
Abstract
Objective and methods
To ascertain the connection between cuproptosis-related genes (CRGs) and the prognosis of hepatocellular carcinoma (HCC) via single-cell RNA sequencing (scRNA-seq) and RNA sequencing (RNA-seq) data, relevant data were downloaded from the GEO and TCGA databases. The differentially expressed CRGs (DE-CRGs) were filtered by the overlaps in differentially expressed genes (DEGs) between HCC patients and normal controls (NCs) in the scRNA-seq database, DE-CRGs between high- and low-CRG-activity cells, and DEGs between HCC patients and NCs in the TCGA database.
Results
Thirty-three DE-CRGs in HCC were identified. A prognostic model (PM) was created employing six survival-related genes (SRGs) (NDRG2, CYB5A, SOX4, MYC, TM4SF1, and IFI27) via univariate Cox regression analysis and LASSO. The predictive ability of the model was validated via a nomogram and receiver operating characteristic curves. Research has employed tumor immune dysfunction and exclusion as a means to examine the influence of PM on immunological heterogeneity. Macrophage M0 levels were significantly different between the high-risk group (HRG) and the low-risk group (LRG), and a greater macrophage level was linked to a more unfavorable prognosis. The drug sensitivity data indicated a substantial difference in the half-maximal drug-suppressive concentrations of idarubicin and rapamycin between the HRG and the LRG. The model was verified by employing public datasets and our cohort at both the protein and mRNA levels.
Conclusion
A PM using 6 SRGs (NDRG2, CYB5A, SOX4, MYC, TM4SF1, and IFI27) was developed via bioinformatics research. This model might provide a fresh perspective for assessing and managing HCC.
Similar content being viewed by others

Background
Hepatocellular carcinoma (HCC) is a common malignancy that is the third most common cause of death caused by cancer worldwide [1, 2]. With a broad range of treatments for patients with HCC, the therapeutic options for advanced HCC have become more varied. However, the relative 5-year survival rate is still low (approximately 18%), highlighting the dismal prognosis of this disease [3]. Therefore, more effective clinical diagnostic and treatment strategies are needed.
Tsvetkov et al. identified a new type of cell death called cuproptosis, which is induced by the presence of copper and affects mitochondria [4]. The process by which the aggregation of proteins promotes cuproptosis is unclear, distinguishing it from apoptosis, ferroptosis, and necroptosis. Over the past century, copper and cancer have been linked in numerous studies, with higher copper levels being observed in tumors than in healthy tissues. Typically, the RNA expression levels of 54 Cu-binding proteins were ascertained from both cancerous and normal tissue samples from 18 distinct types of cancer in the TCGA database [5]. The progression of cancer is linked to elevated levels of Cu inside cells, which promote continuous cell growth, angiogenesis, and metastasis. An increase in oxidative stress resulting from a deficiency in metalation has been associated with HCC. Hep3B human hepatoma cells were subjected to Cu2+ exposure, resulting in hypoacetylation of histones H3 and H4 due to the suppression of histone acetyltransferases [6]. Patients with Wilson’s disease exhibit a loss-of-function mutation in the primary Cu exporter ATP7B, which is the most common type expressed in the liver [7], and patients with neurological Wilson’s disease are subsequently diagnosed with HCC. These findings indicate that abnormal copper buildup can promote the transformation of hepatocytes into malignant cells via a mechanism that is not yet understood [8].
We identified cuproptosis-related genes (CRGs) in this work, examined their associations with the prognosis of patients with HCC via single-cell RNA sequencing (scRNA-seq) and bulk RNA sequencing (RNA-seq) data, and developed a risk diagnostic model. This information is beneficial for understanding the development and molecular process of HCC and offers an appealing alternative for clinical diagnosis and treatment.
Materials and methods
Patients and data collection
The study cohort of HCC patients was obtained from Tianjin Medical University Cancer Institute & Hospital (2012.9–2013.9). A total of 95 paraffin-embedded samples and 10 paired fresh surgical tissue samples from the liver were analyzed and are discussed in this study. Ethical approval was acquired from the Medical Ethics Committee of Tianjin Medical University Cancer Institute & Hospital (ID: bc2023070).
Data source
The transcriptome data and clinical data of HCC patients were downloaded from UCSC Xena (https://xenabrowser.net/hub/), which included 49 normal control (NC) samples and 363 HCC samples. The RNA sequencing datasets (GSE14520) and scRNA-seq datasets (GSE149614) were obtained from the Gene Expression Omnibus (GEO) database at https://www.ncbi.nlm.nih.gHCC/geo/). The GSE14520 research used the GPL3921 [HT_HG-U133A] Affymetrix HT Human Genome U133A Array to encompass 225 HCC samples, considering their survival duration and status. The detection platform of GSE149614 was the GPL24676 Illumina NovaSeq 6000 (Homo sapiens) platform, and the sample set included 10 HCC samples and 8 NC samples.
scRNA-seq analysis
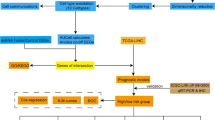
The scRNA-seq data were filtered with the CreateSeuratObject function of the Seurat package (version 4.0.5) (min. cells = 500, min. features = 500) [9]. The cells were screened according to percentage.mt < 5% and percent. HB = 0. Then, NormalizeData was used to standardize the data, and the FindVariableFeatures function was employed to determine genes with highly variable expression between cells. After normalization of the data via ScaleData, principal component analysis (PCA) was performed. JackStraw and ScoreJackStraw were subsequently used for linear dimensionality reduction to identify the availability of the data. After PCA was implemented, we used the FindNeighbors and FindClusters functions from the Seurat package to conduct unsupervised clustering analysis on the cells. The results of clustering were visualized via UMAP and tSNE. The FindAllMarkers function was employed to ascertain the positive marker genes for each cluster. The parameters used were min. pct = 0.2 only.pos = TRUE, and logfc.threshold = 0.1. The marker genes of each subgroup were compared with each cell type marker gene in the CellMarker database to determine the type of cell subgroup. In addition, the SingleR algorithm was used to verify the types of cells.
Differential expression analysis
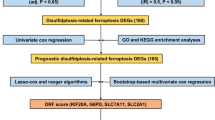
First, the first set of differentially expressed genes (DEGs1) between the HCC and NC samples in each cell subpopulation was identified via FindAllMarkers (p < 0.05). The potential differentiation trajectories of these DEGs were analyzed via monocle2. The CRG was visualized by violin charts on the basis of the literature [10]. The CRG activity of each cell from the scRNA-seq data was subsequently quantified to obtain the AUC value via the AUCell (version 1.12.0) package [11]. The cells were split into categories with high and low CRG activity depending on the optimal threshold identified by the AUCell explore Thresholds function. Through a comparison of gene activity levels in cells with high and low CRG activity, the DE-CRGs were identified. The second set of DEGs of the HCC and NC samples (DEGs2) was ascertained via the limma package (version 3.46.0) in the TCGA-HCC gene expression data [12]. We specified adj. p < 0.05 and |log2FC| > 0.5 as the threshold values to identify DEGs2. The DE-CRGs in HCC were filtered by overlapping DEGs1, CRGs, and DEGs2.
