
Abstract
Background
The hard clam (Mercenaria mercenaria), a marine bivalve distributed along the U.S. eastern seaboard, supports a significant shellfish industry. Overharvest in the 1970s and 1980s led to a reduction in landings. While the transition of industry from wild harvest to aquaculture since that time has enhanced production, it has also exacerbated challenges such as disease outbreaks. In this study, we developed and validated a 66K SNP array designed to advance genetic studies and improve breeding programs in the hard clam, focusing particularly on the development of markers that could be useful in understanding disease resistance and environmental adaptability.
Results
Whole-genome resequencing of 84 individual clam samples and 277 pooled clam libraries yielded over 305 million SNPs, which were filtered down to a set of 370,456 SNPs that were used as input for the design of a 66K SNP array. This medium-density array features 66,543 probes targeting coding and non-coding regions, including 70 mitochondrial SNPs, to capture the extensive genetic diversity within the species. The SNPs were distributed evenly throughout the clam genome, with an average interval of 25,641 bp between SNPs. The array incorporates markers for detecting the clam pathogen Mucochytrium quahogii (formerly QPX), enhancing its utility in disease management. Performance evaluation on 1,904 samples demonstrated a 72.7% pass rate with stringent quality control. Concordance testing affirmed the array's repeatability, with an average agreement of allele calls of 99.64% across multiple tissue types, highlighting its reliability. The tissue-specific analysis demonstrated that some tissue types yield better genotyping results than others. Importantly, the array, including its embedded mitochondrial markers, effectively elucidated complex genetic relationships across different clam groups, both wild populations and aquacultured stocks, showcasing its utility for detailed population genetics studies.
Conclusions
The 66K SNP array is a powerful and robust genotyping tool that offers unprecedented insights into the species’ genomic architecture and population dynamics and that can greatly facilitate hard clam selective breeding. It represents an important resource that has the potential to transform clam aquaculture, thereby promoting industry sustainability and ecological and economic resilience.
Similar content being viewed by others

Background
The hard clam (Mercenaria mercenaria), commonly referred to as the northern quahog, is a bivalve mollusc native to the North American Atlantic coast, with a distribution range extending from Maritime Canada to Florida [1, 2]. Over the years, hard clams have emerged as one of the most economically significant marine resources in the United States. They are a cornerstone of a productive shellfish industry that spans the entire eastern seaboard, with over 3,600 metric tons harvested yearly, valued at around 50 million US dollars [3], and representing the most economically important species in several states. The shift from traditional harvesting of wild stocks to aquaculture represents one of the most transformative trends in the shellfish industry over the last few decades [4]. The growth of hard clam aquaculture has been particularly notable, with annual increases in production due to enhanced techniques and increased hatchery output from Massachusetts to Florida. For example, hard clam production in Florida rose from 87 million clams in 2016 to 115 million in 2021, marking a 31% increase [5].
In addition to their economic value, hard clams play an integral role in the ecosystem, particularly as benthic filter feeders [6]. This species is highly adaptable, thriving in diverse coastal environments with varying temperature and salinity levels, demonstrating significant physiological resilience [7]. Such adaptability not only allows the hard clam to manage stress from environmental changes but also supports its role in nutrient cycling within its habitats [8, 9]. Hard clams are pivotal in benthic-pelagic coupling, a process by which energy and nutrients are transferred from the water column to the benthic (ocean-floor) environment [10]. This ecological function involves filtering vast volumes of water to extract phytoplankton, thereby converting particulate matter into biomass that supports a range of higher trophic levels [11]. Their activities contribute significantly to the improvement of water quality, supporting the health of marine habitats [12]. By filtering algae and suspended particles from the water column, the hard clam helps improve water clarity, which enhances conditions for seagrass growth and helps prevent algal blooms [10, 13]. The ecological benefits extend beyond nutrient cycling, with hard clams also playing a crucial role in bioirrigation—enhancing the oxygenation and thus the overall health of the coastal sediments they inhabit [14].
Despite the ecological and economic benefits associated with hard clams, the industry faces significant challenges due to environmental and biological stressors. The rise in ocean temperatures and changes in salinity levels can lead to adverse effects on the growth, survival, and metamorphosis of bivalve species [15,16,17,18]. Additionally, ocean acidification driven by increased carbon dioxide levels in the ocean poses a particular risk to marine calcifiers like hard clams, threatening their ability to sustain biomineralization processes essential for shell formation and overall survival [19,20,21,22]. The combination of hypoxia and acidification can have additive and synergistic negative effects on the growth and survival of early life stages of bivalves, further exacerbating the challenges faced by these organisms [23]. Additionally, diseases such as QPX disease (an infection caused by Mucochytrim quahogii, formerly Quahog Parasite Unknown) have caused considerable mortality in cultivated hard clam populations, leading to substantial economic losses [24, 25]. Previous research has indicated a genetic basis for resistance to QPX, varying by geographic origin of the clam populations, which suggests that selective breeding for disease resistance could be beneficial [26, 27].
In response to these challenges, the field of genomics offers promising strategies for enhancing clam aquaculture. Genomic techniques can revolutionize selective breeding by enhancing our understanding of molluscan genetics and providing tools for genetic improvement [28]. Traditional selective breeding has been used effectively in the past to improve specific traits in bivalves [29]. For instance, in oysters (Crassostrea virginica and Crassostrea gigas), efforts have concentrated on improving disease resistance [30,31,32], growth rate [33, 34], and salinity tolerance [35]. Similarly, in hard clams, traditional selection has targeted enhancements in survival rates and growth efficiency [36], leveraging hybrid vigor for enhanced traits [37]. The advent of genomic selection (GS) represents a transformative advance in breeding technologies. The use of GS has shown promise in expediting selection for growth performance and disease resistance in various species due to its improved accuracy compared to traditional selection [38,39,40,41]. GS commonly employs SNP arrays, a preferred tool for routine genomic evaluations in major farmed species [42]. This methodology facilitates the accurate estimation of genomic estimated breeding values (GEBVs), predicting an individual’s potential to contribute desirable traits to future generations [43]. SNP arrays are particularly valued for their cost-effectiveness, scalability, and ability to be customized to target specific genetic variations, making them a favored choice over other high-throughput genotyping platforms [44]. SNP arrays have been developed for several aquaculture species including blue mussels [45], eastern oysters [46], Pacific oysters, and European flat oysters [47, 48]. SNP arrays may enable GS and enhance the genetic improvement of hard clams by providing a more precise and efficient means of selecting for traits like disease resistance and environmental resilience. Previous genetic analyses in the hard clam have relied on limited numbers of SNPs or microsatellite markers [27, 49, 50]. The sequencing of the hard clam genome [51, 52] paved the way for the development of advanced genomic tools such as SNP arrays that can empower aquaculture and facilitate population genetic studies.
Considering these advancements, our research aims to develop and validate a SNP genotyping platform for the hard clam. This tool will not only facilitate the effective selection of genetically superior breeding stocks but will also allow for the monitoring of genetic diversity and inbreeding within cultured populations. By integrating genomic tools with traditional aquaculture practices, it is possible to significantly advance the productivity and sustainability of the hard clam industry, ensuring its continued economic viability and ecological contribution.
