
Abstract
Purpose of Review
The review’s main focus centers on the genetics of hereditary cardiac amyloidosis, highlighting the opportunities and challenges posed by the widespread availability of genetic screening and diagnostic cardiac imaging.
Recent Findings
Advancements in cardiac imaging, heightened awareness of the ATTR amyloidosis diagnosis, and greater access to genetic testing have all led to an increased appreciation of the prevalence of ATTR cardiac amyloidosis. Elucidation of the TTR molecular structure and effect of mutations on TTR function have allowed for novel TTR therapy development leading to clinical implementation of transthyretin stabilizers and transthyretin gene silencers.
Summary
The transthyretin amyloidoses are a diverse group of protein misfolding disorders with cardiac and peripheral/autonomic nervous system manifestations due to protein deposition. Genetic screening allows for the early identification of asymptomatic TTR mutation carriers. With the advent of TTR-specific therapeutics, clinical guidance is necessary for the management of individuals with mutations in the TTR gene without evidence of disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction and Nomenclature of Amyloidosis
Amyloidosis is a broad classification for a group of diseases that are characterized by the extracellular deposition of insoluble fibrillar proteins in the form of β-pleated sheets, leading to organ dysfunction [1•]. The presence of the amyloid protein is identified by specific histological techniques employing Congo red staining [2], while the precursor protein that misfolds and forms amyloid fibrils is defined by immunohistochemistry or mass spectroscopy [1•]. The type of amyloidosis is defined by the precursor protein that misfolds. Amyloidogenic light chain (AL) amyloidosis is characterized by organ deposition of monoclonal lambda or kappa free light chains, in the context of a clonal proliferation of bone marrow-derived plasma cells. In contrast, amyloidogenic transthyretin (ATTR) amyloidosis is sub-classified by the genetic sequence of the TTR gene as either genetically normal (wild-type) or variant transthyretin protein. In ATTR amyloidosis, TTR protein, synthesized by the liver, misfolds into amyloid fibrils and causes a specific constellation of organ dysfunction. For the purposes of this review, focused on genetically inherited cardiac amyloidosis, we will restrict our discussion to ATTR amyloidosis with cardiomyopathy.
Transthyretin Protein
Transthyretin (TTR) protein (formerly called prealbumin) is a 127-amino acid plasma transport protein that binds to thyroid hormone and retinol/retinol-binding protein [3], and is encoded by a single-copy gene on the long arm of chromosome 18. Transthyretin has 4 exons that each code for one of four monomers that circulate as a homotetramer (a protein complex made up of four identical subunits that are associated but not covalently bound) [4]. Nearly all TTR is synthesized in the liver but other sites of production include the choroid plexus of the brain and the retinal pigment endothelium of eye [5••]. Despite its name, TTR is not an important contributor of circulating thyroid hormone, while it is an integral protein for transport of retinol (vitamin A).
Organ Involvement
Interestingly, misfolded TTR protein, be it in the context of wild-type or variant genetics, results in significant heterogeneity in the organ manifestations of ATTR amyloidosis. Wild-type ATTR (ATTRwt) amyloidosis most commonly affects the heart and soft tissue, resulting in carpal tunnel syndrome, spinal stenosis [6], and in some cases, spontaneous biceps tendon rupture [7]. The manifestations of hereditary ATTR (hATTR or ATTRv for variant) vary depending on the pathogenic mutation in the TTR gene. The most common manifestations of ATTRv amyloidosis include cardiomyopathy (ATTR-CM), sensorimotor and/or autonomic polyneuropathy, leptomeningeal (and oculoleptomeningeal), and soft tissue involvement.
Polyneuropathy
The peripheral neuropathy of ATTRv amyloidosis begins as a small fiber sensory polyneuropathy in the lower extremities with clinical manifestations as paresthesias and hyperesthesias [8]. There is progression of this sensory neuropathy to more proximal areas of the lower extremities, with eventual involvement of the fingers and hands. There is early impairment of temperature and pain sensation, followed by vibration and position sensation impairment. This sensory neuropathy is subsequently followed by a motor neuropathy resulting in foot drop, wrist drop, and loss of manual dexterity [9].
Autonomic neuropathy may exist alone or in conjunction with a sensorimotor neuropathy, and may be an early sign of the disease. Manifestations of autonomic neuropathy include orthostatic hypotension, GI disturbances including impaired gastric emptying, intractable diarrhea, impotence, anhidrosis, and urinary retention [10, 11]. Autonomic involvement often results in significant morbidity, complicating the management of ATTR-CM related congestive heart failure and may dominate the phenotypic expression of this disease.
Leptomeningeal Involvement
Leptomeningeal amyloidosis is a prominent feature of certain, relatively rare, TTR mutations (Gly47Arg) [12], Asp18Gly [13], Leu12Pro [14], among others, characterized by central nervous system amyloid deposition in the pia and arachnoid membrane and in the subarachnoid space. This results in intracranial hemorrhage, focal neurologic signs, ataxia, visual impairment, dementia, and psychosis. There may be associated vitreous deposits resulting in oculoleptomeningeal involvement [15, 16].
Ocular Involvement
The spectrum of eye involvement includes vitreous opacities, dry eyes, glaucoma, and ocular amyloid angiopathy, and may be seen in a fifth of the patients with the V30M phenotype [5••, 17]. In rare cases, ocular amyloid may be the sole manifestation of the disease [18].
Nephropathy
Renal involvement is variable, and mild renal involvement may be seen with the early-onset Portuguese variant of the V30M mutation, but severe renal dysfunction is less common [19].