Univariate cox regression (UCR) analysis and LASSO analysis
A prognostic model (PM) was developed to determine whether the DE-CRGs in HCC are associated with HCC patient prognosis. We used HCC samples from TCGA for training and GSE14520 for external validation. The expression data of DE-CRGs in HCC were gathered from the training dataset and integrated with clinical information, encompassing overall survival (OS), to generate the clinical expression data for HCC. Survival-related genes (SRGs) were identified via UCR analysis via the survival program (version 3.2-3) (p < 0.1) [13]. The subsequent step involved the development of the PM via LASSO regression analysis of SRGs employing the glmnet package (version 4.1-1) in R [14].
Evaluation and validation of the PM
The prognostic value of the risk model was assessed by calculating the risk score (RS) by employing the SRC expression levels, and the risk coefficients were ascertained via least absolute shrinkage and selection operator (LASSO) regression. The risk score was computed via the following algorithm:
Patients diagnosed with HCC were later categorized as high risk or low risk depending on whether their RS was above or below the median. To assess the survival rates of the high-risk group (HRG) and low-risk group (LRG), we deployed the survminer package (version 0.4.8) to construct Kaplan‒Meier (KM) curves [15]. To determine PM efficacy, we deployed the survival ROC package (version 1.0.3) to generate receiver operating characteristic (ROC) curves, utilizing 1, 3, and 5 years as the survival time nodes [16]. In addition, the GSE14520 dataset was used for the external verification of the risk model.
Subgroup analysis
The subgroup of HCC patients was categorized on the basis of age, sex (female or male), vital status (who died or lived), pathologic stage (stage I, II, III, IV), histologic grade (G1, G2, G3, G4), pathologic M stage (M0, M1, MX), pathologic N stage (N0, N1, NX), and pathologic T stage (T1, T2, T3, T4). The chi-square test was used to examine the patient distribution in various clinical subgroups between the HRG and LRG in the TCGA-HCC database. The Wilcoxon test was used to ascertain variations in risk scores on the basis of distinct clinical data. UCR and multivariate Cox regression (MCR) analyses were conducted to determine the associations between clinical characteristics and the PM in the training dataset. To predict HCC patient survival via the use of SRGs, a nomogram was created. To verify the predictive capacity of the model, a calibration curve was constructed.
Gene set enrichment analysis (GSEA)
To compare the pathway enrichment in the HRG and LRG, GSEA was conducted by deploying the clusterProfiler package (version 3.18.1) [17] and the org.Hs.eg.db package (version 3.12.0) [18]. Initially, the DEGs between the HRG and LRG were ascertained by employing the limma package [12]. GSEA of the GO/KEGG gene set was subsequently performed on the basis of the log2FC values of the DEGs.
Immune infiltration analysis
The percentages of 22 immune cells in the HRG and LRG were computed and compared via the CIBERSORT algorithm [19] with a rank-sum test. Spearman correlation analysis was subsequently performed to ascertain the connection between RS and 22 immune cells; differentially expressed (DE)-immune cells were subsequently identified. Patients were subsequently split into low-level and high-level groups on the basis of the median values of DE-immune cells. An assessment of the connection between DE-immune cells and patient survival was performed via survival analysis. To obtain the immune response results, the tumor immune dysfunction and exclusion (TIDE) score was calculated for each TCGA-HCC sample via TIDE (http://tide.dfci.harvard.edu/). Finally, the variation in programmed death-ligand 1 (PD-L1) expression and its connection with RS were analyzed in the HRG and LRG.
Drug sensitivity analysis
Each cancer cell RS was computed by integrating the expression data acquired from the CellMiner database (https://discover.nci.nih.gov/cellminer/loadDownload.do) with the coefficients. Spearman analysis was conducted between risk scores and medicines, and substances with a significance level of p < 0.05 were chosen. The cell lines were categorized into HRGs and LRGs according to the median value of the RS. The IC50 values of the medicines were compared between the two groups. The IC50 of lenvatinib was compared between the HRG and LRG.
Quantitative real-time polymerase chain reaction (qRT‒PCR)
As previously described, qRT‒PCR was performed [20] with the primers listed in Supplementary Table S1. The qRT‒PCR experiment was performed with a 7500HT Real-Time PCR System (Applied Biosystems, Foster City, CA). Fold changes were determined by relative quantification (2^(-ΔΔCt)), with the internal control GAPDH serving as an endogenous control.
Immunohistochemistry (IHC)
Immunohistochemical analysis was performed on four-thickness tissue sections from representative formalin-fixed and paraffin-embedded (FFPE) blocks that were subjected to immunohistochemical examination via the Dako EnVision method with various antibodies (Supplementary Table S2).
Statistical analysis
The correlations between clinicopathological variables and immunohistochemistry data were analyzed via the χ2 test or Fisher’s exact test. Paired samples were assessed by using the paired Students t -test. We conducted a KM survival analysis to clarify the prognosis. The statistical analyses were conducted via SPSS 27.0.1. The significance level was established as a P value less than 0.05.
Results
Clustering of single cells into 7-cell subsets on the basis of scRNA-seq data
After data filtering was completed, the features of scRNA-seq revealed a significant positive connection between nFeature_RNA and nCount_RNA (Supplementary Fig. 1). Then, data standardization was implemented, and 2000 highly variable genes were selected by deploying “vst” (Supplementary Fig. 2). The data were normalized, and the PCA results are shown in Supplementary Fig. 3. Forty principal components (PCs) were chosen for further analysis (p < 0.05) (Supplementary Fig. 4). The scatter plot revealed that, when the resolution was 0.02, the profile coefficient was 0.27, and the cells were partitioned into seven clusters. Supplementary Fig. 5 reveals that the clustering was better when the resolution was 0.02. The visualization results of clustering via uniform manifold approximation and projection (UMAP) and t-distributed stochastic neighbor embedding (tSNE) are shown in Fig. 1a–b. Figure 1c displays the nine most prominent marker genes for each cluster. We determined that the cell type depended on each cluster marker gene (Fig. 1d; Table 1). Because the marker genes (CD3D, CD3E, and CD3G) of T cells were highly expressed in Cluster 0, it was identified as a T-cell cluster (Fig. 2a). Cluster 1 was identified as a myeloid cell cluster because CD68 and CD163 were highly expressed (Fig. 2b). In addition, the marker genes ALB and SERPINA1 were highly expressed in Cluster 2, so it was identified as a hepatocyte cluster (Fig. 2c). The annotation results for the other clusters are shown in Supplementary Fig. 6. The pie chart depicts the relative abundances of T cells and myeloid cells, which were shown to be the most prevalent (Fig. 2d). Finally, the distributions of the 7 cell subsets were determined via UMAP analysis (Fig. 2e).