Methods
Resequencing and SNP discovery
For SNP discovery, comprehensive whole-genome resequencing was conducted on two groups of hard clams (M. mercenaria) samples (Fig. 1 and Supplementary Table S1): 1) Individual clams (n = 84, wild and aquacultured) with 12 clams from each of seven distinct locations across the Atlantic coast and Gulf of Mexico, and 2) Pooled clam libraries with 277 clams grouped into seven pools, ranging from 28 to 56 clams, based on their geographic source. These pools represent a diverse set of populations and include samples from Maine, Massachusetts, New York (2 locations), Virginia, North Carolina, South Carolina and Florida.

Map of hard clam sampling locations used for the 66K SNP array design and validation. Locations are color-coded by latitude to illustrate the diverse geographic origins of the samples, ranging from Maine to Florida. The map includes an inset for a detailed view of sampling locations in New York
For the individual libraries, DNA was extracted from each clam using a Macherey–Nagel NucleoSpin kit following manufacturer’s instructions. Extracted DNA was then used for Illumina sequencing library synthesis using the NEBNext® Ultra™ DNA Library Prep Kit and samples were sequenced using an Illumina NovaSeq platform (S4 PE150 chemistry). Sequencing effort aimed to achieve an approximate coverage of ~ 30 × per genome, based on the Mercenaria mercenaria reference genome assembly size of 1.86 Gb [52]. For the pooled libraries, DNA was extracted from each individual clam using a standard phenol–chloroform-isoamyl alcohol (PCI) extraction protocol [53]. Equivalent quantities of DNA (~ 100 ng per clam) were used from each clam to create a total of seven DNA pools (1 pool per population). Pooled DNA samples were then used for Illumina library synthesis using an Illumina Truseq Nano DNA library preparation kit and produced libraries were sequenced on a NovaSeq 6000 S4 lane following manufacturer’s protocols. The detailed breakdown of individual and pooled samples, including population codes and specific sources is available in Supplementary Table S2. All the generated reads were aligned to the hard clam genome (GCF_021730395.1) [52] using the BWA (Burrows–Wheeler Aligner) software (bwa mem -t 28 -T 20 -M, v.0.7.17). The aligned bam files were sorted and indexed using Picard-tools (version 2.23.2). Then, variant calling was computed using the Genome Analysis Toolkit (GATK, v4.2.2.0–1-g24a8e02-SNAPSHOT) with default parameters. SNPs were further filtered with parameters “QD < 2, QUAL < 30, FS > 60, SOR > 3, MQ < 40, MQRankSum < -12.5, ReadPosRankSum < -8”.
SNP selection and array design
Initially, the focus was on identifying SNPs within gene regions due to their potentially informative value in genetic studies. Each of the 34,728 annotated protein-coding genes in the chromosome-level assembly of M. mercenaria genome (GCF_021730395.1) was checked for SNP presence. Criteria for selecting biallelic genic SNPs included a minor allele frequency (MAF) greater than 0.05, exclusion of SNPs within 30 base pairs of each other, and avoidance of A/T and C/G transversions. This process was facilitated using PLINK v.1.90 [54], SAMtools v.1.11 [55], BEDTools v.2.31.1 [56], BCFtools v.1.11 [57] and VCFtools v.0.1.16 [58]. The criteria for inclusion of SNPs within genes were stringent and aimed to maximize the power of the selected SNPs. A maximum of three SNPs were selected for each gene following a hierarchical selection process that prioritized coding sequences (CDS), untranslated regions (UTRs), and introns in that order. If more than three SNPs were found within the CDS of a gene, the three SNPs with the highest MAF were chosen. If exactly three SNPs were present in the CDS, all were retained without further filtering. When fewer than three SNPs were identified within the CDS, the selection was expanded to include SNPs in UTRs and introns. To achieve an even distribution across the non-coding regions of the genome, the genome was divided into 1,000 nucleotide windows using BEDTools v.2.31.1. To ensure high confidence and appropriate distribution, windows overlapping with gene coordinates, mitochondrial sequences, or repetitive DNA were eliminated. Within the remaining windows, SNPs were excluded if they had a MAF lower than 0.1, were located within 30 nucleotides of another SNP, were non-biallelic, or were A/T or C/G SNPs. The same filters were applied to collect mitochondrial SNPs. Probes for detecting the pathogen M. quahogii were selected based on the dissimilarity between the QPX genome [59] and the M. mercenaria [52] genome, following ThermoFisher Scientific’s recommendations.
Samples for array evaluation
Samples of wild clams and aquacultured stocks were used to evaluate the array (Fig. 1; Tables 1 and 3). Aquacultured stocks originated from Massachusetts (A1MA), New York (AUSDAD, AUSDAE, and AUSDAF), and Florida (A1FL, A2FL, A3FL). All analyzed clams were adults, except for subsets of juveniles in the aquacultured stocks: 204 out of 307 in AUSDAD, 203 out of 297 in AUSDAE, and 200 out of 298 in AUSDAF. These subsets consisted of juvenile clams (0.1 to 1.3 cm2 in shell surface area) preserved whole (with shell) in ethanol. All analyzed clams were collected between 2019 and 2022 excluding a group that included clams confirmed to be positive for QPX disease. For instance, W03NY (Table 1) consisted of mantle tissues (60 individuals) or DNA (9 samples extracted from mantle tissues) collected from clams harvested in 2003 and included specimens confirmed to be positive for QPX disease using standard histopathology techniques [60]. The same population sampled in 2003 was targeted again in 2022 (W22NY) to evaluate if the genetic composition of the stock has changed over the last two decades.
Before genotyping, the soft tissue of ethanol-fixed juvenile clams was either used as is, or it was dissected to remove the digestive gland before DNA was extracted. This was done to evaluate the effect of removing digestive tissue (which is typically rich in inhibitors) on genotyping outcomes.
Wild samples consisted entirely of adult clams originated from New York, New Jersey, and Florida. The oldest samples used for genotyping were mantle tissues preserved in ethanol and held at -80 °C since 2003 (~ 20 years). Further information about sample types and preservation methods is given in Table 3.
Total genomic DNA was extracted from hard clam samples at the Center for Aquaculture Technologies using a magnetic bead-based protocol. Briefly, samples (~ 10–15 mg) were subsampled and processed using Mag-Bind Blood and Tissue DNA Kits (Omega BioTek, Norcross, GA) according to the manufacturer’s guidelines. Automated processing and liquid handling steps associated with the extraction protocol were performed using PurePrep 96 units (Molgen, San Diego, CA) following the kit and instrument-specific guidelines. The resulting gDNAs were assessed for yield and quality by spectrophotometry (Nanodrop) and 2% agarose gels, targeting a minimum of 35 µl at > 20 ng/µl of largely intact DNA (minimum 5 Kb). Genotyping was performed at Neogen (Lincoln, Nebraska) on custom Axiom 384HT arrays [Axiom HD Array (60 K)_Clam] using processes outlined in the Axiom Assay 384HT Array Format Automated Workflow User Guide. PCA was calculated using PLINK v.1.90 [54].