Cardiovascular Manifestions of ATTR Disease
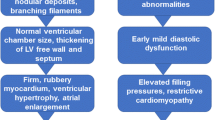
Cardiac amyloidosis is a restrictive cardiomyopathy characterized by extracellular deposition of amyloid fibrils leading to increased biventricular wall thickness and advanced diastolic dysfunction. Cardiac imaging is essential for raising suspicion of diagnosis and as such, echocardiography and cardiac MRI both reflect increased biventricular wall thickness. As amyloid deposits are extracellular, they can be indirectly visualized by imaging, as well. Extracellular amyloid fibril deposition is responsible for the characteristic pattern of late gadolinium enhancement (LGE), prolonged native T1 time, and increased extracellular volume (ECV), noted on cardiac MRI [20, 21]. While left ventricular systolic function as assessed by the left ventricular ejection fraction may appear normal in earlier stages of the disease, deformation imaging through speckle tracking echocardiography often reveals early and characteristic abnormalities in cardiac amyloidosis [22, 23]. Amyloid fibril deposition also leads to a discordance between ventricular wall thickness on cardiac imaging and QRS voltage on ECG, as patients with cardiac amyloidosis may have low QRS voltages on ECG despite increased left ventricular wall thickness [24]. Clinically, this pathophysiology leads to high filling pressures, significant fluid retention, and exercise intolerance due to an inability to augment cardiac output during exertion. Finally, nuclear imaging with bone-avid compounds has dramatically altered the diagnostic approach to ATTR amyloidosis, offering the potential for widespread screening in at-risk patients.
Diffuse atrial infiltration with amyloid fibrils leads to a high prevalence of atrial fibrillation and other atrial arrhythmias [25]. Atrial electromechanical dissociation may also be seen even in the absence of atrial fibrillation and may result in cardioembolic events in sinus rhythm [26, 27]. Often, a diffuse infiltration of the conduction system is present leading to advanced conduction disease that may require permanent pacing [28,29,30]. While pulseless electrical activity is thought to be the most common proximate cause of cardiac death in cardiac amyloidosis, ventricular arrhythmias are common and ICD implantation may be reasonable in select patients for secondary prevention. The benefit of implantation for primary prevention remains less clear [31].
Current Approach to ATTR Amyloidosis Identification
There are eleven different proteins that can misfold into amyloid fibrils and infiltrate the heart [32]. With the advent of gene-silencing therapeutics in ATTRv amyloidosis, accurate typing of amyloid protein is essential for the development of appropriate therapeutic strategies. Of the cardiac-avid amyloid proteins, AL and ATTR are by far the most commonly seen in the USA. Gelsolin amyloidosis is a rare heritable disease that causes well recognized neuropathy and cardiac involvement, but is seen principally in northern Europe [33]. Other precursor proteins that are associated with cardiomyopathy, such as ApoA1, are quite rare.
In the setting of clinical suspicion of cardiac amyloidosis, four main initial tests should be considered: (1) serum free light chains (FLC); (2) serum immunofixation electrophoresis (SIFE); (3) urine immunofixation electrophoresis (UIFE); and (4) 99mTc-pyrophosphate (99mTc PYP) nuclear scan (in the USA) (Fig. 1). The laboratory testing (FLC, SIFE, and UIFE) is essential initial testing elements for AL amyloidosis screening. While bone-avid radiotracers have been applied to cardiac imaging for over 40 years, in the early 2000s, researchers described the application of a similar tracer to PYP (99mTc-3,3-diphosphono-1,2-propanedicarboxylic acid (99mTc DPD), available only in Europe) that not only detected cardiac amyloidosis but differentiated ATTR and AL cardiac amyloidosis with 100% specificity [34, 35]. In 2013, Bokhari and colleagues showed comparable high rates of sensitivity (97%) and specificity (100%) with 99mTc PYP (nuclear tracer available in the USA) in differentiating AL and ATTR cardiac amyloidosis using a heart to contralateral lung (H/CL) quantitative ratio > 1.5 [36••]. Several multicenter evaluations have validated these results with the 99mTc PYP technique and additionally showed H/CL ratio > 1.6 conferred worse survival [37, 38]. With the adoption, awareness, and growing availability of this imaging technique, 99mTc PYP with its remarkable sensitivity and specificity for the ATTR amyloidosis has nearly obviated the need for histological typing of amyloid protein to rule in ATTR cardiac disease. The capacity to diagnose ATTR amyloidosis without biopsy has resulted in a dramatic increase of identified cases over the past 5 years. One important caveat to the nuclear testing is the decreased specificity noted in patients with circulating monoclonal protein and AL amyloidosis; thus, it is imperative to rule out a monoclonal gammopathy with FLC, SIFE, and UIFE as noted above. In cases of clinical ambiguity or equivocal results, obtainment of tissue (usually via endomyocardial biopsy) for typing via immunohistochemistry or with amyloid deposits micro-dissected by laser capture with application of tandem mass spectrometry (gold standard) is recommended [39••]. Once a diagnosis of ATTR amyloidosis is established by imaging with exclusion of AL amyloidosis by serum/urine testing, genetic sequencing of TTR is necessary.

a Key signs commonly encountered in cardiac amyloidosis; b Recommended studies necessary for ruling out light-chain cardiac amyloidosis and diagnosing hereditary ATTR cardiac amyloidosis
Genetic Overview of Hereditary ATTR Disease
There are over 140 known mutations in the transthyretin gene with the majority occurring in exon 2 and 3 [5••]. Mutations in the transthyretin protein structure lead to destabilization of tetrameric formation (the rate limiting step in amyloidogenesis) and accelerate fibril formation. The nomenclature for ATTRv disease places a 1- or 3-letter abbreviation for the normal amino acid at the mutation point followed by the substituted amino acid (e.g., ATTR V122I signifies isoleucine replacing valine at position 122 in the transthyretin amino acid sequence) [40••]. There is often confusion in contemporary genetic reports as the 20-amino acid signal sequence is often included in the numbering of residues. Hence, V122I and pV142I refer to the same substitution. As both wild-type TTR (TTR protein with normal genetic sequence) and mutated TTR can each result in amyloidogensis, non-genetic factors definitively contribute to fibril deposition [5••, 41]. ATTR amyloidosis only requires one mutant TTR allele for disease pathogenesis, thus imparting an autosomal dominant inheritance pattern (50% risk of passage to offspring). Most individuals with the disease phenotype are heterozygous for the pathogenic mutation and express both normal and mutated TTR and as such TTR amyloid fibrils isolated from heterozygous patients contain both normal and variant TTR. When tissue deposits of amyloid are analyzed, 65–75% are composed of variant TTR and the remainder normal TTR [5••].
