
Abstract
Background
Sea cucumbers exhibit a remarkable ability to regenerate damaged or lost tissues and organs, making them an outstanding model system for investigating processes and mechanisms of regeneration. They can also reproduce asexually by transverse fission, whereby the anterior and posterior bodies can regenerate independently. Despite the recent focus on intestinal regeneration, the molecular mechanisms underlying body wall regeneration in sea cucumbers still remain unclear.
Results
In this study, transverse fission was induced in the tropical sea cucumber, Holothuria leucospilota, through constrainment using rubber bands. Histological examination revealed the degradation and loosening of collagen fibers on day-3, followed by increased density but disorganization of the connective tissue on day-7 of regeneration. An Illumina transcriptome analysis was performed on the H. leucospilota at 0-, 3- and 7-days after artificially induced fission. The differential expression genes were classified and enriched by GO terms and KEGG database, respectively. An upregulation of genes associated with extracellular matrix remodeling was observed, while a downregulation of pluripotency factors Myc, Klf2 and Oct1 was detected, although Sox2 showed an upregulation in expression. In addition, this study also identified progressively declining expression of transcription factors in the Wnt, Hippo, TGF-β, and MAPK signaling pathways. Moreover, changes in genes related to development, stress response, apoptosis, and cytoskeleton formation were observed. The localization of the related genes was further confirmed through in situ hybridization.
Conclusion
The early regeneration of H. leucospilota body wall is associated with the degradation and subsequent reconstruction of the extracellular matrix. Pluripotency factors participate in the regenerative process. Multiple transcription factors involved in regulating cell proliferation were found to be gradually downregulated, indicating reduced cell proliferation. Moreover, genes related to development, stress response, apoptosis, and cell cytoskeleton formation were also involved in this process. Overall, this study provides new insights into the mechanisms of whole-body regeneration and uncover potential cross-species regenerative-related genes.
Similar content being viewed by others


Background
Regeneration, a widespread phenomenon in nature, refers to the precise process of reconstructing the injured or lost parts of an organism or its organs [1]. In terms of regenerative capabilities, invertebrates commonly surpass vertebrates [2]. Specially, certain invertebrates like sea cucumbers and sea stars utilize regeneration as a means of asexual reproduction [3, 4]. Nevertheless, despite extensive research, the mechanism behind regeneration remains incompletely comprehended and exhibits variations across diverse species. For example, flatworms possess the astonishing capability to regenerate their entire bodies from minute fragments [5]. Salamanders, on the other hand, can regenerate not only complete limbs and tails but also complicated organs like eyes, nerve and heart [6, 7]. Vertebrates like zebrafish exhibit the ability to regenerate fins following injuries [8], while human livers have demonstrated a regenerative capacity after sustaining damage [9]. Invertebrates such as echinoderms also showcase impressive regenerative prowess, with the ability to regenerate injured or lost organs, as seen in sea urchins [10], sea stars [11] and sea cucumbers [12].
Sea cucumbers, in particular, serve as an exceptional model organism for the study of regeneration. When faced with stressful situations, they have the remarkable ability to expel certain internal organs and subsequently regenerate the lost ones [12]. Recent studies regarding sea cucumber regeneration have primarily focused on the renewal process of diverse organs, including the intestinal tract [13, 14], respiratory tree [15], body wall [16], and tube foot [17]. The body wall tissue of a sea cucumber comprises an epithelial layer, connective tissue layer, muscle layer, and coelomic epithelial layer [16]. Despite extensive observation of sea cucumber body wall regeneration at the level of cellular histology [16], the molecular mechanisms underlying this regenerative process remain unclear.
The sea cucumber Holothuria glaberrima has been widely utilized as a model for investigating regeneration processes [13]. Cellular events involved in H. glaberrima intestinal regeneration encompass an increase in spherule-containing cells, remodeling of the extracellular matrix, formation of spindle-like structures, and robust cellular division, primarily occurring in the coelomic epithelium [16]. During the early stages of intestinal regeneration, there was a significant upregulation in the transcriptional activities of genes, as supported by a transcriptomic analysis [18]. Damage triggers a strong stress response, even during late stages of regeneration, including increased levels of reactive oxygen species (ROSs), activation of antioxidant enzymes, immune system components, and the involvement of extracellular matrix (ECM) remodeling and Wnt signaling [12]. For the pluripotency factors, also known as Yamanaka factors, SoxB1, Myc and Bmi-1 are expressed in the regeneration processes of H. glaberrima radial nerve cord and the digestive tube but lack a coordinated regulation way [19, 20]. Furthermore, differential expression of numerous genes involved in development, ECM formation, and cytoskeletal construction was observed during intestinal regeneration in the sea cucumber Apostichopus japonica [21].
The tropical sea cucumber Holothuria leucospilota, belong to phylum Echinodermata and class Holothuroidea, are primarily distributed in the Indo-Pacific region [22, 23]. H. leucospilota has both sexual and asexual reproduction methods, with transverse fission being the primary asexual one [24, 25]. Studies have shown that after transverse fission, H. leucospilota can regenerate the anterior body out of the posterior body and vice versa [26]. The artificial spawning and culture of H. leucospilota have been reported recently [27]. In addition, the extrinsic (death receptor-mediated) [28, 29] and the intrinsic (mitochondrial-mediated) [30, 31] mechanisms of apoptosis in H. leucospilota coelomocytes under pathogenic and environmental stresses have also been studied. Aim to investigate the molecular mechanism of sea cucumber early body wall regeneration, a transcriptome analysis was performed with the H. leucospilota at 0-, 3- and 7-days after artificially induced fission. The regenerated morphologies and histological characteristics were first clarified. In addition, the signaling pathway and differential expression genes (DEGs) related to body wall regeneration were identified. The involved genes were further determined by in situ hybridization (ISH). This study can provide new insights into the mechanisms of whole-body regeneration and uncover potential cross-species regenerative-related genes.
