and no member of the family had been diagnosed with amyloidosis before, unrecognized amyloid cardiomyopathy can be assumed.
We did not perform sural nerve biopsy, since it is not well suited to rule out amyloid neuropathy due to the proximodistal gradient of nerval TTR amyloid deposition [9]. As the patient exhibits a progressive disease course despite treatment with tafamidis, the detection of his TTR mutation and evaluation of his severe neuropathy enables a change of the treatment strategy considering patisiran and inotersen as potential new options.
In conclusion, our case report underlines the necessity of careful molecular genetic analysis and history taking in every case of TTR amyloidosis independent from the age at onset, particularly in non-endemic areas, to not overlook rare cases of hereditary TTR amyloidosis and offer appropriate disease-modifying treatments.
Availability of data and materials
All data analysed are included in this published article.
Abbreviations
- ACMG:
-
American College of Medical Genetics
- ANA:
-
Antinuclear antibodies
- ANCA:
-
Anti-neutrophil cytoplasmatic antibodies
- ATTR:
-
transthyretin-derived amyloid
- ATTRv:
-
transthyretin-derived amyloid with underlying TTR mutation (v = variant)
- ATTRwt:
-
transthyretin-derived amyloid without underlying TTR mutation (wt = wildype)
- BMI:
-
Body mass index
- CM:
-
Cardiomyopathy
- ExAC:
-
Exome Aggregation Consortium
- gnomAD:
-
Genome Aggregation Database
- HFpEF:
-
Heart failure with preserved ejection fraction
- MRC:
-
Medical Research Council
- NCS:
-
Nerve conduction studies
- PN:
-
Polyneuropathy
- TSH:
-
Thyroid-stimulating hormone
- TTR:
-
Transthyretin
References
Adams D, Koike H, Slama M, Coelho T. Hereditary transthyretin amyloidosis: a model of medical progress for a fatal disease. Nat Rev Neurol. 2019;15:387–404.
Rappezzi C, Quarta CC, Obici L, et al. Disease profile and differential diagnosis of hereditary transthyretin-related amyloidosis with exclusively cardiac phenotype: an Italian perspective. Eur Heart J. 2013;34(7):520–8.
Russo M, Obici L, Bartolomei I, et al. ATTRv amyloidosis Italian Registry: clinical and epidemiological data. Amyloid. 2020;27(4):259–65.
Planté-Bordeneuve V, Said G. Familial amyloid polyneuropathy. Lancet Neurol. 2011;10:1086–97.
Cornwell GG, Murdoch WL, Kyle RA, Westermark P, Pitkänen P. Frequency and distribution of senile cardiovascular amyloid. A clinicopathologic correlation. Am J Med. 1983;75:618–23.
Adams D, Slama M. Hereditary transthyretin amyloidosis: current treatment. Curr Opin Neurol. 2020;33:553–61.
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the American Association for Molecular Pathology. Genet Med. 2015;17(5):405–24.
Online registry for mutations in hereditary amyloidosis. amyloidosismutations.com/mut-attr.php. Accessed 10 Dec 2021.
Kollmer J, Hund E, Hornung B, et al. In vivo detection of nerve injury in familial amyloid polyneuropathy by magnetic resonance neurography. Brain. 2015;183:549–62.
Acknowledgements
Not applicable.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
AT acquired, analyzed and interpreted the patient data and wrote the manuscript. SO, JH, AC, MP, PL, LK, CR, and CR contributed substantially to data acquisition and revised the manuscript for intellectual content. ID contributed molecular genetic analysis data and revised the manuscript for important intellectual content. TR, HS, and CK revised the manuscript for intellectual content. TH contributed to data analysis and interpretation and revised the manuscript for intellectual content. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All investigations were carried out according to the Declaration of Helsinki and the local ethics committee guidelines as part of the clinical routine, not for research purposes. A formal approval for this case report was not required.
Consent for publication
Both patients presented in detail gave their written informed consent for publication. For the deceased family members included in the pedigree their next of kin consented to publication.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Thimm, A., Oubari, S., Hoffmann, J. et al. A novel TTR mutation (p.Ala65Val) underlying late-onset hereditary transthyretin (ATTRv) amyloidosis with mixed cardiac and neuropathic phenotype: a case report. BMC Neurol 22, 469 (2022). https://doi.org/10.1186/s12883-022-02952-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12883-022-02952-3