Key Elements Contributing to Hereditary ATTR Phenotypic Heterogeneity
Type of Mutation
One of the most important determinants of cardiac phenotypic presentation in ATTRv amyloidosis is the mutation. There are five main mutations that are implicated in an exclusive, predominant, or frequently occurring cardiac phenotype worldwide: V122I, T60A, V30M (late-onset), I68L, and L111M [42, 43•, 44, 45] (Table 1). The first three of these mutations are most commonly seen in the USA as geography is an important factor in the epidemiology and mutation prevalence for diagnosed cases of cardiac amyloidosis [46•]. V122I always exhibits a cardiac phenotype and while not classically attributed to this mutation, peripheral neuropathy can co-exist in a small proportion of patients [46•, 47]. V122I is by far the most common mutation seen in the USA with an estimated prevalence of 3.4% among African Americans [48]. T60A, a mutation with origins in the northern part of the Republic of Ireland, nearly always is associated with a cardiac phenotype but often demonstrates significant percentage of mixed disease (both cardiac and neurologic involvement) with autonomic dysfunction seen in 75% of individuals and peripheral neuropathy in 54% [45]. V30M is likely the most common mutation worldwide (outside of the USA) and has many phenotypic presentations heavily influenced by geographic location. Specifically, when evaluating patients with the mutation, outside of endemic geographical loci in specific parts of Japan, Portugal, and northern Sweden (“non-endemic”), the phenotypic presentation has low apparent penetrance that is much more age-dependent, prominently cardiac in expression with a more indolent neuropathy [49, 50].
Age
Age provides crucial effect modification between the association of TTR mutations and phenotypic expression. Interestingly, while TTR mutations are present since birth, ATTR phenotypic expression does not develop until adulthood. With most mutations, penetrance rates increase with age [43•, 49, 51] suggesting that biological processes in addition to mutational considerations, related to the aging process (or environmental/lifetime exposures), over time play a role in disease presentation and timing.
Sex
Sex of the transmitting parent, as well as sex of the carrier, plays importance in phenotypic presentation in hereditary ATTR disease. Many TTR mutations demonstrate equal sex involvement; however, many do show higher incidence in males compared to females, such as V30M non-endemic Japanese cohorts and French V30M carriers [50,51,52,53]. However, sex appears to play a “protective” role in women with less involvement of cardiac disease, often with later onset. For those women with cardiac ATTR disease, less severe cardiac expression with reduced wall thickness and higher LV systolic function has also been observed, at least before menopausal age [54]. In addition, a validation genetic evaluation of Swedish V30M carriers showed higher rates of trait penetrance when inherited from the mother than the father [49]. The mechanism of such sex-specific effects, particularly in regard to earlier-onset disease with maternal inheritance, has not been well-elucidated although some studies have shown differences in mitochondrial DNA haplotypes in these groups [55].
Fibril Composition
There is growing data regarding the association of amyloid fibril composition with organ tropism, and therefore phenotypic presentation of ATTR amyloidosis clinical manifestations. First described by Swedish investigators among carriers of the V30M mutation using subcutaneous fat pad biopsies, full-length TTR (type B fibrils) was associated with earlier-onset disease without a cardiac phenotype with strong affinity for Congo Red and brighter green birefringence. Amyloid deposits composed of TTR fragments (type A fibrils) associated with later onset disease and cardiac involvement and with weaker Congo Red staining and birefringence [56]. Verified at autopsy, all organs displayed the same type of fibril composition in affected organs as noted in the fat pad biopsy. The same group extended this observation to a cohort of non-V30M and noted that type A fibrils were present in nearly all the investigated non-V30M (exception being two ATTR Y114C) carriers with cardiac involvement [57]. In addition, wild-type ATTR has been shown to also have the fragmented type A fibrils [58]. The clinical relevance of fibril composition may inform differential bone-avid nuclear tracer uptake as 97% of patients with type A fibrils and none of the patients with type B fibrils displayed 99mTc DPD uptake. Thus, the specificity of the 99mTc nuclear scans may be partially explained by the fibril type.
Geographic Considerations
As described above, the phenotypes of familial ATTR disease are often varied but important clinical features and characteristics are shared by kinships and individuals from a particular region or singular background, such as V30M described in specific regions in Portugal, northern Sweden, and Japan and have classically been referred to as “endemic V30M” [52, 59,60,61]. With advances in molecular and genetic testing, TTR mutations have been reported worldwide. Harnessing data from a global, multicenter, longitudinal assessment (Transthyretin Amyloidosis Outcomes Survey, (THAOS)), it was demonstrated that TTR amyloidosis type and presentation differ between the USA and the rest of the world (older, more male, more wild-type ATTR disease, with more cardiac phenotypes, V122I mutation most commonly seen in USA) [46•].