SNP array data analyses
The SNP array data were processed using the Axiom Analysis Suite 5.0 software (Thermo Fisher, CA), following the Best Practices Workflow, and using recommended threshold settings (QC ≥ 0.82, QC call rate ≥ 97%, average call rate for passing samples ≥ 98.5%). The marker-conversion rate was calculated as the percentage of polymorphic, QC-compliant, and BestAndRecommended SNPs on the array. The genetic indices were calculated using vcfR 1.15.0 [61], adegenet v2.1.10 [62], and hierfstat [63] packages in R. Pairwise significance of FST values was based on 10,000 iterations of the data. The maximum likelihood tree was constructed using IQ-TREE [64] using the best-fit model of nucleotide substitution based on Akaike information criterion (AIC) in ModelFinder [65], and branch support values were estimated using UFBoot [66].
Results
SNP discovery and array design
Through whole-genome resequencing of 84 individual clams and 277 clams from pooled libraries, we initially identified 305,753,445 SNPs across the M. mercenaria genome. Filtering processes refined this large dataset to identify the most informative SNPs for subsequent analyses. After the initial filtering of genic SNPs, 91,898 SNPs across 32,018 genes were retained. These SNPs included 79,454 within CDSs, 966 in UTRs, and 11,478 in introns. Exploration of intergenic regions yielded an additional 278,452 SNPs. The analysis also yielded 72 mitochondrial SNPs (mtSNPs), 150 SNPs associated with hard clam resistance to QPX disease [27], and 101 markers from the genome of Mucochytrium quahogii (causative agent of QPX disease). The comprehensive SNP collection of 374,463 SNPs was submitted to ThermoFisher Scientific for probe design, including a selection of 3,790 non-polymorphic sequences for design quality control (dQC). All designed probes underwent a thorough evaluation for genomic duplication, interactions with other probes, distance from known polymorphisms, and likelihood of successful probe conversion. This evaluation identified 312,064 SNPs that had a conversion probability greater than 0.6 and were free of genomic duplication or potential interference from other polymorphisms. From this refined pool, SNPs were specifically selected to enhance the array's utility: genic SNPs were prioritized for their functional insights, and intergenic SNPs were included and strategically distributed to ensure comprehensive genome coverage. Importantly, most intergenic SNPs on the array were chosen for having MAF greater than 0.2, a criterion aimed at boosting the analytical power of genetic studies by ensuring sufficient allelic variation. As a result, the final design of the screening array was determined to include 66,543 probes from the hard clam and 101 from QPX, each uniquely targeting a different genetic marker (Table 2).
The array contains 17,492 genic SNPs, 48,981 intergenic SNPs, and 70 mtSNPs of M. mercenaria. Within the category of nuclear genic SNPs, there are 17,385 SNPs located within CDSs, 10 in UTRs, and 97 in introns, representing approximately 50.1% (17,411 genes) of all identified protein-coding genes in the M. mercenaria genome. The nuclear SNPs are evenly distributed across chromosomes, with an average interval of 25,641 bp between SNPs, ranging from 22,481 to 34,102 bp across different chromosomes (Fig. 2, Supplementary Table S3). The distance between the 66 mtDNA SNPs ranged from 35 to 958 bp, with an average of 244 bp.

Chromosomal distribution of SNPs on the hard clam 66K SNP array
Due to the comprehensive coverage of the array and its specific focus on the hard clam, it was named the hard clam 66K SNP array. The official name for this tool, reflecting its broad scope and targeted application, is the Applied Biosystems Axiom Clam_Mm1 Array (384-plate format).
Performance of the SNP array
A genotyping study was conducted on 1,904 wild and aquacultured clams to assess the performance of the 66K SNP array. To assess the repeatability of the genotyping process, 18 individuals were genotyped twice. Of the total number of samples, 1,384 (72.7%) passed the stringent genotyping quality control (QC) standards, which included a DQC threshold greater than 0.82 and a QC call rate exceeding 97%. The average QC call rate for passing samples was 98.8%.
The evaluation revealed notable variations in sample pass rates across different tissue types and preservation methods (Table 3). For example, mantle tissue preserved in ethanol resulted in relatively high pass rates, with populations such as A1MA achieving a 100% success rate. Adductor muscle tissue preserved in ethanol showed variable success rates, ranging from 82.7% in A2FL to 96% in A3FL, the latter being among the highest observed. Conversely, tissues such as juvenile clams preserved in ethanol exhibited markedly lower pass rates. It was also observed that DNA samples stored in water had lower pass rates, typically ranging from 62.5% to 66.7%. This can be attributed to acidic degradation of DNA stored in water over time [67, 68], and the storage durations in this study ranged from three to nineteen years. In contrast, mantle tissue preserved in ethanol at -80℃ for 19 years, which has been shown to be much better for preserving sample integrity, yielded results (85% pass rate) comparable to some of the more recent ethanol-fixed samples (e.g., W1NJ, W2NJ). Tissue (mantle) homogenates preserved in phosphate-buffered saline (PBS) before DNA extraction did not yield any results (6 out of 6 individuals failed; 0% pass rate; Table 3).
Further analysis of juvenile clams genotyped as whole ethanol-fixed animals, with and without digestive tissues, exhibited a clear trend related to the size of the specimens (Table 4). Intact juvenile clams exhibited a negative correlation between size and genotyping success rate, with larger animals exhibiting notably lower success rates. For instance, animals between 0.1 and 0.2 cm2 (shell surface area) had a pass rate of 54.2%, which decreased to as low as 9.8% for those in the 0.6–1.3 cm2 range. However, this trend was not observed in samples where digestive tissue was removed prior to DNA isolation. In these cases, pass rates generally improved and were less variable with size; when the digestive tract was removed, animals sized 0.6–1.3 cm2 showed a 70.5% success rate.
After processing with the Best Practices Workflow for genotype calling, the SNPs from the hard clam 66K SNP array were categorized based on their genotyping clarity and reliability. Overall, 36,153 SNPs were designated as PolyHighResolution, indicating polymorphic SNPs with well-defined genotype clusters. In contrast, 1,133 SNPs were classified as NoMinorHom, where one of the homozygous genotypes was missing. Additionally, 1,221 SNPs were classified as MonoHighResolution, indicating monomorphic SNPs with a single, clear genotype cluster. In the array, SNPs that failed to meet the QC threshold call rate of 97% were categorized as CallRateBelowThreshold, accounting for 7,187 SNPs. OffTargetVariant (OTV) SNPs, which may indicate the presence of an additional cluster, numbered 4,598. Finally, SNPs that presented with multiple issues were grouped into the 'Other' category, which comprised 16,251 SNPs. The total count of BestandRecommended markers, which includes SNPs from PolyHighResolution, NoMinorHom, and MonoHighResolution clusters, was 38,507. This represents 57.87% of the total SNPs on the array (Table 5).
The overall SNP conversion rate, reflecting the number of polymorphic and recommended SNPs, was 56.03%, corresponding to 37,286 SNPs. The remaining markers either fell below the call rate threshold, were identified as OTVs, or had multiple issues preventing them from being classified as reliable markers.