Single-cell characterization of HCC samples via single-cell RNA sequencing. (a–b) The visualization results of clustering via uniform manifold approximation and projection (UMAP) and t-distributed stochastic neighbor embedding (tSNE). (c–d) Expression heatmap and visualization results of the top 9 marker genes in each cluster

Identification of the cell subsets corresponding to each signature in each cluster. (a) Identification and visualization results of T cells by the expression of marker genes (CD3D, CD3E, and CD3G) and UMAP. (b) Identification and visualization results of myeloid cells by the expression of marker genes (CD68 and CD163) and UMAP. (c) Identification and visualization results of hepatocytes by marker genes (ALB and SERPINA1) and UMAP. (d–e) Proportion and distribution of 7 cell subsets determined via pie charts and UMAP. The clusters are assigned different colors on the basis of the cell type, and the picture displays the annotations for each cluster
Identification of DE-CRGs in HCC
A total of 988 DEGs were obtained between the HCC and NC samples in each cell subset in GSE149614 (Fig. 3a). The potential differentiation trajectories of these DEGs are shown in Supplementary Fig. 7. CRG expression in various cell subsets between HCC and NC samples was visualized via a violin plot (Fig. 3b, Supplementary Fig. 8). The results of the quantification of CRG activity for each cell subset in the scRNA-seq data are shown in Fig. 3c. Next, the cells were split into high- and low-CRG activity groups according to the most effective threshold (Fig. 3d–e). The volcano map revealed that 98 DE-CRGs were filtered by overlapping high- and low-CRG activity cells (Fig. 3f). Furthermore, a grand total of 6170 DEGs2 were discovered when the HCC samples were compared with the NC samples. Among these genes, 5320 genes presented increased expression, whereas 850 genes presented decreased expression in the TCGA-HCC gene expression dataset (Fig. 3g). The top 50 DEGs were visualized via a heatmap (Supplementary Fig. 9). Finally, the Venn diagram revealed that 33 DE-CRGs in HCC overlapped with DEGs1, CRGs, and DEGs2 (Fig. 3h).

The determination of differentially expressed gene (DEG)-related CRGs. (a) Volcano map of DEG1 expression in the single-cell sequencing dataset. (b) Expression of CDKN2A in the single-cell sequencing dataset. (c) Scores of CRGs in 7 cell subsets. (d) Selection of the optimal threshold. (e) UMAP diagram showing high- and low-CRG activity cells on the basis of the optimal threshold. (f) Volcano map of the expression of CRGs in the single-cell sequencing dataset. (g) Volcano map of DEG2 expression in the TCGA dataset. (h) Venn diagram of the intersection of DEGs1, CRGs, and DEGs2 in tumor and nontumor samples. p < 0.05
Construction of a PM based on SRGs
UCR analysis revealed 6 SRGs, of which four genes (SOX4, MYC, TM4SF1, and IFI27) were risk factors, and two genes (NDRG2 and CYB5A) were protective factors (Fig. 4a). Subsequently, LASSO regression analysis was deployed to create the PM (Fig. 4b); the model gene coefficients are presented in Table 2. The expression patterns of the 6 SRGs in GSE149614 indicated that these genes were expressed mainly in microglia, fibroblasts, and hepatocytes (Supplementary Fig. 10a–b). To evaluate the interactions among these 6 SRGs, Spearman correlation analysis was conducted. The findings revealed a remarkable inverse relationship between CYB5A and SOX4 and TM4SF1 and a significant direct relationship between CYB5A and NDRG2 and IFI27 (Supplementary Fig. 10c).

Construction of a 6-CRG survival model in HCC patients. (a) The forest plot displays the level of risk associated with each overlapping gene via UCR analysis. (b) According to the minimum criteria, 6 SRGs were selected via the LASSO regression model. The cvfit and lambda curves were generated via the LASSO regression model
Assessment and validation of the prognostic value of the model
HCC patients were split into HRGs and LRGs owing to their RS, and the risk curve is shown in Fig. 5a. The OS curve revealed a significant variation in survival rates between the HRG and LRG groups (p < 0.0001), with the HRG group displaying a lower rate of survival (Fig. 5b). The AUC of the prognostic model for predicting the 1-, 3-, and 5-year survival of HCC patients was 0.665, 0.623, and 0.609, respectively. These values indicate that the performance of the PM was good, as shown in Fig. 5c. The risk model was externally verified with the GSE14520 dataset, and the outcomes were similar to the data presented in Fig. 5d–f.

Prognostic value evaluation of the CRG model. (a) Distributions of the risk score (RS), survival status distributions, and expression heatmap in the TCGA training cohort. (b) OS value stratified by the RS in the TCGA training cohort. (c) Time-dependent ROC curve of the prognostic performance of the RS in the TCGA training cohort. (d) Survival status distributions, RS, and expression heatmap in the external testing cohort. (e) OS value stratified by the RS in the external testing cohort. (f) Time-dependent ROC curve of the prognostic performance of the RS in the external testing cohort
Correlation analysis between the PM and clinical factors
A comparison was made between the number of patients with various clinical subtypes in the HRG and LRG. The analysis revealed significant differences in age, vital status, pathologic stage, histologic grade, and pathologic T stage (Table 3). The Wilcoxon test findings revealed substantial variations in the RS based on several clinical factors, such as age (≤ 60 and ≥ 60), vital status (alive and dead), T stage (T1/T2/T3/T4), and grade (G1/G2/G3/G4) (Fig. 6a). PM was shown to be a valid autonomous predictive factor for HCC patients on the basis of UCR and MCR analyses (Fig. 6b and c). A nomogram was constructed to predict the likelihood of patient survival when it was integrated with clinical parameters. The C-index of the nomogram was 0.6370537, demonstrating its effectiveness (Fig. 6d). The calibration curve of the nomogram established its effectiveness as a model (Fig. 6e).

Prognostic model and clinical correlation analyses of the TCGA data. (a) Connection between the risk score (RS) and clinicopathological features. *p < 0.05, **p < 0.01, and ***p < 0.001; ns.: not significant. (High RS was highly common in patients under 60 years of age, with G3–4, stage III, and T2–3 disease, and a poor prognosis). (b–c) UCR and MCR analyses of clinicopathologic features and RS. (d) Nomogram composed of cuproptosis-related SRG levels for predicting the 1-, 3-, and 5-year overall survival (OS) of HCC patients. (e) Nomogram and calibration curves of the prognostic model
Differences in enriched pathways between the HRG and LRG
We investigated the possible pathways in both the HRG and LRG. The KEGG analysis indicated that retinol metabolism and drug metabolism–cytochrome P450 pathways were considerably more prevalent in the LRG, whereas ribosomes were more prevalent in the HRG (Supplementary Fig. 11a). The GO analysis revealed enrichment of the organic acid catabolic process and carboxylic acid catabolic process in the LRG, whereas ribonucleoprotein complex biogenesis was enriched in the HRG (Supplementary Fig. 11b).