Approach to Genotype-Positive, Phenotype-Negative Individuals
With the widespread availability of genetic testing platforms and overall heightened awareness of ATTR cardiac amyloidosis, pre-symptomatic testing has become a frequent query for clinicians, usually prompted by an index patient in a family. Unfortunately, while there are no evidence-based practice guidelines established to assist in the approach to genotype-positive, phenotype negative carriers, working groups comprised of ATTR experts have convened and developed consensus documents to help guide clinicians for pre-symptomatic testing [62,63,64]. Establishing a monitoring approach that will be high-yield to identify early disease while simultaneously not leading to undue anxiety and increasing the cost of healthcare by unnecessary testing is critically important when considering pre-symptomatic testing. Thus, the predicted age of onset of symptomatic disease (PADO) is important to determine on an individual basis, based on the particular mutation, typical age of onset for that mutation, and onset among family members with established ATTR amyloidosis. Once the PADO has been determined for the individual at-risk, one expert consensus group has proposed monitoring to begin 10 years earlier to establish a baseline and continue annually and perhaps become more frequent as a patient nears PADO, particularly those with mutations that are known to rapidly progress [62]. Through these monitoring visits, a clinical (and not pathological) diagnosis of ATTR amyloidosis can be assumed as described in these consensus guidelines with the following scenarios (assuming comorbidities have been appropriately excluded):
-
1)
One quantified/objective sign or symptom definitely related to onset of ATTR amyloidosis (new sensorimotor neuropathy changed from baseline, autonomic neuropathy or neurogenic/sexual dysfunction, cardiac involvement, or renal/ocular involvement), or
-
2)
Any symptoms/sign likely related to ATTR disease in the absence of objective signs PLUS 1 abnormal test finding, or
-
3)
Absence of symptoms PLUS 2 abnormal test findings
Current Treatment Approaches to Hereditary Cardiac ATTR Amyloidosis
Symptomatic management of heart failure is an important principle of cardiac amyloidosis therapy. Volume reduction with loop diuretics and mineralocorticoid antagonists remains the mainstay of therapy. Standard heart failure management with beta-blockers, ace-inhibitors, and angiotensin receptor blockers is often not tolerated due to worsening of heart failure symptoms and hypotension, especially in the setting of underlying autonomic dysfunction. Calcium channel blockers may be contraindicated due to direct binding of these agents to amyloid fibrils resulting in local toxicity and worsening heart failure [65, 66]. Similarly, digoxin binds irreversibly to AL amyloid fibrils in vitro, and may be associated with local digoxin toxicity in cardiac amyloidosis, although a more recent report suggests that digoxin use may be safe with cautious use in this population [67].
TTR-specific therapy has recently focused primarily on cessation of mutant TTR production by the liver or the stabilization of the TTR tetramers to slow formation of amyloid fibrils. Orthotopic liver transplant (OLT) was the first therapy to target liver production of TTR. Early experience with OLT in hereditary ATTR with the V30M mutation was promising with no evidence of disease progression at 6 months [68] and with some clinical improvement at 2 years post-transplant [69]. Long-term registry data suggested an 82% 5-year survival with OLT in ATTR V30M as compared to a 59% 5-year survival in ATTRv patients without the V30M mutation [70]. However, longer-term follow-up identified progression of ATTR cardiomyopathy even after OLT [71]. Fibril composition appears to play an important role in disease progression, as patients with type A fibrils (mixture of truncated and full-length fibrils) having a higher propensity for development or progression of cardiomyopathy after OLT [72]. Furthermore, it appears that type A fibrils are likely to complex with ATTRwt fibrils leading to progressive cardiomyopathy after OLT, despite removal of mutant protein [73, 74]. Thus, while OLT may be promising in selected patients with V30M ATTRv disease without significant cardiomyopathy, this therapy is not an optimal long-term option for all patients with ATTRv disease. Additionally, OLT will not benefit patients with wild-type ATTR amyloidosis.
The discovery of a Portuguese family who were complex heterozygotes with V30M/T119M TTR phenotype and had no manifestation of ATTRv disease led to the realization that natural TTR stabilization (in the form of a hetero tetramer in this case) could prevent disease development [75, 76]. The subsequent search for synthetic TTR stabilizers led ultimately to the repurposing of the NSAID diflunisal and the development of the novel compound tafamidis. In patients with ATTRv neuropathy, tafamidis reduces the rate of progression of neuropathy [77••] and is associated with improved survival [78]. Likewise, in a placebo-controlled randomized trial, diflunisal reduced the progression of neurologic symptoms at 2 years in patients with ATTRv peripheral and autonomic neuropathy and was associated with preserved quality of life [79••]. For ATTR-CM, TTR stabilization with diflunisal and tafamidis was also associated with improved survival in a retrospective cohort study with a mixed cohort of patients with ATTRwt and ATTRv cardiomyopathy (with most patients in the ATTRv group expressing the V122I mutation) [80]. Most recently, a randomized controlled trial of tafamidis in patients with ATTRwt and ATTRv cardiomyopathy revealed a lower rate of the primary composite outcome of all-cause mortality and cardiovascular hospitalizations in the tafamidis group compared to placebo. Tafamidis was associated with lower all-cause mortality [81••]. Based on this pivotal trial, the FDA approved tafamidis for patients with ATTR cardiomyopathy, irrespective of mutation status, in May 2019. Tafamidis is also approved for ATTR polyneuropathy in Europe, Japan, and South America.
While TTR stabilization prevents TTR tetrameric dissociation and subsequent amyloid fibrillogenesis, there is also interest in inhibiting the production of TTR by the liver through TTR gene silencing. The small interfering RNA (siRNA) agent, patisiran, improved neurologic function and quality of life in patients with ATTRv peripheral neuropathy in a randomized, placebo-controlled clinical trial [82••]. In the subgroup of patients with cardiac amyloidosis, patisiran was associated with reduced all-cause hospitalizations and mortality, with a trend towards improvement in left ventricular wall thickness, global longitudinal strain, and cardiac biomarkers [83]. Similarly, the anti-sense oligodeoxynucleotide inotersen improved neurologic outcomes and quality of life in patients with ATTRv peripheral neuropathy in another randomized trial [84••]. Each of these two gene-silencing agents was approved by US FDA for ATTR polyneuropathy in hereditary amyloidosis only. Neither of these agents is currently approved for ATTRv without neuropathy (i.e., largely excluding V122I carriers) nor approved for ATTRwt amyloidosis. While published reports primarily assessed the effect of TTR gene-silencing on neurologic outcomes, clinical trials of gene-silencing agents that include primary cardiovascular end points are ongoing.