Among the BestandRecommended markers identified, 12,262 SNPs were classified as genic, involving 12,223 distinct protein-coding genes. This represents a significant portion (35.2%) of the 34,728 protein-coding genes identified within the M. mercenaria genome. In addition, 26,179 intergenic SNPs were identified. This underscores the extensive coverage of the genomic landscape provided by the array. The inclusion of 66 mtSNPs highlights the comprehensive approach to capturing the complete genetic diversity of the hard clam, from nuclear to organellar DNA.
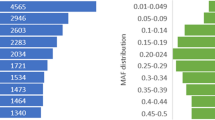
Among the 1,384 clam samples that passed the genotyping QC, a subset of BestandRecommended SNPs showed low MAF, with 2,138 SNPs (5.73%) having a MAF less than 0.05, and 3,123 SNPs (8.38%) with a MAF less than 0.1 (Fig. 3). SNPs with a lower MAF are less prevalent within the population but may still be of significance for certain traits and genetic diversity studies. Overall, 30,621 (82.12%) of the SNPs had MAF > = 0.2, making them highly informative.

Distribution of minor allele frequency of SNP markers on the hard clam 66K SNP array based on 1,384 samples passing genotyping QC
Genotyping was repeated for 18 individual hard clams to assess the reproducibility of the results. The average concordance rate across these repeated measures was high at 99.64%. This indicates that the SNP calls were consistent, with only 0.36% of the SNPs showing discordant genotypes on retesting (Table 6). Further analysis of the discordant SNPs identified by repeated genotyping shows a high degree of reproducibility. Of the total 2,134 discordant SNPs, 1,887 occurred in only one sample, indicating that most observed discrepancies were isolated events (Supplementary Table S4). Lower frequencies of discordance involving multiple individuals were observed, with 208 discordant SNP genotypes appearing in two samples, 27 in three, and progressively fewer in four to twelve samples, indicating a minimal systematic error.
Hemocyte samples showed particularly high concordance rates, ranging from 99.64% to 99.85%. Samples from juvenile clams with digestive tissues removed had slightly lower concordance rates of 99.16% to 99.34%. This observed variation in concordance could be attributed to differences in DNA quality, which is a common and understandable occurrence in samples representing more complex biological matrices.
Exploring genetic structures in hard clams using a 66K SNP array
To confirm the utility of the SNP array in elucidating clam population genetic structure, a Principal Component Analysis (PCA) was conducted. The analysis encompassed wild and aquacultured clams sampled from different geographic locations (Fig. 4A). The resulting PCA highlighted a distinct clustering pattern. For instance, aquacultured clam samples from the Northeast (AUSDAD, AUSDAE, and AUSDAF) clustered separately from Florida samples (A1FL, A2FL, and A3FL). Wild clams formed a single cluster on the PCA plot and showed little dispersion, indicating a genetic coherence among these groups. However, a closer look at wild clams showed that the clams from Florida (WFL) formed a separate cluster from the northeastern clams (W03NY, W22NY, W1NJ, W2NJ) (Fig. 4B). The PCA substantiates the capability of the hard clam 66K SNP array to resolve complex genetic relationships within and across wild and aquacultured populations.

Principal component analysis of (A) wild and aquacultured hard clams with genotype data from markers on the 66K SNP array. B Detailed view of the genetic clustering among wild populations from Northeastern and Florida regions. Abbreviations: A1MA (population 1 in MA), A1FL (population 1 in FL), A2FL (population 2 in FL), A3FL (population 3 in FL), AUSDAD (USDA strain D), AUSDAE (USDA strain E), AUSDAF (USDA strain F), W03NY (clams from NY, 2003), W22NY (clams from NY, 2022), W1NJ (population 1 in NJ), W2NJ (population 2 in NJ), WFL (wild clams from FL). For full names and additional details, see Table 1
We analyzed the genetic structure and diversity of the hard clam populations using various genetic metrics, including observed heterozygosity (Ho), expected heterozygosity (He), inbreeding coefficients (FIS), and fixation indices (FST). The overall genetic structure revealed an FST value of 0.024 for the total dataset, indicating a low to moderate level of genetic differentiation among the populations (Supplementary Table S5). The overall inbreeding coefficient was 0.029, reflecting minimal inbreeding within the total sample set. Observed heterozygosity was 0.396, while expected heterozygosity was 0.408, suggesting that genetic variation is slightly higher than what is observed.
The FIS values for aquacultured populations were found to be low (from 0.000 to 0.050), indicating the implementation of effective genetic management practices that minimize the occurrence of inbreeding (Supplementary Table S6). The Ho values for these populations ranged from 0.380 to 0.403, and He ranged from 0.393 to 0.402, indicating a healthy level of genetic diversity within these populations. The wild populations (W03NY, W1NJ, W22NY, and W2NJ) exhibited slightly elevated but still low FIS values of 0.042, 0.041, 0.039, and 0.042, respectively. The observed heterozygosity for these populations was approximately 0.400, and the expected heterozygosity was approximately 0.418 (Supplementary Table S6). Pairwise FST values (Supplementary Table S7) indicated significant genetic differentiation between certain locations. The FST values for the wild populations from New York and New Jersey (W03NY, W1NJ, W22NY, W2NJ) were found to be close to zero (p-value > 0.05), indicating that there is no significant genetic differentiation among them. However, these populations exhibited significant divergence from the wild Florida population (WFL), underscoring the influence of geography and the environment on genetic differentiation. Similarly, all aquacultured populations (A1MA, AFL, AUSDA) displayed pronounced genetic distinctness.
Mitochondrial diversity in hard clam populations
Four mtSNPs were excluded from the original set of 70 due to genotyping quality control deficiencies, and one was monomorphic across all evaluated samples. The absence of heterozygotes in any of the mtSNPs confirms the haploid nature of these markers. Moreover, this observation indicates a lack of heteroplasmy, which refers to the presence of multiple mitochondrial DNA types within the cells of an organism [69], in the studied populations.
Further analyses were conducted using the remaining mtSNPs to explore phylogenetic relationships and haplotype distributions among the hard clam populations. The phylogenetic analysis identified three distinct mitochondrial haplogroups, labeled Hap1, Hap2, and Hap3, (Fig. 5A, Supplementary Fig. S1).

Phylogenetic analysis and haplotype distribution of mitochondrial SNPs in hard clam populations. A Maximum likelihood phylogenetic tree of 1,384 hard clams based on mitochondrial SNPs. Branches represent identified haplogroups (Hap1, Hap2, Hap3), illustrating genetic relationships derived from the K hard clam 66K SNP array. B Distribution of mitochondrial haplotypes among various hard clam populations
Phylogenetic clustering revealed distinct patterns of haplotype distribution across both wild and aquacultured samples (Fig. 5B, Supplementary Table S8). Notably, Hap1 was exclusively found in aquacultured Florida samples. Hap2 showed a broader geographic distribution, it was present in all samples except A3FL and AUSDAF, indicating its widespread occurrence across diverse geographic and breeding backgrounds. Hap3 was present at high frequency in nearly all samples including 100% of AUSDAF samples and 93.3% of the wild Florida (WFL) samples. In contrast, Hap3 was present at lower frequency in A2FL and A3FL samples (28.7% and 7.1% respectively).