Effects of risk models on immune heterogeneity
The percentages of immune cells were determined via the rank-sum test. The results revealed that there were variations (p < 0.05) between the HRG and LRG in terms of the quantity of naive B cells, activated T cells with memory CD4 T cells, follicular helper T cells, regulatory T cells (Tregs), resting NK cells, monocytes, M0 macrophages, and mast cells (Fig. 7a). A significant positive correlation was observed between Tregs and M0 macrophages and the RS (Cor > 0.2, p < 0.05), as determined by Spearman correlation analysis between the RS and immune cells. Conversely, a significant negative connection was observed between resting mast cells, monocytes, and the RS (Cor < -0.2, p < 0.05) (Fig. 7b). The HCC patients were subsequently categorized into high- and low-level groups according to the median number of Tregs, M0 macrophages, resting mast cells and monocyte proportion, respectively. Patient survival differed significantly only between teams with high and low M0 macrophage counts. Additionally, higher levels of macrophages were associated with poorer patient prognosis, as shown by OS curve analysis (Fig. 7c). These findings suggest that macrophages could have a significant effect on HCC. Furthermore, the elevated TIDE scores observed in the HRG indicate that this group may be more vulnerable to immunological resistance (Fig. 7d). The results of the correlation analysis revealed a significant positive correlation (Cor = 0.3, p < 0.05) between TIDE scores and the RS (Fig. 7e). The results of the distinction analysis revealed that PD-L1 expression did not significantly differ between the HRG and LRG (p > 0.05). The scatter diagram (Fig. 7f) demonstrated a significant inverse connection (Cor = -0.12, p < 0.05) between PD-L1 and the RS. In conclusion, the HRG exhibited a significantly greater number of immunological nonresponders than the LRG did, and patients who did not respond had greater RS (p < 0.05) (Fig. 7g and h).

Differential analysis of immune characteristics between HRGs and LRGs in the TCGA cohort. (a) Analysis of the differences in immunocyte proportions between the two risk groups. *p < 0.05, **p < 0.01, ***p < 0.001. (b) Spearman correlation analysis of the risk score (RS) and immune cells. (c) KM curves of patients with different proportions of macrophages. (d) Differential analysis of TIDE scores between the HRG and LRG. (e) Correlations between the RS and TIDE scores. (f) Correlation between RS and the expression of PD-L1. (g) Analysis of the immunotherapy response between the two risk groups. (h) Differential analysis of RS between immunological nonresponders and responders
The IC50 values of idarubicin and rapamycin were different between the two groups
We used the CellMiner database to investigate antitumor drugs that are sensitive to SRG expression. Spearman correlation analysis of the RS and drug IC50 values revealed that the IC50 values of cobimetinib and trametinib were negatively correlated with the RS (Cor < -0.2), and the other 10 drugs were positively correlated with the RS (Cor > 0.2) (Supplementary Fig. 12). Analysis of the four drugs (Cor > 0.3) revealed that the IC50 values of idarubicin and rapamycin significantly differed between the high- and low-RS groups (Supplementary Fig. 13).
Validation of CRGs
First, we quantified the mRNA expression of FDX1, DLAT, LIAS, and CDKN2A between fresh HCC surgical tissues and adjacent tissues (Fig. 8a). The FDX1, DLAT, LIAS, and CDKN2A expression levels in 10 HCC samples were markedly greater than those in paired neighboring tissues. Moreover, we detected the expression of CDKN2A via IHC. Compared with NC samples, HCC samples presented elevated levels of CDKN2A protein expression (Fig. 8b). To further confirm these outcomes, IHC and qRT‒PCR were carried out to compare the expression levels of six SRGs between HCC tissues and normal liver tissues. At the protein level, the SOX4, MYC, TM4SF1, and IFI27 expression levels were greater in HCC samples than in NC samples, and the NDRG2 and CYB5A expression levels were lower in HCC samples than in NC samples (Fig. 9). In our cohort, the expression of each gene associated with cuproptosis significantly affected patient survival. High-risk individuals had worse outcomes than low-risk patients did (Fig. 10a). High expression of CD68 and CD163 was observed in high-RS patients, but low expression was observed in patients with low-RS in the TCGA database (Fig. 10b) and in our own cohort (Fig. 10c and d). In summary, the data analysis and important discoveries in this investigation were highly dependent.

Validation of cuproptosis-related SRGs in HCC. (a) mRNA expression of FDX1, DLAT, LIAS, and CDKN2A between fresh HCC surgical tissues and adjacent tissues. (b) Results of CDKN2A immunohistochemical staining and relative expression of paraffin-embedded normal and tumor samples

Immunohistochemical staining and qRT‒PCR results for NDRG2 (a), CYB5A (b), SOX4 (c), MYC (d), TM4SF1 (e) and IFI27 (f) in normal and tumor tissues. The black box in the upper left corner represents normal tissues

(a) Outcomes of the KM survival analysis revealed that HRGs had a lower survival rate than did the LRGs in our cohort. The red line represents the HRG, and the blue line represents the LRG. (b) The expression of CD68 and CD163 in the HRG and the LRG in the TCGA cohort. (c) Immunohistochemical staining of CD68 and CD163 in our cohort. (d) The expression of CD68 and CD163 in the HRG and LRG in our cohort. The data represent the mean ± SD. (*p < 0.05, **p < 0.01, ***p < 0.001). Original magnification, ×400
Discussion
Globally, HCC is the primary cause of cancer-related fatalities. The improved prognosis for the majority of patients with HCC persists as a result of its high metastatic rate. Cu homeostasis dysfunction is implicated in several pathologies, including Menkes disease and hepatic Wilson’s disease [21], and in cancer progression-related processes, including proliferative immortality, angiogenesis, and metastasis [22,23,24,25,26]. The development of efficient therapeutic and prognostic biomarkers is crucial for enhancing the prognosis and increasing the survival rate of HCC patients. Cuproptosis is a type of mitochondrial cell death caused by Cu that is not related to apoptosis, ferroptosis, or necroptosis [4, 27]. In contrast, the hallmark signal for cuproptosis and the relationship between cuproptosis and HCC remain unknown. Therefore, studying the prognostic signature of CRGs in HCC is highly important. Using the TCGA and GEO databases, we developed and verified a prognostic CRG model and examined its potential correlation with clinicopathological characteristics.