Conclusions
Transthyretin amyloidosis can occur in the context of genetically normal or mutant TTR protein. While ATTRwt is almost exclusively a cardiac-restricted phenotype, the ATTRv amyloidoses are a group of diverse autosomal dominant disorders caused by point mutations in the TTR gene. These diseases are characterized by varying degrees of a sensorimotor and autonomic neuropathy and a restrictive cardiomyopathy, with phenotypic expression varying by mutation, sex, and age of onset. The development of critical diagnostic tools, including Tc99m pyrophosphate cardiac imaging and genetic testing, has led to a heightened awareness of these diseases. Widespread genetic testing raises questions of penetrance and the management of patients with genotype-positive, phenotype-negative disease. While treatment with TTR gene silencers and TTR stabilizers has been effective in the management of nervous system and cardiac manifestations of this disease, further studies are needed to explore the role of these agents in preventing the onset of disease in individuals who are mutant allele carriers, as well as to assess the role of biomarkers in tracking disease progression. In addition, practice guidelines are urgently needed to guide the clinical approach and to ensure that adequate genetic considerations are entertained when treating patient with ATTRv and their family members.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
• Benson MD, Buxbaum JN, Eisenberg DS, Merlini G, MJM S, Sekijima Y, et al. Amyloid nomenclature 2018: recommendations by the International Society of Amyloidosis (ISA) nomenclature committee. Amyloid. 2018;25(4):215–9 An important paper published in 2018 by the International Society of Amyloidosis nomenclature committee regarding updates and consensus recommendations for nomenclature regarding the various types amyloidosis.
Libbey CA, Skinner M, Cohen AS. Use of abdominal fat tissue aspirate in the diagnosis of systemic amyloidosis. Arch Intern Med. 1983;143(8):1549–52.
Robbins J. Thyroxine-binding proteins. Prog Clin Biol Res. 1976;5:331–55.
Hou X, Aguilar MI, Small DH. Transthyretin and familial amyloidotic polyneuropathy. Recent progress in understanding the molecular mechanism of neurodegeneration. FEBS J. 2007;274(7):1637–50.
•• Benson MD, Kincaid JC. The molecular biology and clinical features of amyloid neuropathy. Muscle Nerve. 2007;36(4):411–23 A seminal paper reviewing the molecular and clinical features of ATTR variant disease and neuropathy.
Aus dem Siepen F, Hein S, Prestel S, Baumgartner C, Schonland S, Hegenbart U, Rocken C, Katus HA, Kristen AV. Carpal tunnel syndrome and spinal canal stenosis: harbingers of transthyretin amyloid cardiomyopathy? Clin Res Cardiol. 2019 April 5. https://doi.org/10.1007/s00392-019-01467-1.
Geller HI, Singh A, Alexander KM, Mirto TM, Falk RH. Association between ruptured distal biceps tendon and wild-type transthyretin cardiac amyloidosis. Jama. 2017;318(10):962–3.
Ando Y, Nakamura M, Araki S. Transthyretin-related familial amyloidotic polyneuropathy. Arch Neurol. 2005;62(7):1057–62.
Suanprasert N, Berk JL, Benson MD, Dyck PJ, Klein CJ, Gollob JA, et al. Retrospective study of a TTR FAP cohort to modify NIS+7 for therapeutic trials. J Neurol Sci. 2014;344(1–2):121–8.
Gonzalez-Duarte A. Autonomic involvement in hereditary transthyretin amyloidosis (hATTR amyloidosis). Clin Auton Res. 2019;29(2):245–51.
Vita G, Mazzeo A, Di Leo R, Ferlini A. Recurrent syncope as persistently isolated feature of transthyretin amyloidotic polyneuropathy. Neuromuscul Disord. 2005;15(3):259–61.
Uehara T, Kakuda K, Sumi-Akamaru H, Yamauchi A, Mochizuki H, Naka T. An autopsy case of leptomeningeal amyloidosis associated with transthyretin Gly47Arg mutation. Rinsho Shinkeigaku. 2016;56(11):777–80.
Vidal R, Garzuly F, Budka H, Lalowski M, Linke RP, Brittig F, et al. Meningocerebrovascular amyloidosis associated with a novel transthyretin mis-sense mutation at codon 18 (TTRD 18G). Am J Pathol. 1996;148(2):361–6.
McColgan P, Viegas S, Gandhi S, Bull K, Tudor R, Sheikh F, et al. Oculoleptomeningeal amyloidosis associated with transthyretin Leu12Pro in an African patient. J Neurol. 2015;262(1):228–34.
Petersen RB, Goren H, Cohen M, Richardson SL, Tresser N, Lynn A, et al. Transthyretin amyloidosis: a new mutation associated with dementia. Ann Neurol. 1997;41(3):307–13.
Blevins G, Macaulay R, Harder S, Fladeland D, Yamashita T, Yazaki M, et al. Oculoleptomeningeal amyloidosis in a large kindred with a new transthyretin variant Tyr69His. Neurology. 2003;60(10):1625–30.
Connors LH, Lim A, Prokaeva T, Roskens VA, Costello CE. Tabulation of human transthyretin (TTR) variants, 2003. Amyloid. 2003;10(3):160–84.
Yazaki M, Connors LH, Eagle RC Jr, Leff SR, Skinner M, Benson MD. Transthyretin amyloidosis associated with a novel variant (Trp41Leu) presenting with vitreous opacities. Amyloid. 2002;9(4):263–7.
Lobato L, Beirao I, Silva M, Fonseca I, Queiros J, Rocha G, et al. End-stage renal disease and dialysis in hereditary amyloidosis TTR V30M: presentation, survival and prognostic factors. Amyloid. 2004;11(1):27–37.
Duca F, Kammerlander AA, Panzenbock A, Binder C, Aschauer S, Loewe C, et al. Cardiac magnetic resonance T1 mapping in cardiac amyloidosis. J Am Coll Cardiol Img. 2018;11(12):1924–6.
Maceira AM, Joshi J, Prasad SK, Moon JC, Perugini E, Harding I, et al. Cardiovascular magnetic resonance in cardiac amyloidosis. Circulation. 2005;111(2):186–93.
Bellavia D, Abraham TP, Pellikka PA, Al-Zahrani GB, Dispenzieri A, Oh JK, et al. Detection of left ventricular systolic dysfunction in cardiac amyloidosis with strain rate echocardiography. J Am Soc Echocardiogr. 2007;20(10):1194–202.