Results
Morphological and histological changes of the body wall during regeneration
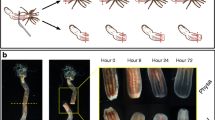
Artificially induced transverse fission was performed on the sea cucumber H. leucospilota (Fig. 1a). Within 48 h after skin strangulation, all individuals underwent splitting into two parts, and the internal organs were observed through the fracture (Fig. 1b). The wound completely healed within 7 days, indicating the completion of early body wall regeneration (Fig. 1c).

a Schematic diagram demonstrating the experimental procedure. The precise position for middle strangulation wound was confirmed in the sea cucumber in natural state, and subsequently, the wound was tightly secured using a leather band. b The wound at the head end of the sea cucumber after the occurrence of the strangulation injury was divided into two. c The growth of the anus at the head end of the sea cucumber was carried out 9 days following the strangulation (7 days post-wound formation)
Histological examination of the wound site on day-3 and day-7 of regeneration revealed no significant changes in the epithelial layer of the body wall (Fig. 2a-c). The connective tissue collagen fibers of normal sea cucumber are closely arranged, and a few cells are scattered in the connective tissue (Fig. 2d). However, degradation and loosening of collagen fibers were observed in the connective tissue layer on day-3, resulting in its laxity (Fig. 2e). The connective tissue appeared denser but exhibited disorganization compared to normal tissue on day-7 (Fig. 2f). Myocyte de-differentiation was evident in the vicinity of the injury site during both day-3 and day-7 of regeneration, with disorganized, attenuated, or complete absence of muscle layers or bundles observed at these two time points (Fig. 2g-i). Notably, the coelomic epithelial layer was absent within the inner part of the muscle layer during day-3 and day-7 of regeneration.

Histological sections of the body wall stained by HE. a-c The epithelial layer of the body wall at the wound site on day-0 (a), -3 (b), and -7 (c) of regeneration. d-f The connective tissue layer of the body wall at the wound site on day-0 (d), -3 (e), and -7 (f) of regeneration. g-i The muscle layer of the body wall on day-0 (g), -3 (h), and -7 (i) of regeneration. el: epithelial layer; ctl: connective tissue layer; ml: muscle layer; cel: coelomic epithelial layer
Sequence mapping and transcript assembly
After collecting the raw transcriptomic sequencing data, the clean reads were obtained by filtering them using the fastp software. The clean reads generated for day-0, day-3 and day-7 samples of body wall regeneration ranged from 41,794,852 to 58,307,640, the sequencing quality was excellent, with Q20 values exceeding 95% (Table 1). TopHat2 was employed to align the clean reads to the reference genome, resulting in unique mappings. The unique mappings for day-0, day-3 and day-7 samples ranged from 72.0% to 75.18% (Table 2). Finally, transcript assembly was performed, resulting in a total of 75,710 transcripts. The length distribution of transcripts exhibits considerable variation. The dataset consists of 16,535 transcripts within the 0–400 bp range, 18,053 transcripts within the 400–1000 bp range, and 13,326 transcripts within the 1000–1800 bp range. Notably, the largest subset comprises transcripts larger than 1800 bp, totaling 27,796 in count (Fig. 3a).

Transcriptome information and inter-sample correlation analysis. a Distribution profile of transcript lengths. The x-axis represents the range of transcript length, while the y-axis indicates the number of transcripts within each length range. b Histogram demonstrating the annotation of gene functions. The x-axis denotes the name of the database, while the y-axis represents the number of sequences annotated to each respective database. c Venn diagram depicting the annotation of gene functions. Circles of different colors signify the number of genes annotated to various databases, with the overlapping sections indicating genes simultaneously annotated in multiple libraries. d Correlation analysis for the gene expression of body wall tissue samples from day-0, -3, and -7 of regeneration. The values within the graph represent correlation coefficients between the two samples, where a higher value indicates a greater similarity
Functional annotation of genes
To annotate the genes, BLASTx was employed using various protein databases. The proteins with the highest sequence similarity were selected for annotation. Out of the total number of genes, 31,343 (63.08%) were successfully annotated. Specifically, among the annotated genes, 25,594, 13,343, 16,627, 18,646, 18,281 and 20,319 were annotated with the NR, KEGG, SWISS-PROT, GO, COG and PFAM databases, respectively (Fig. 3b). The Venn diagram illustrates the number of expressed genes that were annotated in different combinations of databases. A total of 9,200 genes were simultaneously annotated in multiple libraries, while 1,483 and 501 genes were exclusively annotated in the NR and PFAM databases, respectively (Fig. 3c).
Differentially expressed genes (DEGs)
By correlation analysis, a high correlation coefficient indicated a strong resemblance in transcript expression between biological replicate samples (Fig. 3d). Differentially expressed genes (DEGs) were identified as adjusted P < 0.05 and |log2FC |≥ 1 (FC means fold change here). Volcano plots were employed to analyze the distinctively expressed genes between the day-3 (Fig. 4a) and day-7 (Fig. 4b) groups compared to the day-0 group. In the day-3 group, a total of 2,238 significantly differentially expressed genes were observed, with 1,079 genes being up-regulated and 1,159 genes being down-regulated (Fig. 4a). Similarly, in the day-7 group, 2,717 significantly differentially expressed genes were identified, with 1,293 genes being up-regulated and 1,424 genes being down-regulated (Fig. 4b). Furthermore, a total of 1,007 genes were found to be co-expressed in both the day-3 and day-7 groups, consisting of 453 up-regulated genes and 554 down-regulated genes (Fig. 4c).