Analysis of QPX presence
The assessment of the efficacy of the hard clam 66K SNP array's QPX probes included six histologically confirmed QPX-positive controls and 35 QPX-negative controls. The median log2 ratio is a measure of the relative abundance of specific SNP markers detected by the Affymetrix SNP Array. A high median log2 ratio indicates a significant increase in detection signal, which is used to infer the presence of QPX pathogen-related markers. Among the positive controls, only a single sample demonstrated a significantly high median log2 ratio (Fig. 6). This indicates that while the probes can identify QPX presence, their effectiveness may vary with the pathogen load within the sample.

Median log2 ratios across hard clam samples from different regions and infection statuses. Blue points indicate clams from the Northeast (NJ, NY, MA). Orange points represent clams from Florida, where QPX is not known to exist. Green points are from histologically confirmed QPX-positive samples. Pink points are from QPX-negative samples. The red dashed line marks the threshold for positive detection
In contrast, all histologically validated QPX-negative samples showed very low median log2 ratios. This consistent result across the negative controls underscores the specificity of the probes under these test conditions. Furthermore, the samples originating from Florida, where QPX is not known to be present, also had low median log2 ratios, further underlining the lack of “false-positive” signals. In fact, the array may be able to only detect intense infections as shown for some northeastern clams (where QPX is enzootic) and for one of the histologically QPX-positive clams. It should be noted that QPX disease in clams is typically focal, and a biopsy sample collected for genotyping may not contain parasite cells even if it is derived from a clam microscopically confirmed to be infected. Despite these limitations, the current findings indicate that the hard clam 66K SNP array can detect QPX in clams, but a more elaborate study that includes samples with broad range of known concentrations of QPX DNA is needed to provide a more comprehensive assessment of the sensitivity of the array for parasite detection.
Discussion
Array design and performance
The design of the 66K SNP array for M. mercenaria represents a significant advance in genomic tool development, tailored to meet both the ecological and aquacultural demands of the hard clam industry. This customized array includes SNPs derived from an expansive pool of genomic data encompassing a large portion of the native geographical range. Approximately 305 million raw SNPs were identified through whole-genome resequencing, showcasing the vast genetic diversity inherent to M. mercenaria. The subsequent filtering and validation process refined this number, ensuring that only the most reliable and informative SNPs were included. The SNP selection was strategically conducted to balance between genic and intergenic regions, enhancing the utility of the array for various genetic studies. The final selection included 12,262 genic SNPs, mapped to 12,223 genes, representing approximately 35.2% of all protein-coding genes in the clam's genome. This extensive coverage allows the identification of gene-specific markers linked to traits such as disease resistance, stress tolerance, and growth rates. The inclusion of 26,179 intergenic SNPs provides a broader genomic landscape, facilitating the investigation of neutral processes across different clam populations.
Mitochondrial SNPs were also carefully chosen for their known relevance in tracing maternal lineages and assessing population dynamics and historical demography. The meticulous selection of these 66 mitochondrial markers underscores the array's design philosophy—precision in genetic representation to support robust ecological and evolutionary studies.
The array's architecture also considered the physical distribution of SNPs across the clam genome. SNPs were evenly spaced to maximize genomic coverage and minimize bias in genetic linkage analyses. This spatial arrangement is crucial for conducting genome-wide association studies (GWAS), which rely on the comprehensive genomic representation to accurately identify associations between genetic variants and phenotypic traits. The even distribution of SNP markers ensures thorough genomic coverage, facilitating detailed genetic analysis and supporting the advancement of genomic selection in hard clam aquaculture.
The performance of the hard clam 66K SNP array, with its concordance rate of 99.64%, places it on the upper end of the range for first-generation SNP arrays in other bivalves, typically between 96.6% and 99.8% [46, 47]. These figures demonstrate the array's robust design, comparable to or exceeding that of many initial bivalve arrays designed for similar purposes. The marker conversion rate (56.03%) is somewhat low with a large proportion of loci being classified as “Other”. This result highlights the challenge in array development for high polymorphism species, and the conversion rate may be improved by a two-step process [46].
The evaluation revealed significant variability in sample pass rates across different tissue types and preservation methods, underscoring the importance of sample handling in genetic studies. It should be noted that we purposely included a small number of samples generated from other standard workflows (e.g., tissue homogenates in PBS which is routinely used for DNA extraction for qPCR diagnostics of QPX [70]) to evaluate their usefulness and complementarity to genotyping with the hard clam SNP array. The findings show that protocol adaptations are needed to make such samples useful. Overall, mantle and adductor muscle tissue preserved in ethanol showed high pass rates, demonstrating the effectiveness of ethanol preservation in maintaining DNA integrity, including for samples preserved for 19 years. This finding is consistent with best practices across genomic studies in aquatic species, where ethanol preservation is often recommended to ensure the stability and quality of DNA samples [71]. Conversely, DNA samples stored in water showed markedly lower pass rates, which could be attributed to DNA degradation over time [72]. This aspect of the study highlights the critical role of preservation methods in genetic research, suggesting that maintaining optimal preservation conditions is essential for maximizing the success of genotyping efforts. Juvenile clams can also be used as biomaterial for genotyping. The study revealed a clear trend related to the size of the juvenile clams, with larger animals generally having lower genotyping success rates when digestive tissues were included during DNA isolation. This observation suggests the potential complications of including larger amounts of digestive tissue, which may contain inhibitors or contaminants that affect DNA quality [73]. When digestive tissues were removed, success rates improved significantly, suggesting that careful tissue selection and handling can mitigate some challenges associated with genotyping larger or more complex tissue samples.