Cuproptosis is associated with ten key genes according to the published literature. The detection of these key genes in our cohort confirmed the occurrence of copper-induced cell death. On the basis of comprehensive bioinformatics analysis, we screened six SRGs: NDRG2, CYB5A, SOX4, MYC, TM4SF1, and IFI27. N-myc downstream-regulated gene family 2 (NDRG2) functions as a tumor suppressor gene, hampers HCC cell proliferation [28], and decreases CD24 expression in HCC [29]. Recent reports indicate that elevated levels of NDRG2 expression suppress epithelial‒mesenchymal transition (EMT) in HBV-linked HCC [30]. Cytochrome B5 type A (CYB5A) suppresses the metastasis of HCC by controlling the process of autophagy and enhancing the responsiveness to ruxolitinib [31]. Regrettably, the anticancer impact of CYB5A in HCC has only been briefly addressed. The expression of SRY-box transcription factor 4 (SOX4) is significantly elevated in various types of malignancies [32, 33], and SOX4 overexpression promotes metastasis in HCC [34, 35]. The oncogenic potential of c-MYC in HCC has been proven in vivo and in vitro during hepatocarcinogenesis [36]. Higher extracellular copper concentrations enhance HCC cell growth and invasion by regulating the MYC/CTR1 axis [37]. When the expression of Axin2 and cyclin D1 is upregulated, the overexpression of transmembrane-4 L-six family member-1 (TM4SF1) enhances the proliferation and motility of HCC cells [38]. Interferon alpha-inducible protein 27 (IFI27) is elevated in lesional and nonlesional psoriatic skin and some cancers, and AC10364 was found to inhibit cell viability and proliferation partially by downregulating the expression of IFI27 in HCC [39,40,41]. The model showed better performance in predicting the clinical outcomes of patients with HCC after surgical resection. In Spearman correlation analysis of 6 SRGs, there was a significant direct relationship between CYB5A, NDRG2 and IFI27. Our rationale primarily encompasses the following points: Firstly, co-expressed genes are likely to participate in the same biological pathways or functional modules, indicating intricate molecular mechanisms. Preserving these genes is essential for a comprehensive understanding of their biological functions and interactions in HCC. Secondly, in HCC, CYB5A, NDRG2, and IFI27 are potentially co-involved in critical biological processes, including cell proliferation, apoptosis, and metabolic regulation [28, 31, 42]. This makes them crucial for predictive models. Model stability should be prioritized during construction. Lastly, co-expressed genes can offer complementary information, enhancing both stability and predictive performance. During data analysis, the removal of co-expressed genes can result in significant information loss, particularly when the sample size is constrained. Retaining these genes is crucial for maintaining the integrity of the data and ensuring the comprehensiveness of the analysis. Our IHC validation results revealed that high-risk patients had a worse prognosis than low-risk patients did. In summary, the six SRGs play important roles in tumor carcinogenesis. Among these six genes, only MYC has been reported to be associated with copper metabolism. Nevertheless, the precise involvement of MYC in cuproptosis in HCC has yet to be clarified. Owing to the lack of relevant hallmarks of cuproptosis, the six SRGs may also be potential targets that can be exploited to elucidate the mechanism of cuproptosis.
Research has shown that cancer cells often display distinct changes in metabolism to sustain their proliferation and growth [43,44,45]. Different members of the cytochrome P450 (CYP450) family have emerged as significant factors in the response to cancer treatments and resensitization to anticancer drugs [46]. The involvement of ribosome-related activities in the genesis and progression of different malignancies is well documented. The process of O-GlcNAcylation of RACK1 leads to increased stability of RACK1, attachment to ribosomes, binding to PKCβII, and ultimately stimulating the development of hepatocellular carcinogenesis [47]. C1QBP enhances the survival, migration, and invasion of HCC cells and is linked to genes that encode ribosomal RPL-linked proteins and mitochondrial MRPL-linked proteins in HCC patients [48]. TCOF1, a factor involved in ribosome biogenesis, has oncogenic functions through the regulation of KRAS-activated and EMT genes in HCC. It is essential for the elevated production of ribosomal RNA (rRNA), a characteristic of cancer [49]. Momordica anti-HIV protein of 30 kDa (MAP30) induces the apoptosis of HCC cells [50]. In this study, the potential pathways in the HRG and LRG according to the RS of the six SRGs were analyzed via KEGG and GO analyses. Significant variation was detected in the involved pathways between the HRG and LRG. The results revealed that ribosome and ribonucleoprotein complex biogenesis was enriched in the HRG, whereas retinol metabolism, drug metabolism-CYP450, organic acid catabolic process and carboxylic acid catabolic process were significantly enriched in the LRG. These findings indicate that the six SRGs are involved primarily in regulating ribosomes during tumor progression, which further indicates that cuproptosis may be involved in this process. These findings provide insight into the molecular mechanism of cuproptosis in HCC.
The cellular components of the tumor immune microenvironment (TIME) are highly complex and are involved in cancer immune evasion, inflammation, and response to immunotherapy treatment [51]. The TIME of HCC is poorly characterized, and the key components include cancer-associated fibroblasts (CAFs), tumor-associated macrophages (TAMs), Tregs, tumor-associated neutrophils (TANs), and myeloid-derived suppressor cells (MDSCs) [52]. CAFs secrete high levels of CXCL11 and CCL5 into the tumor microenvironment to enhance HCC metastasis through multiple pathways [53, 54]. HCC-CAFs protect chemotactic neutrophils from spontaneous apoptosis through an IL6-STAT3-PDL1 signaling cascade [55]. Tumor cells release Wnt ligands to increase the M2-like polarization of TAMs by activating the canonical Wnt/β-catenin signaling pathway. This polarization leads to immunosuppression inside HCC tumors [56]. GDF15 triggers immunosuppression in HCC by interacting with CD48 on Tregs [57]. A recent bioinformatics study using four CRGs (CDKN2A, GLS, LIPT1, and DLAT) demonstrated that HRGs presented a greater percentage of macrophages and Tregs than did LRGs [58]. TANs recruit macrophages and Tregs, which enhance HCC growth, metastasis and sorafenib resistance [59]. These results are consistent with our data. However, the screened CRGs and the constructed PM are different. This finding also proves the importance of introducing macrophages and Tregs in cuproptosis. We used CD68 in combination with CD163 to identify TAMs in our cohort. The proportion of TAMs in the HRG was greater than that in the LRG. In addition, the other key components of the tumor microenvironment may be involved in the screened CRGs in our model. Compared with the LRG, the HRG had a greater percentage of Tregs. An increase in TAMs was linked to a worse prognosis in HCC patients, confirming the predictive ability of the model. This notion requires validation via further study. This phenomenon might be one of the specific mechanisms of cuproptosis in HCC. Additionally, the HRG had a high TIDE score, which was linked to improved immune evasion, confirming the model’s prediction accuracy. Patients with elevated levels of immune checkpoints in malignancies may have increased response to therapy with immune checkpoint inhibitors (ICIs). However, the precise mechanism remains unknown. GOLM1 in HCC promotes CD8 + T-cell suppression by stabilizing PD-L1 and delivering it into TAMs via exosomes [60]. Our findings revealed that patients in the high-RS group had lower PD-L1 expression, indicating that treatment with ICIs may have poor efficacy for patients in the HRG. In our model, the negative correlation between the RS and immune checkpoint expression implied that the poor prognosis in the HRG might be due in part to the lower immune checkpoint expression of PD-L1 in this group than in the LRG group. The constructed PM might be employed to identify patients at risk for poor ICI treatment and poor outcomes, allowing for the better tailoring of individualized treatment. The above findings also indicate that the mechanism of HCC cuproptosis and the mechanism of cuproptosis involved in immune regulation are complex. Prospective studies are needed to confirm these findings.