a & b Differentially expressed genes (DEGs) in the day-3 (a) and day-7 (b) compared to the day-0 regenerated groups. Volcano plots illustrating DEGs in the day-3 and day-7 groups, respectively. The x-axis represents the fold change in gene expression, while the y-axis represents the statistical test value of the gene expression variation. The values on both axes are logarithmically normalized. Each point on the plot corresponds to a specific gene; red points indicate significantly up-regulated genes, blue points indicate significantly down-regulated genes, and gray points represent genes with no significant difference. c Venn diagram presenting the number of commonly and significantly differentially up-regulated and down-regulated genes across different regeneration time points
Functional classification of DEGs
The Gene Ontology (GO) terms derived from DEGs were exhibited in Fig. 5a. In the upregulated genes, significant enrichment was observed on day-3 for terms related to the extracellular region, collagen trimer, peptidase and endopeptidase activities, calcium ion binding, lysozyme activity and immune-related functions. On the other hand, terms associated with ribosomal subunit, ribosome, focal adhesion, cell-substrate junction, protein synthesis and metabolism significantly enriched on day-7. For downregulated genes, enrichment was observed on both days for terms related to motile cilium, cell projection, plasma membrane bounded cell projection, cilium, movement of cell or subcellular components, microtubule-based movement and metallopeptidase activity.

a GO classification results of DEGs in the day-3 and day-7 groups in comparison to the day-0 group are summarized into three main GO categories, namely cellular components (CC), molecular functions (MF), and biological processes (BP). The Y-axis represents the GO ontology, while the X-axis indicates the significance level of enrichment. b KEGG classification of DEGs in the day-3 and day-7 groups in comparison to the day-0 group. The y-axis represents the pathway name, while the x-axis indicates the Rich factor. A larger Rich factor suggests a more significant enrichment. The size of the bubbles corresponds to the number of genes in the pathway, and the color of the bubbles indicates different P value ranges. Only the top 15 enrichment results are displayed
To further illustrate the pathways linked to body wall regeneration, the DEGs enrichment were further analyzed with the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (Fig. 5b). On day-3, upregulated genes demonstrated significant enrichment in the pathways of ECM-receptor interaction, Antigen processing and presentation and Protein digestion and absorption. Conversely, on day-7, the enriched pathways among the upregulated genes were Signaling pathways regulating pluripotency of stem cells, MAPK signaling pathway-fly and Retinol metabolism. Notably, the Toll-like receptor signaling pathway exhibited significant enrichment on both days. Among the downregulated genes, there was enrichment of both the TGF beta signaling pathway and Protein digestion and absorption pathways on both days. Specifically, on day-3, the Proteasome, RNA degradation and Axon regeneration pathways displayed enrichment, while on day-7, the ECM-receptor interaction and Focal adhesion pathways were enriched.
Expression analysis of genes related to regeneration
Based on GO category and KEGG enrichment analysis, the genes crucial for body wall regeneration can be clarified into four distinctive groups: ECM-associated genes (Fig. 6a), pluripotency factors (Fig. 6b), signaling pathways (Fig. 6c) and other genes related to the process of regeneration (Fig. 6d).

Heat map illustrating the change in expression trends of DEGs related to extracellular matrix reconstruction (a), pluripotency factors (b), signaling pathway (c) and other regenerated factors (d) during genes the non-regeneration phase (D0) and regeneration at day-3 (D3) and day-7 (D7). The genes related to extracellular matrix reconstruction include ECM component and ECM-modifying protein; the pluripotency factors include Myc, Klf2, Sox2 and Oct1; the genes related to signaling pathway include Wnt, Hippo, TGF-β and MAPK; The genes related to other regenerated factors include development, stress response, apoptosis and cytoskeleton
Among the ECM-associated genes, several were found to be upregulated during one or multiple stages of regeneration. These genes include COLs, GPC1, FBN1, PRS2, CATL, ADAMTS7, 9 and 20, FBLN1 and TENR (Fig. 6a). In contrast, the expression levels of FBN2 and 3, FBLN2, LAMs, TSPs, PRS3, CATD, ADAMTS3 and 6, MMPs and FBP1 were found to be downregulated during the process of regeneration (Fig. 6a).
Four Pluripotency factors were identified, namely Myc, Klf2, Sox2 and PO2F1 (Oct1), as shown in Fig. 6b. Among these factors, Sox2 exhibited upregulation on day 7 of the regeneration, whereas the expression levels of Myc, Klf2 and Oct1 were observed to be downregulated.
As shown in Fig. 6c, the analysis revealed differential expression patterns of various transcription factors within the Wnt, Hippo, TGF-β and MAPK signaling pathways. While Wnt5A and CSK exhibited upregulation in the Wnt signaling pathway, the transcription factors Wnt1, FZD2, CTNB, BAMBI, TF7L2, LRPs and PPARA showed downregulation progressively. Similarly, in the Hippo signaling pathway, YAP1A was downregulated, whereas BMP2B, SMAD6 and BAMBI in the TGF-β signaling pathway, as well as RRAS2 and MAPK1 in the MAPK signaling pathway, were found to be downregulated during the process of regeneration.
Other genes associated with the regeneration process are presented in Fig. 6d. Among these differentially expressed genes, we found the greatest difference in up-regulation of SAA and down-regulation of GELS1 during regeneration. The expression of the development-related gene Hox-B1 was found to be upregulated, while immune-related genes such as HSP70 and SAA and several antioxidant enzymes including SODC, CATA and GST4 showed upregulation during regeneration. Conversely, the expression levels of apoptosis-related genes CASP7 and CASP8 were upregulated, whereas BIRC5 exhibited downregulation. Furthermore, cytoskeleton-related genes TBA1A and ACT displayed upregulation, while GELS1 and MYO3B were observed to be downregulated.
Validation of regeneration-associated genes by ISH
ISH was conducted to investigate the spatial and temporal distributions of regeneration-associated genes Klf2, Sox2, MMP14 and TGFR1 during early body wall regeneration of H. leucospilota. The results revealed that Klf2 displayed extensive expression in the connective tissues of the day-0 sample, but limited positive signals were observed in the day-3 and day-7 samples (Fig. 7a-c). MMP14 exhibited widespread expression in the connective tissues of the day-0 sample, but minimal expression in regenerating connective tissue (Fig. 7d-f), which aligns with the down-regulation trend of Klf2 and MMP14 indicated by the RNA sequencing data on day-3 and day-7 of regeneration. Sox2 gene expression was notably high in the coelomic epithelial layer and the positive signal for Sox2 intensified during the early regeneration of the somatic wall (Fig. 7g-i), supporting the RNA sequencing data indicating an increase of Sox2 expression during early regeneration. Moreover, no positive signal for TGFR1 was observed in the epithelial layer of the day-0 sample, but there was an increase in positive signal on day-3 followed by a decrease on day-7 (Fig. 7j-l), supporting the RNA sequencing data that demonstrated an up-regulation of TGFR1 on day-3 and a return to normal levels on day-7.