Schiano-Lomoriello V, Galderisi M, Mele D, Esposito R, Cerciello G, Buonauro A, et al. Longitudinal strain of left ventricular basal segments and E/e’ ratio differentiate primary cardiac amyloidosis at presentation from hypertensive hypertrophy: an automated function imaging study. Echocardiography. 2016;33(9):1335–43.
Carroll JD, Gaasch WH, McAdam KP. Amyloid cardiomyopathy: characterization by a distinctive voltage/mass relation. Am J Cardiol. 1982;49(1):9–13.
Mints YY, Doros G, Berk JL, Connors LH, Ruberg FL. Features of atrial fibrillation in wild-type transthyretin cardiac amyloidosis: a systematic review and clinical experience. ESC Heart Fail. 2018;5(5):772–9.
Dubrey S, Pollak A, Skinner M, Falk RH. Atrial thrombi occurring during sinus rhythm in cardiac amyloidosis: evidence for atrial electromechanical dissociation. Br Heart J. 1995;74(5):541–4.
Feng D, Edwards WD, Oh JK, Chandrasekaran K, Grogan M, Martinez MW, et al. Intracardiac thrombosis and embolism in patients with cardiac amyloidosis. Circulation. 2007;116(21):2420–6.
Barbhaiya CR, Kumar S, Baldinger SH, Michaud GF, Stevenson WG, Falk R, et al. Electrophysiologic assessment of conduction abnormalities and atrial arrhythmias associated with amyloid cardiomyopathy. Heart Rhythm. 2016;13(2):383–90.
Anzai N, Akiyama K, Tsuchida K, Yamada M, Kito S, Yamamura Y. Treatment by pacemaker in familial amyloid polyneuropathy. Chest. 1989;96(1):80–4.
Milner J, Teixeira RN, Marinho AV, Silva N, Calretas S, Ferrao J, Furtado E, Telo MJ, Ventura M, Cristovao J, Elvas L, Pego GM, Antonia N. Pacemaker implantation in familial amyloid polyneuropathy: when and for whom? J Interv Card Electrophysiol. 2019;55(2):207–211.
Hamon D, Algalarrondo V, Gandjbakhch E, Extramiana F, Marijon E, Elbaz N, et al. Outcome and incidence of appropriate implantable cardioverter-defibrillator therapy in patients with cardiac amyloidosis. Int J Cardiol. 2016;222:562–8.
Maleszewski JJ. Cardiac amyloidosis: pathology, nomenclature, and typing. Cardiovasc Pathol. 2015;24(6):343–50.
Lehmonen L, Kaasalainen T, Atula S, Mustonen T, Holmstrom M. Myocardial tissue characterization in patients with hereditary gelsolin (AGel) amyloidosis using novel cardiovascular magnetic resonance techniques. Int J Cardiovasc Imaging. 2019;35(2):351–8.
Perugini E, Guidalotti PL, Salvi F, Cooke RM, Pettinato C, Riva L, et al. Noninvasive etiologic diagnosis of cardiac amyloidosis using 99mTc-3,3-diphosphono-1,2-propanodicarboxylic acid scintigraphy. J Am Coll Cardiol. 2005;46(6):1076–84.
Puille M, Altland K, Linke RP, Steen-Muller MK, Kiett R, Steiner D, et al. 99mTc-DPD scintigraphy in transthyretin-related familial amyloidotic polyneuropathy. Eur J Nucl Med Mol Imaging. 2002;29(3):376–9.
•• Bokhari S, Castano A, Pozniakoff T, Deslisle S, Latif F, Maurer MS. (99m)Tc-pyrophosphate scintigraphy for differentiating light-chain cardiac amyloidosis from the transthyretin-related familial and senile cardiac amyloidoses. Circ Cardiovasc Imaging. 2013;6(2):195–201 This is the first article depicting the use of Tc-pyrophosphate scanning specifically for differentiating between light-chain and ATTR cardiac amyloidosis with incorporation of the heart-to-contralateral ratio.
Gillmore JD, Maurer MS, Falk RH, Merlini G, Damy T, Dispenzieri A, et al. Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation. 2016;133(24):2404–12.
Treglia G, Glaudemans A, Bertagna F, Hazenberg BPC, Erba PA, Giubbini R, et al. Diagnostic accuracy of bone scintigraphy in the assessment of cardiac transthyretin-related amyloidosis: a bivariate meta-analysis. Eur J Nucl Med Mol Imaging. 2018;45(11):1945–55.
•• Wechalekar AD, Gillmore JD, Hawkins PN. Systemic amyloidosis. Lancet. 2016;387(10038):2641–54 This is a very comprehensive and up-to-date-review of systemic amyloidosis and the various clinical treatments and outcomes associated with each type of amyloidosis.
•• Ruberg FL, Grogan M, Hanna M, Kelly JW, Maurer MS. Transthyretin amyloid cardiomyopathy: JACC State-of-the-Art Review. J Am Coll Cardiol. 2019;73(22):2872–91 A very recent review of ATTR cardiac amyloidosis reviewing pathophysiology, diagnosis, management, and clinical treatment algorithms for ATTR disease.
Westermark P, Sletten K, Johansson B, Cornwell GG 3rd. Fibril in senile systemic amyloidosis is derived from normal transthyretin. Proc Natl Acad Sci U S A. 1990;87(7):2843–5.
Almeida MR, Hesse A, Steinmetz A, Maisch B, Altland K, Linke RP, et al. Transthyretin Leu 68 in a form of cardiac amyloidosis. Basic Res Cardiol. 1991;86(6):567–71.
• Buxbaum JN, Ruberg FL. Transthyretin V122I (pV142I)* cardiac amyloidosis: an age-dependent autosomal dominant cardiomyopathy too common to be overlooked as a cause of significant heart disease in elderly African Americans. Genet Med. 2017;19(7):733–42 A comprehensive assessment of ATTRV122I cardiac amyloidosis disussing the available data on genetic penetrance, prevalence, and outcomes seen in this specific and most frequent ATTR genetic mutation seen in the United States.