HCC cuproptosis has been studied subsequently. The SRGs were also subjected to drug sensitivity analysis, and several candidates were identified. Similar findings to those of the current study were reported in prior studies, irrespective of whether the scoring models utilized cuproptosis-related miRNAs, cuproptosis-linked lncRNAs, or cuproptosis-linked CRGs. Patients with greater cuproptosis-related scores had a lower OS rate [58, 61, 62]. Moreover, drug sensitivity analyses were conducted by Lei Ding et al. [10]. and Qiongyue Zhang et al. [63]. reported lower IC50 values for samples with high cuproptosis-linked scores. Patients with high cuproptosis-linked scores may benefit more from certain forms of chemotherapy. In this study, the IC50 values for idarubicin and rapamycin were lower for specimens with high cuproptosis-related scores than for those with low cuproptosis-related scores. This finding is partially consistent with previous research results. However, the IC50 predictions indicated a positive correlation between sensitivity to most targeted drugs and RS. These findings indicate that high-risk patients might not benefit from most targeted drugs. We analyzed the RS that could help identify patients most likely to benefit from antitumor drugs. This research is anticipated to offer evidence-based information that is crucial for clinical treatment.
However, the current investigation is subject to several limitations. The investigation was predicated on a publicly accessible database. While every effort was made to incorporate a multitude of datasets to enhance the rigor of this result verification, further functional and mechanistic analyses of these genes, both individually and in combination, are necessary to substantiate the clinical applicability of our model. Second, further investigations are needed to clarify the molecular mechanisms underlying cuproptosis and CRG, given the limited understanding of cuproptosis. Finally, experimental investigations into the correlation between RS and immune activity are lacking, necessitating further research. One notable strength of this research is the development of a six-gene model via algorithmic means; this model has the capacity to significantly inform clinical decision-making. Furthermore, in addition to utilizing publicly available datasets to develop the model, this research also employed clinical tissues and public datasets to validate the findings, significantly bolstering the study’s credibility.
In brief, we created and verified a model for the diagnosis of HCC risk using six CRGs that have independent prognostic significance. The risk evaluation model offers a noteworthy immunological perspective on the cuproptosis mechanisms that govern the clinical prognosis of HCC.
Data availability
The data that support the findings of this study are openly available on TCGA website, UCSC Xena and GEO database.
References
Llovet JM, Kelley RK, Villanueva A, Singal AG, Pikarsky E, Roayaie S, Lencioni R, Koike K, Zucman-Rossi J, Finn RS. Hepatocellular carcinoma. Nat Rev Dis Primers. 2021;7(1):6.
Vogel A, Meyer T, Sapisochin G, Salem R, Saborowski A. Hepatocellular carcinoma. Lancet. 2022;400(10360):1345–62.
Nagtegaal ID, Odze RD, Klimstra D, Paradis V, Rugge M, Schirmacher P, Washington KM, Carneiro F, Cree IA, Board WHOCTE. The 2019 WHO classification of tumours of the digestive system. Histopathology. 2020;76(2):182–8.
Tsvetkov P, Coy S, Petrova B, Dreishpoon M, Verma A, Abdusamad M, Rossen J, Joesch-Cohen L, Humeidi R, Spangler RD, et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science. 2022;375(6586):1254–61.
Blockhuys S, Celauro E, Hildesjo C, Feizi A, Stal O, Fierro-Gonzalez JC, Wittung-Stafshede P. Defining the human copper proteome and analysis of its expression variation in cancers. Metallomics. 2017;9(2):112–23.
Kang J, Lin C, Chen J, Liu Q. Copper induces histone hypoacetylation through directly inhibiting histone acetyltransferase activity. Chem Biol Interact. 2004;148(3):115–23.
Tsang T, Davis CI, Brady DC. Copper biology. Curr Biol. 2021;31(9):R421–7.
Gunjan D, Shalimar, Nadda N, Kedia S, Nayak B, Paul SB, Gamanagatti SR, Acharya SK. Hepatocellular Carcinoma: an unusual complication of Longstanding Wilson Disease. J Clin Exp Hepatol. 2017;7(2):152–4.
Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol. 2018;36(5):411–20.
Ding L, Li W, Tu J, Cao Z, Li J, Cao H, Liang J, Liang Y, Yu Q, Li G. Identification of cuproptosis-related subtypes, cuproptosis-related gene prognostic index in hepatocellular carcinoma. Front Immunol. 2022;13:989156.
Aibar S, Gonzalez-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, Rambow F, Marine JC, Geurts P, Aerts J, et al. SCENIC: single-cell regulatory network inference and clustering. Nat Methods. 2017;14(11):1083–6.
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47.
Zhou RS, Zhang EX, Sun QF, Ye ZJ, Liu JW, Zhou DH, Tang Y. Integrated analysis of lncRNA-miRNA-mRNA ceRNA network in squamous cell carcinoma of tongue. BMC Cancer. 2019;19(1):779.
Friedman J, Hastie T, Tibshirani R. Regularization paths for generalized Linear models via Coordinate Descent. J Stat Softw. 2010;33(1):1–22.
Liu TT, Li R, Huo C, Li JP, Yao J, Ji XL, Qu YQ. Identification of CDK2-Related Immune Forecast Model and ceRNA in Lung Adenocarcinoma, a Pan-cancer Analysis. Front Cell Dev Biol. 2021;9:682002.
Zheng Y, Wen Y, Cao H, Gu Y, Yan L, Wang Y, Wang L, Zhang L, Shao F. Global characterization of Immune Infiltration in Clear Cell Renal Cell Carcinoma. Onco Targets Ther. 2021;14:2085–100.
Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–7.