In situ hybridization (ISH) illustrating the spatial expression patterns of Klf2 (a-c), Sox2 (d-f), MMP14 (g-i) and TGFR1 (j-l) during the early regeneration phase of the body wall. The three rows of horizontal images represent tissue sections at 0 (a, d, j, g), 3 (b, e, h, k) and 7 (c, f, i, l) days of body wall regeneration, respectively. Insets provide a high magnification view of the boxed areas in the corresponding main micrographs. el: epithelial layer; ctl: connective tissue layer; ml: muscle layer
Discussion
Asexual reproduction has been documented in 16 holothurian species, occurring through transverse fission (architomy) and fragmentation in adult sea cucumbers [4]. Previous studies have demonstrated the ability of sea cucumbers Cladolabes schmeltzii, Colochirus robustus, Pseudocolochirus violaceus and Holothuria scabra to regenerate their anterior and posterior portions following transverse surgery [26]. Natural asexual reproduction through fission has been observed as a common phenomenon in H. leucospilota [32]. In this study, we present a novel method for artificially inducing transverse fission in H. leucospilota by gradually constraining its middle section using rubber bands (Fig. 1a). Based on this approach, Histological and transcriptomic analyses were conducted to explore the underlying mechanisms of whole-body regeneration in sea cucumbers.
The body wall of sea cucumbers primarily consists of connective tissue, which enables fission by transforming the extracellular matrix [33]. This specific connective tissue, referred to as mutable collagenous tissue (MCT) [34, 35] or catch connective tissue [36], possesses the unique capability to modify its mechanical properties [37]. The stiffness of MCT is determined by the interaction of three protein groups: matrix metalloproteinases (MMPs), tissue inhibitors of metalloproteinases (TIMPs), and cross-link complexes that interconnect collagen fibrils [38]. An increase in MMPs concentration or activity in the connective tissue leads to the degradation of cross-link complexes. This breakdown enables collagen fibrils to slide along one another, leading to the compliant pattern of MCT [12]. Consequently, local changes in the properties of the body wall’s connective tissue facilitate the sea cucumbers divided into two parts.
Sea cucumbers possess the remarkable capability of undergoing visceral evisceration and regenerating lost organs as a response to environmental deterioration [39] or various human impacts [40]. After evisceration, rapid wound healing and activation of the immune system occur [12]. The regenerated tissue layers of the intestine originate from the relevant components of the mesentery, esophageal stump, and cloacal stump [13]. Upon division, the anterior fragment contains the aquapharyngeal bulb, gonad, Polian vesicles and the front half of the intestinal tube, while the posterior fragment retains the rear gut portion, cloaca and respiratory trees [12]. Both fragments undergo wound healing, followed by the regeneration of missing structures, such as the aquapharyngeal bulb in the posterior fragment and the cloaca in the anterior fragment [4]. Additionally, the remaining intestine extends either forwards or backwards until a complete intestinal tube is formed.
The present transcriptomic analysis has revealed the differential expression of ECM-related genes during the body wall regeneration of H. leucospilota. Upregulated expression was observed for ECM-related genes such as COLs, GPC1, FBN1, PRS2, CATL, ADAMTS7, 9 and 20, FBLN1 and TENR during regeneration, while downregulated expression was observed for FBN2 and 3, FBLN2, LAMs, TSPs, PRS3, CATD, ADAMTS3 and 6, MMPs, and FBP1 (Fig. 6a). ECM is an essential component of connective tissues, providing physical support and regulates cellular processes [41]. Previous studies have demonstrated the crucial role of ECM reconstruction in the regeneration of the intestine [21], central nervous system [20], and body wall [16] of sea cucumbers. Matrix metalloproteases (MMPs) are enzymes that play integral functions in the degradation of specific ECM components and promote tissue regeneration by facilitating cell proliferation, migration, differentiation and apoptosis [42]. Additionally, tissue inhibitors of metalloproteinases (TIMPs) regulate the degradation of ECM components and tissue remodeling by interacting with MMPs [43]. In a transcriptomic analysis of intestine regeneration in A. japonicas, upregulation of all MMPs except MMP14 was observed, in contrast to our findings during body wall regeneration [21]. During the body wall regeneration process, the downregulation of multiple MMPs and TIMPs suggests a potential reduction in ECM degradation. Furthermore, the differential expression of genes involved in ECM components and ECM-modifying proteins at day-3 and day-7 highlights the significance of ECM reconstruction. These genes likely contribute to the remodeling of connective tissues and the restoration of tissue architecture following injury.
The activation of regeneration-related signaling pathways at the transcript level was observed during the early stage (day-0 and day-3). By day-7 of regeneration, downregulation of Wnt1, Wnt5A, FZD2, LRPs and CTNB were appeared in the upstream pathway, resulted in the downregulation of TF7L2, Myc, and PPARA in the downstream pathway, subsequently impacting cell proliferation (Fig. 6c). The Wnt/β-catenin signaling pathway is primarily responsible for regulating cell proliferation [44]. However, our findings differ from previous reports regarding the expression levels of this pathway in intestine regeneration [21]. In addition, YAP1A, a transcription factor in the Hippo pathway, also exhibited downregulation. The overexpression of YAP in the nucleus promotes cell proliferation through its interaction with β-catenin [45]. In our analysis, the MAPK signaling pathways were found to be enriched with upregulated genes, while the TGF-β signaling pathway was enriched with downregulated genes. The TGF-β pathway regulates various cellular activities, including cell proliferation, apoptosis, and differentiation [46]. The downregulation of BMP2B, SMAD6 and BAMBI, as well as the upregulation of TGFR1, was observed, suggesting their involvement in inhibiting cell proliferation during early body wall regeneration. Furthermore, the downregulation of transcription factors RRAS2 and MAPK1, components of the MAPK family, implies their potential role in regulating cell proliferation and differentiation during regeneration stages [47].