Ranlov I, Alves IL, Ranlov PJ, Husby G, Costa PP, Saraiva MJ. A Danish kindred with familial amyloid cardiomyopathy revisited: identification of a mutant transthyretin-methionine111 variant in serum from patients and carriers. Am J Med. 1992;93(1):3–8.
Sattianayagam PT, Hahn AF, Whelan CJ, Gibbs SD, Pinney JH, Stangou AJ, et al. Cardiac phenotype and clinical outcome of familial amyloid polyneuropathy associated with transthyretin alanine 60 variant. Eur Heart J. 2012;33(9):1120–7.
• Maurer MS, Hanna M, Grogan M, Dispenzieri A, Witteles R, Drachman B, et al. Genotype and phenotype of transthyretin cardiac amyloidosis: THAOS (Transthyretin Amyloid Outcome Survey). J Am Coll Cardiol. 2016;68(2):161–72 Utilizing an international registry of ATTR amyloidosis, this study describes the genotype and phenotype of ATTR cardiomyopathy in the US with comparison to the rest of the world.
Connors LH, Prokaeva T, Lim A, Theberge R, Falk RH, Doros G, et al. Cardiac amyloidosis in African Americans: comparison of clinical and laboratory features of transthyretin V122I amyloidosis and immunoglobulin light chain amyloidosis. Am Heart J. 2009;158(4):607–14.
Jacobson DR, Alexander AA, Tagoe C, Buxbaum JN. Prevalence of the amyloidogenic transthyretin (TTR) V122I allele in 14 333 African-Americans. Amyloid. 2015;22(3):171–4.
Hellman U, Alarcon F, Lundgren HE, Suhr OB, Bonaiti-Pellie C, Plante-Bordeneuve V. Heterogeneity of penetrance in familial amyloid polyneuropathy, ATTR Val30Met, in the Swedish population. Amyloid. 2008;15(3):181–6.
Rapezzi C, Quarta CC, Riva L, Longhi S, Gallelli I, Lorenzini M, et al. Transthyretin-related amyloidoses and the heart: a clinical overview. Nat Rev Cardiol. 2010;7(7):398–408.
Plante-Bordeneuve V, Carayol J, Ferreira A, Adams D, Clerget-Darpoux F, Misrahi M, et al. Genetic study of transthyretin amyloid neuropathies: carrier risks among French and Portuguese families. J Med Genet. 2003;40(11):e120.
Ikeda S, Nakazato M, Ando Y, Sobue G. Familial transthyretin-type amyloid polyneuropathy in Japan: clinical and genetic heterogeneity. Neurology. 2002;58(7):1001–7.
Misu K, Hattori N, Nagamatsu M, Ikeda S, Ando Y, Nakazato M, et al. Late-onset familial amyloid polyneuropathy type I (transthyretin Met30-associated familial amyloid polyneuropathy) unrelated to endemic focus in Japan. Clinicopathological and genetic features. Brain. 1999;122(Pt 10):1951–62.
Rapezzi C, Riva L, Quarta CC, Perugini E, Salvi F, Longhi S, et al. Gender-related risk of myocardial involvement in systemic amyloidosis. Amyloid. 2008;15(1):40–8.
Olsson M, Hellman U, Plante-Bordeneuve V, Jonasson J, Lang K, Suhr OB. Mitochondrial haplogroup is associated with the phenotype of familial amyloidosis with polyneuropathy in Swedish and French patients. Clin Genet. 2009;75(2):163–8.
Ihse E, Ybo A, Suhr O, Lindqvist P, Backman C, Westermark P. Amyloid fibril composition is related to the phenotype of hereditary transthyretin V30M amyloidosis. J Pathol. 2008;216(2):253–61.
Ihse E, Rapezzi C, Merlini G, Benson MD, Ando Y, Suhr OB, et al. Amyloid fibrils containing fragmented ATTR may be the standard fibril composition in ATTR amyloidosis. Amyloid. 2013;20(3):142–50.
Bergstrom J, Gustavsson A, Hellman U, Sletten K, Murphy CL, Weiss DT, et al. Amyloid deposits in transthyretin-derived amyloidosis: cleaved transthyretin is associated with distinct amyloid morphology. J Pathol. 2005;206(2):224–32.
Conceicao I, De Carvalho M. Clinical variability in type I familial amyloid polyneuropathy (Val30Met): comparison between late- and early-onset cases in Portugal. Muscle Nerve. 2007;35(1):116–8.
Connors LH, Richardson AM, Theberge R, Costello CE. Tabulation of transthyretin (TTR) variants as of 1/1/2000. Amyloid. 2000;7(1):54–69.
Suhr OB, Svendsen IH, Andersson R, Danielsson A, Holmgren G, Ranlov PJ. Hereditary transthyretin amyloidosis from a Scandinavian perspective. J Intern Med. 2003;254(3):225–35.
Conceicao I, Damy T, Romero M, Galan L, Attarian S, Luigetti M, et al. Early diagnosis of ATTR amyloidosis through targeted follow-up of identified carriers of TTR gene mutations. Amyloid. 2019;26(1):3–9.
Obici L, Kuks JB, Buades J, Adams D, Suhr OB, Coelho T, et al. Recommendations for presymptomatic genetic testing and management of individuals at risk for hereditary transthyretin amyloidosis. Curr Opin Neurol. 2016;29(Suppl 1):S27–35.
Schmidt HH, Barroso F, Gonzalez-Duarte A, Conceicao I, Obici L, Keohane D, et al. Management of asymptomatic gene carriers of transthyretin familial amyloid polyneuropathy. Muscle Nerve. 2016;54(3):353–60.
Gertz MA, Skinner M, Connors LH, Falk RH, Cohen AS, Kyle RA. Selective binding of nifedipine to amyloid fibrils. Am J Cardiol. 1985;55(13 Pt 1):1646.
Gertz MA, Falk RH, Skinner M, Cohen AS, Kyle RA. Worsening of congestive heart failure in amyloid heart disease treated by calcium channel-blocking agents. Am J Cardiol. 1985;55(13 Pt 1):1645.