Array applications
Principal component analysis performed on the SNP array provided deep insights into the genetic structures of M. mercenaria populations, highlighting its utility in delineating genetic variation across different geographic and management contexts. This analysis, which included wild and aquacultured populations from different geographic regions, revealed distinct genetic clustering patterns critical for understanding the impact of regional culture practices and environmental factors on the genetic diversity of hard clams. The PCA results highlighted significant differences in genetic clustering between aquacultured populations in the Northeast (AUSDA: AUSDAD, AUSDAE, and AUSDAF) and clams in Florida (AFL: A1FL, A2FL, and A3FL). This clear separation highlights the influence of region-specific selective breeding practices and localized environmental adaptations. The ability to assess this variation is critical because it reflects not only adaptive responses to local conditions but also potential neutral divergence due to limited gene flow, as well as the results of human intervention in breeding strategies aimed at optimizing traits beneficial to aquaculture productivity and sustainability. Our analysis revealed clear genetic differentiation between wild and aquacultured populations of M. mercenaria in Florida, in contrast to the minimal genetic differences observed previously using microsatellite markers [74]. The wild populations from the Northeast (W03NY, W22NY, W1NJ, W2NJ) showed less genetic dispersion compared to the Florida wild population (WFL) and the aquacultured populations, indicating a relatively homogeneous genetic makeup within this regional group. This homogeneity is supported by the lack of significant genetic differentiation among these northeastern wild populations, as evidenced by FST values close to zero, indicating strong genetic connectivity. However, the significant genetic differentiation between these northeastern populations and the wild Florida population (WFL) suggests the existence of factors leading to distinct genetic structures. These factors could be geographic barriers that limit gene flow and/or distinct environmental pressures and natural selection processes in different habitats. Such genetic isolation between northeastern and southern hard clam populations has been previously reported using other genetic and genomic methods [50]. For instance, using a genotyping-by-sequencing approach, Ropp et al. [48] also showed clear genetic segregation between clams collected from New York and New Jersey (which were genetically similar) and those sourced from North and South Carolina (the Ropp study did not include clams from Florida). These findings have important implications for conservation and management strategies, as they underscore the need to consider local genetic specificities when planning breeding and conservation efforts to ensure the preservation of genetic diversity and adaptability [75]. The PCA revealed that certain individuals from the aquacultured populations A3FL and AUSDAF exhibited unique placements on the PCA plot. This unusual distribution could be attributed to several factors. One possible explanation is the presence of unique genetic traits that have arisen due to localized breeding strategies and specific environmental conditions [76]. Another critical factor to consider is ascertainment bias in SNP arrays. Ascertainment bias occurs when the SNPs included in the array are identified and selected based on a discovery panel that does not represent the entire population's genetic diversity [77]. This bias can result in skewed allele frequency distributions and may influence the observed population structure [78]. The distinct placement of some individuals from A3FL and AUSDAF on the PCA plot might be partially due to such bias. To fully understand the genetic structure of these populations and the potential impacts of ascertainment bias, further analysis should be conducted.
The ability to analyze mtSNPs in M. mercenaria increases the potential applications of the hard clam SNP array for mitochondrial DNA analysis in both research and aquaculture breeding programs. In this context, phylogenetic analysis of mtSNPs identified three mitochondrial haplogroups within M. mercenaria, illustrating the hard clam 66K SNP array’s utility in elucidating mitochondrial genetic structures. The delineation of these groups provides insight into the historical and adaptive narratives that have shaped the genetic landscape of M. mercenaria. Our results complement previous mitochondrial DNA studies of the hard clams, which described M. mercenaria stocks in the Atlantic as a single evolutionary unit divided into at least three closely related populations, noting regional adaptive differences among northern, central, and southern populations [2]. Our findings also reflect the importance of mitochondrial markers in revealing phylogenetic divisions within bivalve species, as demonstrated in previous studies [79].
The use of the hard clam SNP array for the detection of QPX in hard clams exemplifies how advanced genomic tools are becoming increasingly important in the management of disease in marine aquaculture. Despite the promising application of this approach, our findings echo challenges with the detection of some pathogens using SNP arrays for the eastern oyster [46], where specificity and sensitivity issues were encountered, particularly at lower pathogen concentrations. Our finding suggests that while the SNP array can detect the presence of QPX, the pathogen load likely influences the detection thresholds. This could be due to variability in the ability to detect different stages of infection (e.g., genome replication within a parasite cell [24] or different DNA extraction efficiencies between different parasite life stages) or to the overall pathogen load present in the tissue samples, which could affect the probe's ability to detect the pathogen [80], potentially leading to a high rate of type II error. Additional research is needed to define QPX detection thresholds for confirmation of infection status and to clarify the relationship between parasite load and signal strength for potential quantification of parasite infections. This preliminary assessment indicates that detection of positive controls is possible, but comprehensive testing and validation are necessary before the arrays can be reliably used for diagnosis.
Conclusions
The development of the 66K SNP array for M. mercenaria marks a significant step forward in the integration of genomic tools into hard clam aquaculture. By facilitating detailed genetic analyses, supporting breeding for desirable traits, and potentially aiding in disease management and environmental adaptation strategies, this tool enhances the ability to manage hard clam populations more effectively. Future research should focus on expanding the applicability of the array to hard clam populations, further refining SNP selection and array design methodologies, and integrating these genomic tools with ecological and conservation approaches. This new array is expected to be a reliable tool for genome-wide association studies of clam resistance to various biological and environmental stressors and for genomic selection, thus taking clam aquaculture to the next level.
Availability of data and materials
Data generated or analyzed during this study are included in this published article and its Supplementary Files or are available on NCBI under BioProject PRJNA1125906. The Applied Biosystems Axiom Clam_Mm1 Array can be ordered from ThermoFisher following request approval by the corresponding authors.
References
Kraeuter JN, Castagna M. Biology of the hard clam. 1st ed. Amsterdam: Elsevier; 2001.
Baker P, Austin JD, Bowen BW, Baker SM. Range-wide population structure and history of the northern quahog (Merceneria merceneria) inferred from mitochondrial DNA sequence data. ICES J Mar Sci. 2008;65:155–63.
National Marine Fisheries Service. Fisheries Economics of the United States. 2018.
Brown AR, Lilley M, Shutler J, Lowe C, Artioli Y, Torres R, et al. Assessing risks and mitigating impacts of harmful algal blooms on mariculture and marine fisheries. Rev Aquac. 2020;12:1663–88.
Florida Aquaculture Surveys. https://shellfish.ifas.ufl.edu/industry/florida-aquaculture-survey. Accessed 12 Jul 2024.
Gili J-M, Coma R. Benthic suspension feeders: their paramount role in littoral marine food webs. Trends Ecol Evol. 1998;13:316–21.
Dickinson GH, Matoo OB, Tourek RT, Sokolova IM, Beniash E. Environmental salinity modulates the effects of elevated CO2 levels on juvenile hard shell clams, Mercenaria mercenaria. J Exp Biol . 2013. https://doi.org/10.1242/jeb.082909.
Doering PH, Kelly JR, Oviatt CA, Sowers T. Effect of the hard clam Mercenaria mercenaria on benthic fluxes of inorganic nutrients and gases. Mar Biol. 1987;94:377–83.
Gobler CJ, Doall MH, Peterson BJ, Young CS, DeLaney F, Wallace RB, et al. Rebuilding a collapsed bivalve population, restoring seagrass meadows, and eradicating harmful algal blooms in a temperate lagoon using spawner canctuaries. Front Mar Sci. 2022;9:911731.
Doering P, Oviatt C. Application of filtration rate models to field populations of bivalves: an assessment using experimental mesocosms. Mar Ecol Prog Ser. 1986;31:265–75.
Peterson CH, Summerson HC, Duncan PB. The influence of seagrass cover on population structure and individual growth rate of a suspension-feeding bivalve. Mercenaria mercenaria J Mar Res. 1984;42:123–38.
Newell RIE. Ecosystem influences of natural and cultivated populations of suspension-feeding bivalve molluscs: a review. J Shellfish Res. 2004;23:51–62.
Galimany E, Lunt J, Freeman CJ, Segura-García I, Mossop M, Domingos A, et al. Bivalve feeding on the brown tide Aureoumbra lagunensis in a shallow coastal environment. Front Mar Sci. 2021;8:714816.
Norkko J, Shumway SE. Bivalves as bioturbators and bioirrigators. In: Shumway SE, editor. Shellfish aquaculture and the environment. 1st ed. Wiley; 2011. p. 297–317.