Differential expression of the pluripotency factors Myc, Klf2, Sox2 and Oct1 was observed in the transcriptome of H. leucospilota. Specifically, Sox2 showed upregulation, whereas the other genes exhibited downregulation. Results of ISH revealed a decrease in the Klf2-positive signal in the regenerating body wall connective tissue (Fig. 7a-c), while an increase in the Sox2-positive signal was observed in the regenerating body wall coelomic cell layer (Fig. 7d-f). Pluripotent stem cells can be induced from mouse embryonic or adult fibroblasts by introducing Oct3/4, Sox2, c-Myc and Klf4 [48]. Studies have reported the involvement of Myc, SoxB1, and Klf13 genes in intestinal regeneration in sea cucumbers H. glaberrima and A. japonicus [19, 49]. Myc has exhibited differential expression in the central nervous system regeneration of H. glaberrima [20]. Differential expression of Oct4, Sox2, and c-Myc has been reported in the regeneration process of the earthworm Eisenia foetida [50].
The development-related gene Hox-B1 displayed upregulated expression in the early phases of body wall regeneration in H. leucospilota (Fig. 6d). Following injury, several genes associated with stress response were activated, including HSP70, SAA, SODC, CATA and GST4. Additionally, apoptosis-related genes such as CASP7, CASP8, and BIRC5 exhibited differential expression (Fig. 6d). In addition to regeneration, stress-related antioxidant [51, 52] and apoptosis [29,30,31, 53] also play crucial roles in responses to the pathogen infection in sea cucumbers. Furthermore, muscle dedifferentiation is observed during the regeneration of visceral organs in echinoderms, including H. leucospilota [54]. Furthermore, Histological analysis revealed muscle cell dedifferentiation on day-3 and day-7 (Fig. 2). Transcriptome analysis revealed alterations in gene expression linked to TBA1A, ACT, GELS1 and MYO3B during the process of body wall regeneration (Fig. 6d). These findings suggest the involvement of cell cytoskeleton formation, which is consistent with previous studies on intestinal regeneration in A. japonicus [55].
Conclusion
An Illumina transcriptome analysis was conducted on H. leucospilota at 0-, 3-, and 7-days post-induced fission to examine gene expression patterns during early regeneration. The functional annotation of DEGs not only validates previous reported cellular events [16] but also opens up potential new avenues for future research. The genes implicated in early body wall regeneration can be categorized into four groups: genes associated with extracellular matrix reconstruction, pluripotency factors, signaling pathways, and other genes involved in regeneration. This study enhances our understanding of the molecular mechanisms involved in regeneration in echinoderms, and may also provide insights into the regenerative mechanisms of higher vertebrates.
Methods
Animals and artificially induced transverse fission
Healthy H. leucospilota were collected from Daya Bay, Shenzhen, Guangdong Province, China. Prior to the experiment, they were acclimated in glass tanks with aerated seawater for a week. Nine sea cucumbers were randomly selected and divided into three groups, with three individuals in each group. By gradually constraining the middle section of the sea cucumbers using rubber bands, they were eventually subjected to induce transverse fission (Fig. 1a). Morphological changes in the regenerating regions of the sea cucumbers after fracture were observed through a stereomicroscope (Fig. 1b). The body wall tissues from the 0-day, 3-day, and 7-day groups after transverse fission, were promptly frozen using liquid nitrogen following dissection.
RNA extraction and qualification
Total RNA was extracted from the body wall tissue samples of H. leucospilota using TRIzol Reagent (Invitrogen, USA). The concentration and purity of the extracted RNA were determined using Nanodrop2000 (Thermo Fisher Scientific Inc., USA), while its integrity was assessed through 1% agarose gel electrophoresis. The RNA Integrity Number (RIN) values were determined using Agilent2100 (Agilent, USA). For the construction of a single library, a minimum of ≥ 1ug of total RNA was required, with a concentration of ≥ 35 ng/μL, OD260/280 ≥ 1.8, and OD260/230 ≥ 1.0.
Library construction and sequencing
For the sequencing experiment, the Illumina TruSeqTM RNA sample prep Kit method (Illumina Inc., USA) was utilized for library construction, and sequencing was performed using the Illumina Novaseq 6000 sequencing platform (Illumina Inc.). In brief, mRNA was isolated from total RNA using magnetic beads with Oligo (dT). The mRNA was randomly fragmented, and the resulting small fragments of approximately 300 bp were isolated through magnetic bead screening. Subsequently, the double-stranded cDNA was synthesized using mRNA as a template, with the addition of six random base primers, and ligated with an adapter. The sequencing process was then accomplished on the Illumina platform (Illumina Inc.).
Sequencing data quality control and sequence mapping
To obtain high-quality sequencing data (clean data), the software fastp (https://github.com/OpenGene/fastp) was used to filter out sequencing connector sequences, low-quality read segments, sequences with a high uncertain base information rate (N), and sequences with a length that was too short in the original sequencing data. Subsequently, the clean data (reads) were compared with the H. leucospilota reference genome [22] using the software TopHat2 (http://tophat.cbcb.umd.edu/) to obtain mapped data (reads) for further analysis.
Transcript assembly and functional annotation
The assembled transcripts were compared with known transcripts to obtain new transcripts without annotation information. Then, functional annotation was carried out on these new transcripts. In order to obtain comprehensive gene or transcript annotation information, all the genes and transcripts obtained from transcriptome assembly were compared with various databases. In this case, the databases included NR, Swiss-Prot, Pfam, Clusters of Orthologous Groups of proteins (COG), Gene Ontology (GO), and Kyoto Encyclopedia of Genes and Genomes (KEGG).