Wu H, Jiang W, Ji G, Xu R, Zhou G, Yu H. Exploring microRNA target genes and identifying hub genes in bladder cancer based on bioinformatic analysis. BMC Urol. 2021;21(1):90.
Chen B, Khodadoust MS, Liu CL, Newman AM, Alizadeh AA. Profiling Tumor infiltrating Immune cells with CIBERSORT. Methods Mol Biol. 2018;1711:243–59.
Wang Y, Sun B, Zhao X, Zhao N, Sun R, Zhu D, Zhang Y, Li Y, Gu Q, Dong X, et al. Twist1-related miR-26b-5p suppresses epithelial-mesenchymal transition, migration and invasion by targeting SMAD1 in hepatocellular carcinoma. Oncotarget. 2016;7(17):24383–401.
Czlonkowska A, Litwin T, Dusek P, Ferenci P, Lutsenko S, Medici V, Rybakowski JK, Weiss KH, Schilsky ML. Wilson disease. Nat Rev Dis Primers. 2018;4(1):21.
Denoyer D, Masaldan S, La Fontaine S, Cater MA. Targeting copper in cancer therapy: ‘Copper that Cancer’. Metallomics. 2015;7(11):1459–76.
Ge EJ, Bush AI, Casini A, Cobine PA, Cross JR, DeNicola GM, Dou QP, Franz KJ, Gohil VM, Gupta S, et al. Connecting copper and cancer: from transition metal signalling to metalloplasia. Nat Rev Cancer. 2022;22(2):102–13.
Zhang X, Yang Q. Association between serum copper levels and lung cancer risk: a meta-analysis. J Int Med Res. 2018;46(12):4863–73.
Aubert L, Nandagopal N, Steinhart Z, Lavoie G, Nourreddine S, Berman J, Saba-El-Leil MK, Papadopoli D, Lin S, Hart T, et al. Copper bioavailability is a KRAS-specific vulnerability in colorectal cancer. Nat Commun. 2020;11(1):3701.
Saleh SAK, Adly HM, Abdelkhaliq AA, Nassir AM. Serum levels of Selenium, Zinc, Copper, Manganese, and Iron in prostate Cancer patients. Curr Urol. 2020;14(1):44–9.
Tang D, Chen X, Kroemer G. Cuproptosis: a copper-triggered modality of mitochondrial cell death. Cell Res. 2022;32(5):417–8.
Guo Y, Li X, Sun X, Wang J, Yang X, Zhou X, Liu X, Liu W, Yuan J, Yao L, et al. Combined aberrant expression of NDRG2 and LDHA Predicts Hepatocellular Carcinoma Prognosis and mediates the Anti-tumor Effect of Gemcitabine. Int J Biol Sci. 2019;15(9):1771–86.
Zheng J, Li Y, Yang J, Liu Q, Shi M, Zhang R, Shi H, Ren Q, Ma J, Guo H, et al. NDRG2 inhibits hepatocellular carcinoma adhesion, migration and invasion by regulating CD24 expression. BMC Cancer. 2011;11:251:251–9.
Yu K, Mei Y, Wang Z, Liu B, Deng M. LncRNA LINC00924 upregulates NDRG2 to inhibit epithelial-mesenchymal transition via sponging mir-6755-5p in hepatitis B virus-related hepatocellular carcinoma. J Med Virol. 2022;94(6):2702–13.
Guo H, Liang S, Wang Y, Zhou S, Yin D, Zhang S, Wang J, Wu D, Ma K, Liu Y, et al. Cytochrome B5 type a alleviates HCC metastasis via regulating STOML2 related autophagy and promoting sensitivity to ruxolitinib. Cell Death Dis. 2022;13(7):623.
Wang B, Li Y, Tan F, Xiao Z. Increased expression of SOX4 is associated with colorectal cancer progression. Tumour Biol. 2016;37(7):9131–7.
Song GD, Sun Y, Shen H, Li W. SOX4 overexpression is a novel biomarker of malignant status and poor prognosis in breast cancer patients. Tumour Biol. 2015;36(6):4167–73.
Tsai CN, Yu SC, Lee CW, Pang JS, Wu CH, Lin SE, Chung YH, Tsai CL, Hsieh SY, Yu MC. SOX4 activates CXCL12 in hepatocellular carcinoma cells to modulate endothelial cell migration and angiogenesis in vivo. Oncogene. 2020;39(24):4695–710.
Hao F, Wang N, Zhang Y, Xu W, Chen Y, Fei X, Wang J. E2F7 enhances hepatocellular carcinoma growth by preserving the SP1/SOX4/Anillin axis via repressing miRNA-383-5p transcription. Mol Carcinog. 2022;61(11):975–88.
Xu K, Ding J, Zhou L, Li D, Luo J, Wang W, Shang M, Lin B, Zhou L, Zheng S. SMYD2 promotes Hepatocellular Carcinoma Progression by Reprogramming glutamine metabolism via c-Myc/GLS1 Axis. Cells. 2022;12(1).
Porcu C, Antonucci L, Barbaro B, Illi B, Nasi S, Martini M, Licata A, Miele L, Grieco A, Balsano C. Copper/MYC/CTR1 interplay: a dangerous relationship in hepatocellular carcinoma. Oncotarget. 2018;9(10):9325–43.
Zhu C, Luo X, Wu J, Liu Y, Liu L, Ma S, Xie R, Wang S, Ji W. TM4SF1, a binding protein of DVL2 in hepatocellular carcinoma, positively regulates beta-catenin/TCF signalling. J Cell Mol Med. 2021;25(5):2356–64.
Wenzel J, Tomiuk S, Zahn S, Kusters D, Vahsen A, Wiechert A, Mikus S, Birth M, Scheler M, von Bubnoff D, et al. Transcriptional profiling identifies an interferon-associated host immune response in invasive squamous cell carcinoma of the skin. Int J Cancer. 2008;123(11):2605–15.
Li S, Xie Y, Zhang W, Gao J, Wang M, Zheng G, Yin X, Xia H, Tao X. Interferon alpha-inducible protein 27 promotes epithelial-mesenchymal transition and induces ovarian tumorigenicity and stemness. J Surg Res. 2015;193(1):255–64.
Kim YK, Ha HH, Lee JS, Bi X, Ahn YH, Hajar S, Lee JJ, Chang YT. Control of muscle differentiation by a mitochondria-targeted fluorophore. J Am Chem Soc. 2010;132(2):576–9.
Wang T, Guo K, Yang S, Zhang D, Cui H, Yin J, Yuan S, Wang Y, Qi Y, Wu W. Identification and validation of SLCO4C1 as a biological marker in hepatocellular carcinoma based on anoikis classification features. Aging. 2024;16(2):1440–62.
Currie E, Schulze A, Zechner R, Walther TC, Farese RV Jr. Cellular fatty acid metabolism and cancer. Cell Metab. 2013;18(2):153–61.