Muchtar E, Gertz MA, Kumar SK, Lin G, Boilson B, Clavell A, et al. Digoxin use in systemic light-chain (AL) amyloidosis: contra-indicated or cautious use. Amyloid. 2018;25(2):86–92.
Holmgren G, Steen L, Ekstedt J, Groth CG, Ericzon BG, Eriksson S, et al. Biochemical effect of liver transplantation in two Swedish patients with familial amyloidotic polyneuropathy (FAP-met30). Clin Genet. 1991;40(3):242–6.
Holmgren G, Ericzon BG, Groth CG, Steen L, Suhr O, Andersen O, et al. Clinical improvement and amyloid regression after liver transplantation in hereditary transthyretin amyloidosis. Lancet. 1993;341(8853):1113–6.
Wilczek HE, Larsson M, Ericzon BG. Long-term data from the Familial Amyloidotic Polyneuropathy World Transplant Registry (FAPWTR). Amyloid. 2011;18(Suppl 1):193–5.
Okamoto S, Zhao Y, Lindqvist P, Backman C, Ericzon BG, Wijayatunga P, et al. Development of cardiomyopathy after liver transplantation in Swedish hereditary transthyretin amyloidosis (ATTR) patients. Amyloid. 2011;18(4):200–5.
Gustafsson S, Ihse E, Henein MY, Westermark P, Lindqvist P, Suhr OB. Amyloid fibril composition as a predictor of development of cardiomyopathy after liver transplantation for hereditary transthyretin amyloidosis. Transplantation. 2012;93(10):1017–23.
Dubrey SW, Davidoff R, Skinner M, Bergethon P, Lewis D, Falk RH. Progression of ventricular wall thickening after liver transplantation for familial amyloidosis. Transplantation. 1997;64(1):74–80.
Garcia-Herola A, Prieto M, Pascual S, Berenguer M, Lopez-Viedma B, Mir J, et al. Progression of cardiomyopathy and neuropathy after liver transplantation in a patient with familial amyloidotic polyneuropathy caused by tyrosine-77 transthyretin variant. Liver Transpl Surg. 1999;5(3):246–8.
Coelho T, Carvalho M, Saraiva MJ, Alves I, Almeida MR, Costa PP. A strikingly benign evolution of FAP in an individual compound heterozygote for two TTR mutations: TTR Met30 and TTR Met119. J Rheumatol. 1993;20:179.
Hammarstrom P, Schneider F, Kelly JW. Trans-suppression of misfolding in an amyloid disease. Science. 2001;293(5539):2459–62.
•• Coelho T, Maia LF, Martins da Silva A, Waddington Cruz M, Plante-Bordeneuve V, Lozeron P, et al. Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology. 2012;79(8):785–92 A randomized-controlled trial of 128 patients with ATTRv neuropathy randomized to tafamadis (TTR stabilizer) or placebo which did not meet primary end-point of trial but in secondary analyses showed significant delay in neurologic impairment.
Coelho T, Ines M, Conceicao I, Soares M, de Carvalho M, Costa J. Natural history and survival in stage 1 Val30Met transthyretin familial amyloid polyneuropathy. Neurology. 2018;91(21):e1999–2009.
•• Berk JL, Suhr OB, Obici L, Sekijima Y, Zeldenrust SR, Yamashita T, et al. Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. Jama. 2013;310(24):2658–67 The randomized-controlled trial of 130 patients with ATTRv polyneuropathy randomized to diflunisal or placebo showing efficacy of diflunisal in reducing neurologic progression and preserving quality of life at 2 years.
Rosenblum H, Castano A, Alvarez J, Goldsmith J, Helmke S, Maurer MS. TTR (transthyretin) stabilizers are associated with improved survival in patients with TTR cardiac amyloidosis. Circ Heart Fail. 2018;11(4):e004769.
•• Maurer MS, Schwartz JH, Gundapaneni B, Elliott PM, Merlini G, Waddington-Cruz M, et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med. 2018;379(11):1007–16 A recent randomized-controlled trial evaluating tafamadis vs. placebo in ATTR cardiac amyloidosis resulting in reduced all-cause mortality and cardiovascular-related hospitalizations in tafamadis treated patients, particularly in ATTRwt and NYHA I-II patients.
•• Adams D, Gonzalez-Duarte A, O'Riordan WD, Yang CC, Ueda M, Kristen AV, et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med. 2018;379(1):11–21 This phase 3 randomized-controlled trial evaluated intravenous patisiran vs. placebo in ATTR amyloidosis with polyneuropathy showed statistically significant improvements in multiple neurologic clinical metrics in patisiran-treated arm compared to placebo.
Solomon SD, Adams D, Kristen A, Grogan M, Gonzalez-Duarte A, Maurer MS, et al. Effects of Patisiran, an RNA interference therapeutic, on cardiac parameters in patients with hereditary transthyretin-mediated amyloidosis. Circulation. 2019;139(4):431–43.
•• Benson MD, Waddington-Cruz M, Berk JL, Polydefkis M, Dyck PJ, Wang AK, et al. Inotereson treatment for patients with hereditary transthyretic amyloidosis. N Engl J Med. 2018;379(11):22–31 This phase 3 randomized-controlled trial evaluated subcutaneous inotersen vs. placebo in ATTR amyloidosis with polyneuropathy showing improved course of neurologic disease in patients treated with inotersen compared to placebo controls.
Funding
Dr. Gopal is supported by a career development grant from the American Heart Association (FTF 17FTF33670369). Dr. Ruberg is supported by the National Institutes of Health (R01 HL139671).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Dr. Ruberg and Dr. Gopal receive research funding from Eidos Therapeutics. Dr. Ruberg receives research and consulting income from Pfizer. Dr. Siddiqi has no conflicts to disclose.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Biomarkers of Heart Failure
Rights and permissions
About this article

Cite this article
Gopal, D.M., Ruberg, F.L. & Siddiqi, O.K. Impact of Genetic Testing in Transthyretin (ATTR) Cardiac Amyloidosis. Curr Heart Fail Rep 16, 180–188 (2019). https://doi.org/10.1007/s11897-019-00436-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11897-019-00436-z