Analysis of differentially expressed genes
To quantitatively analyze the overall expression level of genes, RSEM was utilized and obtain the quantitative index Transcripts Per Million reads (TPM). DESeq2 was used to analyze the differential expression of genes between two groups (n = 3). Genes were considered differentially expressed genes (DEGs) with significance levels of adjusted P < 0.05 and |log2FC|≥ 1.
GO and KEGG enrichment analysis of DEGs
The Gene Ontology (GO) enrichment of DEGs was performed using the Goattools Python package (https://github.com/tanghaibao/GOatools). Upon mapping gene IDs to GO terms, P < 0.05 were used to determine statistically significant enrichment. Similarly, For the Kyoto Encyclopedia of Genes and Genomes (KEGG)[56, 57] enrichment analysis of DEGs, the Gene IDs were mapped to KEGG pathways, and statistical significance was determined by P < 0.05.
Tissue sectioning and hematoxylin/eosin (HE) staining
The body wall tissues fixed with 4% paraformaldehyde were subjected to a series of steps, including dehydration, clarification, wax dipping and embedding. Subsequently, the wax blocks were sliced at a thickness of 3 μm using a paraffin slicer. The tissue slices were placed on a water bath at 40℃ to flatten, and then delicately transferred onto glass slides. These slides, containing the tissue sections, were then baked in a 60℃ oven. The dewaxed and washed paraffin sections were subsequently stained with hematoxylin and eosin, followed by dehydration and sealing. Finally, the sections were examined under a microscope.
In situ hybridization
To validate the reliability of the transcriptomic data, ISH was performed to conduct cellular localization and relative expression of selected genes. Specifically, the PCR DIG Probe Synthesis Kit (Roche, Switzerland) was used to synthesize DNA probes labeled with digoxigenin (DIG). For ISH, body wall tissue samples from sea cucumber were first collected and overnight fixed in a 4% paraformaldehyde fixation solution (Sangon Biotech, China). After fixation, the tissues were dehydrated in gradient ethanol and embedded in paraffin. The wax block was cut into sections of 3 μm in thickness. Subsequently, the sections were immersed in xylene and gradually transferred to ethanol for the purpose of dewaxing. Once dewaxed, the sections were incubated in Proteinase K (20 μg/mL) for 30 min. Endogenous peroxidase was blocked, followed by dropwise addition of prehybridization solution. The sections were then subjected to hybridization by dropwise addition of the probe-containing hybridization solution. Subsequently, the sections were washed sequentially with 2 × SSC, 1 × SSC, and 0.5 × SSC solutions. After that, the sections were undergoing a series of sequential treatments, including blocking with BSA, addition of mouse anti-digoxigenin antibody labeled with horseradish peroxidase (anti-DIG-HRP), and introduction of the 3,3’-Diaminobenzidine (DAB) chromogenic solution. After completion of the DAB color development, the nuclei were stained with Harris Hematoxylin. The sections were dehydrated in a gradient ethanol series and xylene, and finally sealed with neutral resin. The sections were observed under a microscope and photographed for further analysis.
Availability of data and materials
The datasets presented in this study can be found in NCBI with accession number: PRJNA1007557.
Abbreviations
- CO1A2:
-
Collagen alpha-2(I) chain
- CO4A1:
-
Collagen alpha-1(IV) chain
- CO6A3:
-
Collagen alpha-3 (VI) chain
- GPC1:
-
Glypican-1
- FBN1:
-
Fibrillin-1
- FBN2:
-
Fibrillin-2
- FBN3:
-
Fibrillin-3
- FBLN1:
-
Fibulin-1
- FBLN2:
-
Fibulin-2
- LAMB1:
-
Laminin subunit beta-1
- LAMA3:
-
Laminin subunit alpha-3
- TSP1:
-
Thrombospondin-1
- TSP4:
-
Thrombospondin-4
- PRS2:
-
Serine protease 23
- PRS3:
-
Serine protease 33
- CATD:
-
Cathepsin D
- CATL:
-
Cathepsin L
- ADAMTS:
-
A disintegrin and metalloproteinase with thrombospondin motifs
- MMP:
-
Matrix metalloproteinase
- TIMP:
-
Tissue inhibitors of metalloproteinase
- FBP1:
-
Fibropellin-1
- TENR:
-
Tenascin-R
- KLF2:
-
Krüeppel-like factor 2
- KLF13:
-
Krüeppel-like factor 13
- SOX2:
-
Transcription factor SOX-2
- SOXB1:
-
Transcription factor SOX-B1
- OCT1 (PO2F1):
-
POU domain, class 2, transcription factor 1;OCT4 (PO5F1): POU domain, class 5, transcription factor 1
- WNT1:
-
Protein Wnt-1
- WNT5A:
-
Protein Wnt-5a
- FZD2:
-
Frizzled2
- CTNB:
-
Catenin beta
- BAMBI:
-
BMP and activin membrane-bound inhibitor homolog
- CSK:
-
Tyrosine-protein kinase CSK
- TF7L2:
-
Transcription factor 7-like 2
- LRP:
-
Low-density lipoprotein receptor-related protein
- PPARA:
-
Peroxisome proliferator-activated receptor alpha
- YAP1A:
-
Yes association protein 1-A
- TGFR1:
-
TGF-beta receptor type-1
- BMP2B:
-
Bone morphogenetic protein 2-B
- SMAD6:
-
Mothers against decapentaplegic homolog 6
- RRAS2:
-
Ras-related protein R-Ras2
- MAPK1:
-
Mitogen-activated protein kinase 1
- HXB1:
-
Homeobox protein Hox-B1
- Hox:
-
Homeobox protein
- BIRC5:
-
Baculoviral IAP repeat-containing protein 5
- SAA:
-
Serum amyloid A protein
- HSP70:
-
Heat shock 70 kDa protein
- SODC:
-
Superoxide dismutase [Cu–Zn]
- CATA:
-
Catalase
- GST4:
-
Glutathione S-transferase 4
- CASP7:
-
Caspase-7
- CASP8:
-
Caspase-8
- TBA1A:
-
Tubulin alpha-1A chain
- ACT:
-
Actin
- GELS1:
-
Gelsolin-like protein 1
- MYO3B:
-
Myosin-IIIb
- ECM:
-
Extracellular matrix
- BMPs:
-
Bone morphogenetic proteins
- BMI-1:
-
Polycomb complex protein BMI-1
- DEGs:
-
Differential expression genes
- ISH:
-
In situ Hybridization
- MCT:
-
Mutable collagenous tissue
- RIN:
-
RNA Integrity Number
- NR:
-
Non-Redundant Protein Sequence Database
- GO:
-
Gene Ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- COG:
-
Clusters of Orthologous Groups of proteins
- RSEM:
-
RNA-seq by expectation maximization
- DAB:
-
Diaminobenzidine
References
Carlson BM. An Introduction to Regeneration. In: Principles of Regenerative Biology. Edited by Carlson BM. Burlington: Academic Press; 2007. p. 1–29.