Ivanina AV, Dickinson GH, Matoo OB, Bagwe R, Dickinson A, Beniash E, et al. Interactive effects of elevated temperature and CO2 levels on energy metabolism and biomineralization of marine bivalves Crassostrea virginica and Mercenaria mercenaria. Comp Biochem Physiol A: Mol Integr Physiol. 2013;166:101–11.
Miller C, Waldbusser G. A post-larval stage-based model of hard clam Mercenaria mercenaria development in response to multiple stressors: temperature and acidification severity. Mar Ecol Prog Ser. 2016;558:35–49.
Matoo OB, Ivanina AV, Ullstad C, Beniash E, Sokolova IM. Interactive effects of elevated temperature and CO2 levels on metabolism and oxidative stress in two common marine bivalves (Crassostrea virginica and Mercenaria mercenaria). Comp Biochem Physiol A: Mol Integr Physiol. 2013;164:545–53.
Liang J, Masanja F, Li Y, Guo Y, Deng Y, Zhao L. Hard clam resilience to marine heatwaves in the face of climate change. Front Mar Sci. 2024;11:1382825.
Gobler CJ, Clark HR, Griffith AW, Lusty MW. Diurnal fluctuations in acidification and hypoxia reduce growth and survival of larval and juvenile bay scallops (Argopecten irradians) and hard clams (Mercenaria mercenaria). Front Mar Sci. 2017;3. https://doi.org/10.3389/fmars.2016.00282.
Schwaner C, Farhat S, Barbosa M, Boutet I, Tanguy A, Pales Espinosa E, et al. Molecular features associated with resilience to ocean acidification in the northern quahog, Mercenaria mercenaria. Mar Biotechnol. 2023;25:83–99. https://doi.org/10.1007/s10126-022-10183-3.
Schwaner C, Barbosa M, Connors P, Park T-J, De Silva D, Griffith A, et al. Experimental acidification increases susceptibility of Mercenaria mercenaria to infection by Vibrio species. Mar Environ Res. 2020;154:104872.
Schwaner C, Farhat S, Haley J, Pales Espinosa E, Allam B. Proteomic and transcriptomic responses enable clams to correct the pH of calcifying fluids and sustain biomineralization in acidified environments. IJMS. 2022;23:16066.
Gobler CJ, DePasquale EL, Griffith AW, Baumann H. Hypoxia and acidification have additive and synergistic negative effects on the growth, survival, and metamorphosis of early life stage bivalves. PLoS ONE. 2014;9:e83648.
Geraci-Yee S, Brianik CJ, Rubin E, Collier JL, Allam B. Erection of a new genus and species for the pathogen of hard clams ‘Quahog Parasite Unknown’ (QPX): Mucochytrium quahogii gen. nov., sp. nov. Protist. 2021;172:125793.
Smolowitz R. A review of QPX disease in the northern quahog (= hard clam) Mercenaria mercenaria. J Shellfish Res. 2018;37:807–19.
Guo X, Ford SE. Infectious diseases of marine molluscs and host responses as revealed by genomic tools. Phil Trans R Soc B. 2016;371:20150206.
Farhat S, Tanguy A, Pales Espinosa E, Guo X, Boutet I, Smolowitz R, et al. Identification of variants associated with hard clam, Mercenaria mercenaria, resistance to Quahog Parasite Unknown disease. Genomics. 2020;112:4887–96.
Hollenbeck CM, Johnston IA. Genomic tools and selective breeding in molluscs. Front Genet. 2018;9:253.
Tan K, Zhang H, Zheng H. Selective breeding of edible bivalves and its implication of global climate change. Rev Aquac. 2020;12:2559–72.
Ragone Calvo LM, Calvo GW, Burreson EM. Dual disease resistance in a selectively bred eastern oyster, Crassostrea virginica, strain tested in Chesapeake Bay. Aquaculture. 2003;220:69–87.
Proestou DA, Vinyard BT, Corbett RJ, Piesz J, Allen SK, Small JM, et al. Performance of selectively-bred lines of eastern oyster, Crassostrea virginica, across eastern US estuaries. Aquaculture. 2016;464:17–27.
Allen SK, Small JM, Kube PD. Genetic parameters for Crassostrea virginica and their application to family-based breeding in the mid-Atlantic. USA Aquaculture. 2021;538: 736578.
Zhang J, Li Q, Xu C, Han Z. Response to selection for growth in three selected strains of the Pacific oyster Crassostrea gigas. Aquaculture. 2019;503:34–9.
Guo X. Genetics in shellfish culture. Molluscan Shellfish Aquaculture: A Practical Guide. In: Shumway SE, editor. 5M Books Ltd; 2021. p. 393–413. https://scholarship.libraries.rutgers.edu/esploro/outputs/bookChapter/Genetics-in-shellfishculture/991031744442504646.
Griffiths JS, Johnson KM, Sirovy KA, Yeats MS, Pan FTC, La Peyre JF, et al. Transgenerational plasticity and the capacity to adapt to low salinity in the eastern oyster. Crassostrea virginica Proc R Soc B. 2021;288:20203118.
Hadley NH, Dillon RT, Manzi JJ. Realized heritability of growth rate in the hard clam Mercenaria mercenaria. Aquaculture. 1991;93:109–19.
Camara MD, Allen SK, Carnegie RB, Reece KS. Out-crossing among commercial strains of the northern quahog, Mercenaria mercenaria: survival, growth and implications for selective breeding. J Shellfish Res. 2006;25:403–16.
Robledo D, Matika O, Hamilton A, Houston RD. Genome-wide association and genomic selection for resistance to amoebic gill disease in Atlantic salmon. G3 Genes|Genomes|Genetics. 2018;8:1195–203.
Nascimento-Schulze JC, Bean TP, Houston RD, Santos EM, Sanders MB, Lewis C, et al. Optimizing hatchery practices for genetic improvement of marine bivalves. Rev Aquac. 2021;13:2289–304.
Vu SV, Gondro C, Nguyen NTH, Gilmour AR, Tearle R, Knibb W, et al. Prediction accuracies of genomic selection for nine commercially important traits in the Portuguese oyster (Crassostrea angulata) using DArT-Seq technology. Genes. 2021;12:210.
Wang Y, Sun G, Zeng Q, Chen Z, Hu X, Li H, et al. Predicting growth traits with genomic selection methods in Zhikong scallop (Chlamys farreri). Mar Biotechnol. 2018;20:769–79.
Yáñez JM, Barría A, López ME, Moen T, Garcia BF, Yoshida GM, et al. Genome-wide association and genomic selection in aquaculture. Rev Aquac. 2023;15:645–75.
Song H, Dong T, Yan X, Wang W, Tian Z, Sun A, et al. Genomic selection and its research progress in aquaculture breeding. Rev Aquac. 2023;15:274–91.
Gunderson KL, Steemers FJ, Lee G, Mendoza LG, Chee MS. A genome-wide scalable SNP genotyping assay using microarray technology. Nat Genet. 2005;37:549–54.
Nascimento-Schulze JC, Bean TP, Peñaloza C, Paris JR, Whiting JR, Simon A, et al. SNP discovery and genetic structure in blue mussel species using low coverage sequencing and a medium density 60 K SNP-array. Evol Appl. 2023;16:1044–60.