Han J, Han ML, Xing H, Li ZL, Yuan DY, Wu H, Zhang H, Wang MD, Li C, Liang L, et al. Tissue and serum metabolomic phenotyping for diagnosis and prognosis of hepatocellular carcinoma. Int J Cancer. 2020;146(6):1741–53.
Yun SH, Han SH, Park JI. Peroxisome Proliferator-Activated Receptor gamma and PGC-1alpha in Cancer: Dual Actions as Tumor Promoter and Suppressor. PPAR Res. 2018; 2018:6727421.
Rodriguez-Antona C, Ingelman-Sundberg M. Cytochrome P450 pharmacogenetics and cancer. Oncogene. 2006;25(11):1679–91.
Duan F, Wu H, Jia D, Wu W, Ren S, Wang L, Song S, Guo X, Liu F, Ruan Y, et al. O-GlcNAcylation of RACK1 promotes hepatocellular carcinogenesis. J Hepatol. 2018;68(6):1191–202.
Hou G, Lu Z, Wang Z, Yang X. The mitochondrial protein C1QBP promotes Hepatocellular Carcinoma Progression by Enhancing Cell Survival, Migration and Invasion. J Cancer. 2022;13(8):2477–89.
Wu C, Xia D, Wang D, Wang S, Sun Z, Xu B, Zhang D. TCOF1 coordinates oncogenic activation and rRNA production and promotes tumorigenesis in HCC. Cancer Sci. 2022;113(2):553–64.
Zhou Y, Yang D, Qiang Z, Meng Y, Li R, Fan X, Zhao W, Meng Y. Ribosome-inactivating protein MAP30 isolated from Momordica charantia L. induces apoptosis in hepatocellular carcinoma cells. Recent Pat Anticancer Drug Discov. 2022.
Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, Coussens LM, Gabrilovich DI, Ostrand-Rosenberg S, Hedrick CC, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. 2018;24(5):541–50.
Rizvi S, Wang J, El-Khoueiry AB. Liver Cancer immunity. Hepatology. 2021;73(1):86–103.
Liu G, Sun J, Yang ZF, Zhou C, Zhou PY, Guan RY, Sun BY, Wang ZT, Zhou J, Fan J, et al. Cancer-associated fibroblast-derived CXCL11 modulates hepatocellular carcinoma cell migration and tumor metastasis through the circUBAP2/miR-4756/IFIT1/3 axis. Cell Death Dis. 2021;12(3):260.
Xu H, Zhao J, Li J, Zhu Z, Cui Z, Liu R, Lu R, Yao Z, Xu Q. Cancer associated fibroblast-derived CCL5 promotes hepatocellular carcinoma metastasis through activating HIF1alpha/ZEB1 axis. Cell Death Dis. 2022;13(5):478.
Cheng Y, Li H, Deng Y, Tai Y, Zeng K, Zhang Y, Liu W, Zhang Q, Yang Y. Cancer-associated fibroblasts induce PDL1 + neutrophils through the IL6-STAT3 pathway that foster immune suppression in hepatocellular carcinoma. Cell Death Dis. 2018;9(4):422.
Yang Y, Ye YC, Chen Y, Zhao JL, Gao CC, Han H, Liu WC, Qin HY. Crosstalk between hepatic tumor cells and macrophages via Wnt/beta-catenin signaling promotes M2-like macrophage polarization and reinforces tumor malignant behaviors. Cell Death Dis. 2018;9(8):793.
Wang Z, He L, Li W, Xu C, Zhang J, Wang D, Dou K, Zhuang R, Jin B, Zhang W et al. GDF15 induces immunosuppression via CD48 on regulatory T cells in hepatocellular carcinoma. J Immunother Cancer. 2021;9(9).
Yang C, Guo Y, Wu Z, Huang J, Xiang B. Comprehensive Analysis of Cuproptosis-Related Genes in Prognosis and Immune Infiltration of Hepatocellular Carcinoma Based on Bulk and Single-Cell RNA Sequencing Data. Cancers (Basel). 2022;14(22).
Zhou SL, Zhou ZJ, Hu ZQ, Huang XW, Wang Z, Chen EB, Fan J, Cao Y, Dai Z, Zhou J. Tumor-Associated Neutrophils Recruit Macrophages and T-Regulatory cells to promote progression of Hepatocellular Carcinoma and Resistance to Sorafenib. Gastroenterology. 2016;150(7):1646–e16581617.
Chen J, Lin Z, Liu L, Zhang R, Geng Y, Fan M, Zhu W, Lu M, Lu L, Jia H, et al. GOLM1 exacerbates CD8(+) T cell suppression in hepatocellular carcinoma by promoting exosomal PD-L1 transport into tumor-associated macrophages. Signal Transduct Target Ther. 2021;6(1):397.
Zhang Z, Zeng X, Wu Y, Liu Y, Zhang X, Song Z. Cuproptosis-related risk score predicts prognosis and characterizes the Tumor Microenvironment in Hepatocellular Carcinoma. Front Immunol. 2022;13:925618.
Zhang G, Sun J, Zhang X. A novel cuproptosis-related LncRNA signature to predict prognosis in hepatocellular carcinoma. Sci Rep. 2022;12(1):11325.
Zhang Q, Huang Y, Xia Y, Liu Y, Gan J. Cuproptosis-related lncRNAs predict the prognosis and immune response in hepatocellular carcinoma. Clin Exp Med. 2022.
Acknowledgements
None.
Funding
This work was supported by the Tianjin Health Research Project (KJ20043); the Foundation of Administration of Foreign Experts Affairs of Tianjin (2016005); the Tianjin Key Medical Discipline (Pathology) Construction Project (TJYXZDXK-012A); and the Project of Precision Diagnosis and Drug Treatment of Tumor of Tianjin Cancer Institute and Hospital (ZLJZZDYYWZL14).
Author information
Authors and Affiliations
Contributions
W.Y. Conceptualization; Z.FL. Formal analysis; W.Y. and Z.FL. Funding acquisition; W.Y., S.B., and G.YN., Investigation; Y.HC., G.YH. and D.TT., Methodology; W.Y. Writing—original draft; S.BC. and Z.FL. Writing—review & editing. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Human tissues and human data in this study have been performed in accordance with the Declaration of Helsinki and approved by the Medical Ethics Committee of Tianjin Medical University Cancer Institute & Hospital (ID: bc2023070). Informed consents were written by participants in this study.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, Y., Zang, F., Shao, B. et al. From bioinformatics to clinical applications: a novel prognostic model of cuproptosis-related genes based on single-cell RNA sequencing data in hepatocellular carcinoma. BMC Immunol 25, 59 (2024). https://doi.org/10.1186/s12865-024-00649-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12865-024-00649-5