Goss RJ. Introduction. In: Principles of Regeneration. Edited by Goss RJ: Academic Press; 1969. p. 1–7.
Shibata D, Hirano Y, Komatsu M. Life cycle of the multiarmed sea star Coscinasterias acutispina (Stimpson, 1862) in laboratory culture: sexual and asexual reproductive pathways. Zoolog Sci. 2011;28(5):313–7.
Dolmatov IY. Asexual reproduction in holothurians. ScientificWorldJournal. 2014;2014: 527234.
Agata K, Tanaka T, Kobayashi C, Kato K, Saitoh Y. Intercalary regeneration in planarians. Dev Dyn. 2003;226(2):308–16.
Joven A, Elewa A, Simon A. Model systems for regeneration: salamanders. Development. 2019;146(14):dev167700.
Dwaraka VB, Voss SR. Towards comparative analyses of salamander limb regeneration. J Exp Zool B Mol Dev Evol. 2021;336(2):129–44.
Rabinowitz JS, Robitaille AM, Wang Y, Ray CA, Thummel R, Gu H, Djukovic D, Raftery D, Berndt JD, Moon RT. Transcriptomic, proteomic, and metabolomic landscape of positional memory in the caudal fin of zebrafish. Proc Natl Acad Sci U S A. 2017;114(5):E717–26.
Michalopoulos GK, DeFrances MC. Liver regeneration. Science. 1997; 276(5309):60–6.
Dubois P, Ameye L. Regeneration of spines and pedicellariae in echinoderms: a review. Microsc Res Tech. 2001;55(6):427–37.
Byrne M, Mazzone F, Elphick MR, Thorndyke MC, Cisternas P. Expression of the neuropeptide SALMFamide-1 during regeneration of the seastar radial nerve cord following arm autotomy. Proc Biol Sci. 1901;2019(286):20182701.
Dolmatov IY. Molecular Aspects of Regeneration Mechanisms in Holothurians. Genes. 2021;12(2):250.
Quispe-Parra D, Valentin G, Garcia-Arraras JE: A roadmap for intestinal regeneration. Int J Dev Biol 2021, 65(4–5–6):427–437.
Garcia-Arraras JE, Bello SA, Malavez S. The mesentery as the epicenter for intestinal regeneration. Semin Cell Dev Biol. 2019;92:45–54.
Eisapour M, Salamat N, Salari MA, Bahabadi MN, Salati AP. Post-autotomy regeneration of respiratory tree in sea cucumber Holothuria parva. J Exp Zool B Mol Dev Evol. 2022;338(3):155–69.
San Miguel-Ruiz JE, Garcia-Arraras JE. Common cellular events occur during wound healing and organ regeneration in the sea cucumber Holothuria glaberrima. BMC Dev Biol. 2007;7:115.
Diaz-Balzac CA, Abreu-Arbelo JE, Garcia-Arraras JE. Neuroanatomy of the tube feet and tentacles in Holothuria glaberrima (Holothuroidea, Echinodermata). Zoomorphology. 2010;129(1):33–43.
Quispe-Parra DJ, Medina-Feliciano JG, Cruz-Gonzalez S, Ortiz-Zuazaga H, Garcia-Arraras JE. Transcriptomic analysis of early stages of intestinal regeneration in Holothuria glaberrima. Sci Rep. 2021;11(1):346.
Mashanov VS, Zueva OR, Garcia-Arraras JE. Expression of pluripotency factors in echinoderm regeneration. Cell Tissue Res. 2015;359(2):521–36.
Mashanov VS, Zueva OR, García-Arrarás JE. Transcriptomic changes during regeneration of the central nervous system in an echinoderm. BMC Genomics. 2014;15:357.
Sun L, Yang H, Chen M, Ma D, Lin C: RNA-Seq reveals dynamic changes of gene expression in key stages of intestine regeneration in the sea cucumber Apostichopus japonicus. [corrected]. PLoS One 2013;8(8):e69441.
Chen T, Ren C, Wong NK, Yan A, Sun C, Fan D, Luo P, Jiang X, Zhang L, Ruan Y, et al. The Holothuria leucospilota genome elucidates sacrificial organ expulsion and bioadhesive trap enriched with amyloid-patterned proteins. Proc Natl Acad Sci U S A. 2023;120(16): e2213512120.
Wu XF, Chen T, Huo D, Yu ZH, Ruan Y, Cheng CH, Jiang X, Ren CH. Transcriptomic analysis of sea cucumber (Holothuria leucospilota) coelomocytes revealed the echinoderm cytokine response during immune challenge. BMC genomics. 2020;21(1):306.
Purwati P. Fissiparity in Holothuria leucospilota from tropical Darwin waters, northern Australia. SPC Beche-de-Mer Information Bulletin. 2004;20:26–33.
Purwati P, Luong-Van JT. Sexual reproduction in a fissiparous holothurian species, Holothuria leucospilota Clark 1920 (Echinodermata: Holothuroidea). SPC Beche-de-Mer Information Bulletin. 2003;18:33–8.