Guo X, Puritz JB, Wang Z, Proestou D, Allen S, Small J, et al. Development and evaluation of high-density SNP arrays for the eastern oyster Crassostrea virginica. Mar Biotechnol. 2023;25:174–91.
Qi H, Song K, Li C, Wang W, Li B, Li L, et al. Construction and evaluation of a high-density SNP array for the Pacific oyster (Crassostrea gigas). PLoS ONE. 2017;12:e0174007.
Gutierrez AP, Turner F, Gharbi K, Talbot R, Lowe NR, Peñaloza C, et al. Development of a medium density combined-species SNP array for Pacific and European oysters (Crassostrea gigas and Ostrea edulis). G3 (Bethesda). 2017;7:2209–18.
Wang Y, Wang A, Guo X. Development and characterization of polymorphic microsatellite markers for the northern quahog Mercenaria mercenaria (Linnaeus, 1758). J Shellfish Res. 2010;29:77–82.
Ropp AJ, Reece KS, Snyder RA, Song J, Biesack EE, McDowell JR. Fine-scale population structure of the northern hard clam ( Mercenaria mercenaria ) revealed by genome-wide SNP markers. Evol Appl. 2023;16:1422–37.
Song H, Guo X, Sun L, Wang Q, Han F, Wang H, et al. The hard clam genome reveals massive expansion and diversification of inhibitors of apoptosis in Bivalvia. BMC Biol. 2021;19:15.
Farhat S, Bonnivard E, Pales Espinosa E, Tanguy A, Boutet I, Guiglielmoni N, et al. Comparative analysis of the Mercenaria mercenaria genome provides insights into the diversity of transposable elements and immune molecules in bivalve mollusks. BMC Genomics. 2022;23:192.
Sambrook J, Fritsch EF, Maniatis T, Russell DW, Green MR. Molecular cloning: a laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–9.
Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–2.
Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27:2987–93.
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–8.
Garcia-Vedrenne AE, Groner M, Page-Karjian A, Siegmund G-F, Singhal S, Sziklay J, et al. Development of genomic resources for a thraustochytrid pathogen and investigation of temperature influences on gene expression. PLoS ONE. 2013;8:e74196.
Dahl SF, Thiel J, Allam B. Field performance and QPX disease progress in cultured and wild-type strains of Mercenaria mercenaria in New York waters. J Shellfish Res. 2010;29:83–90.
Knaus BJ, Grünwald NJ. vcfr : a package to manipulate and visualize variant call format data in R. Mol Ecol Resour. 2017;17:44–53.
Jombart T. adegenet : a R package for the multivariate analysis of genetic markers. Bioinformatics. 2008;24:1403–5.
Goudet J. hierfstat, a package for r to compute and test hierarchical F -statistics. Mol Ecol Notes. 2005;5:184–6.
Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32:268–74.
Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 2017;14:587–9.
Hoang DT, Chernomor O, von Haeseler A, Minh BQ, Vinh LS. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol Biol Evol. 2018;35:518–22.
Matange K, Tuck JM, Keung AJ. DNA stability: a central design consideration for DNA data storage systems. Nat Commun. 2021;12:1358.
Schaudien D, Baumgärtner W, Herden C. High preservation of DNA standards diluted in 50% glycerol. Diagn Mol Pathol. 2007;16:153–7.
Parakatselaki M-E, Ladoukakis ED. mtDNA heteroplasmy: origin, detection, significance, and evolutionary consequences. Life. 2021;11:633.
Geraci-Yee S, Allam B, Collier JL. A nested quantitative PCR assay for detection of the hard clam pathogen Mucochytrium quahogii (=QPX) in environmental samples. Front Mar Sci. 2022;9:988918.
Stein ED, White BP, Mazor RD, Miller PE, Pilgrim EM. Evaluating ethanol-based sample preservation to facilitate use of DNA barcoding in routine freshwater biomonitoring programs using benthic macroinvertebrates. PLoS ONE. 2013;8:e51273.
Röder B, Frühwirth K, Vogl C, Wagner M, Rossmanith P. Impact of long-term storage on stability of standard DNA for nucleic acid-based methods. J Clin Microbiol. 2010;48:4260–2.
Schrader C, Schielke A, Ellerbroek L, Johne R. PCR inhibitors - occurrence, properties and removal. J Appl Microbiol. 2012;113:1014–26.
Hargrove JS, Sturmer L, Scarpa J, Austin JD. Assessment of genetic diversity in wild and aquaculture stocks of Mercenaria mercenaria in Florida. J Shellfish Res. 2015;34:355–65.
Hofmann EE, Klinck JM, Kraeuter JN, Powell EN, Grizzle RE, Buckner SC, et al. Population dynamics model of the hard clam, Mercenaria mercenaria: development of the age- and length-frequency structure of the population. J Shellfish Res. 2006;25:417–44.
Garant D, Forde SE, Hendry AP. The multifarious effects of dispersal and gene flow on contemporary adaptation. Funct Ecol. 2007;21:434–43.
Lachance J, Tishkoff SA. SNP ascertainment bias in population genetic analyses: Why it is important, and how to correct it. BioEssays. 2013;35:780–6.
Geibel J, Reimer C, Weigend S, Weigend A, Pook T, Simianer H. How array design creates SNP ascertainment bias. PLoS ONE. 2021;16:e0245178.
Foighil DÓ, Hilbish TJ, Showman RM. Mitochondrial gene variation in Mercenaria clam sibling species reveals a relict secondary contact zone in the western Gulf of Mexico. Mar Biol. 1996;126:675–83.
Call DR. Challenges and opportunities for pathogen detection using DNA microarrays. Crit Rev Microbiol. 2005;31:91–9.
Acknowledgements
The authors are very thankful to the many people who helped secure and process biological samples. These include Md. Wasif Alam, Sabrina Geraci-Yee, Ellen Biesack, Ann Ropp, and Dale Parsons. Expert technical assistance was provided by Maria Krutkina and Kateryna Pantiukh of SciBear (genomic data analysis and SNP selection), ThermoFisher (array design), the Center for Aquaculture Technologies and Neogen (genotyping services).
Funding
This work was primarily supported by the National Oceanic and Atmospheric Administration under the National Sea Grant Advanced Aquaculture Collaborative Program via New York Sea Grant (NA18OAR4170096). Additional support was provided by the New York State Department of Environmental Conservation.
Author information
Authors and Affiliations
Contributions
D.G., X.G., E.P.E., K.R., J.M., H.Y., G.R., J.R., A.C., A.T. and B.A. designed the study and secured the funding. X.G., E.P.E., K.R., J.M., H.Y., G.R., J.R. and B.A. collected biological samples. and S.F. performed genomic analysis. D.G. and B.A. analyzed data and drafted the paper. All authors contributed to the editing of the manuscript and approved the final version of the paper.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Grouzdev, D., Farhat, S., Guo, X. et al. Development and validation of a 66K SNP array for the hard clam (Mercenaria mercenaria). BMC Genomics 25, 847 (2024). https://doi.org/10.1186/s12864-024-10756-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-024-10756-7