Dolmatov IY, Khang NA, Kamenev YO. Asexual reproduction, evisceration, and regeneration in holothurians (Holothuroidea) from Nha Trang Bay of the South China Sea. Russ J Mar Biol. 2012;38(3):243–52.
Huang W, Huo D, Yu Z, Ren C, Jiang X, Luo P, Chen T, Hu C. Spawning, larval development and juvenile growth of the tropical sea cucumber Holothuria leucospilota. Aquaculture. 2018;488:22–9.
Li HP, Chen T, Sun HY, Wu XF, Jiang X, Ren CH. The first cloned echinoderm tumor necrosis factor receptor from Holothuria leucospilota: Molecular characterization and functional analysis. Fish Shellfish Immun. 2019;93:542–50.
Yan AF, Ren CH, Chen T, Jiang X, Sun HY, Huo D, Hu CQ, Wen J. The first tropical sea cucumber caspase-8 from Holothuria leucospilota: Molecular characterization, involvement of apoptosis and inducible expression by immune challenge. Fish Shellfish Immun. 2018;72:124–31.
Li XM, Chen T, Wu XF, Jiang X, Luo P, Zixuan E, Hu CQ, Ren CH. Apoptosis-Inducing Factor 2 (AIF-2) Mediates a Caspase-Independent Apoptotic Pathway in the Tropical Sea Cucumber (Holothuria leucospilota). Int J Mol Sci. 2022;23(6):3008.
Li XM, Chen T, Wu XF, Li ZB, Zhang X, Jiang X, Luo P, Hu CQ, Wong NK, Ren CH. Evolutionarily Ancient Caspase-9 Sensitizes Immune Effector Coelomocytes to Cadmium-Induced Cell Death in the Sea Cucumber, Holothuria leucospilota. Front Immunol. 2022;13:927880.
Conand C, Morel CM, Mussard R. A new study of asexual reproduction in holothurians: Fission in Holothuria leucospilota populations on Reunion Island in the Indian Ocean. SPC Beche-de-Mer Informa tion Bulletin. 1997;9:5–11
Liu Y-X, Zhou D-Y, Ma D-D, Liu Y-F, Li D-M, Dong X-P, Tan M-Q, Du M, Zhu B-W. Changes in collagenous tissue microstructures and distributions of cathepsin L in body wall of autolytic sea cucumber (Stichopus japonicus). Food Chem. 2016;212:341–8.
Wilkie IC. Variable tensility in echinoderm collagenous tissues: A review. Mar Behav Physiol. 1984;11(1):1–34.
Bonneel M, Hennebert E, Aranko AS, Hwang DS, Lefevre M, Pommier V, Wattiez R, Delroisse J, Flammang P. Molecular mechanisms mediating stiffening in the mechanically adaptable connective tissues of sea cucumbers. Matrix Biol. 2022;108:39–54.
Motokawa T. Connective tissue catch in echinoderms. Biol Rev. 2008;59:255–70.
Wilkie IC. Mutable collagenous tissue: overview and biotechnological perspective. Prog Mol Subcell Biol. 2005;39:221–50.
Ribeiro AR, Barbaglio A, Oliveira MJ, Ribeiro CC, Wilkie IC, Candia Carnevali MD, Barbosa MA. Matrix metalloproteinases in a sea urchin ligament with adaptable mechanical properties. PLoS ONE. 2012;7(11): e49016.
Byrne M. The morphology of autotomy structures in the sea cucumber Eupentacta quinquesemita before and during evisceration. J Exp Biol. 2001;204(Pt 5):849–63.
García-Arrarás JE, Estrada-Rodgers L, Santiago R, Torres II, Díaz-Miranda L, Torres-Avillán I. Cellular mechanisms of intestine regeneration in the sea cucumber, Holothuria glaberrima Selenka (Holothuroidea:Echinodermata). J Exp Zool. 1998;281(4):288–304.
Theocharis AD, Skandalis SS, Gialeli C, Karamanos NK. Extracellular matrix structure. Adv Drug Deliv Rev. 2016;97:4–27.
Bellayr IH, Mu X, Li Y. Biochemical insights into the role of matrix metalloproteinases in regeneration: challenges and recent developments. Future Med Chem. 2009;1(6):1095–111.
Dolmatov IY, Kalacheva NV, Tkacheva ES, Shulga AP, Zavalnaya EG, Shamshurina EV, Girich AS, Boyko AV, Eliseikina MG. Expression of Piwi, MMP, TIMP, and Sox during Gut Regeneration in Holothurian Eupentacta fraudatrix (Holothuroidea, Dendrochirotida). Genes. 2021;12(8):1292.
Hayat R, Manzoor M, Hussain A. Wnt signaling pathway: A comprehensive review. Cell Biol Int. 2022;46(6):863–77.
Jiang L, Li J, Zhang C, Shang Y, Lin J. YAP-mediated crosstalk between the Wnt and Hippo signaling pathways (Review). Mol Med Rep. 2020;22(5):4101–6.
Zhang Y, Alexander PB, Wang XF. TGF-beta Family Signaling in the Control of Cell Proliferation and Survival. Cold Spring Harbor Perspectives in Biology. 2017;9(4):a022145.
Zhang W, Liu HT. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002;12(1):9–18.
Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–76.
Chen L, Yao F, Qin Y, Shao Y, Fang L, Yu X, Wang S, Hou L. The potential role of Krüppel-like factor 13 (Aj-klf13) in the intestine regeneration of sea cucumber Apostichopus japonicus. Gene. 2020;735: 144407.
Zheng P, Shao Q, Diao X, Li Z, Han Q. Expression of stem cell pluripotency factors during regeneration in the earthworm Eisenia foetida. Gene. 2016;575(1):58–65.
Ren CH, Chen T, Jiang X, Wang YH, Hu CQ. Identification and functional characterization of a novel ferritin subunit from the tropical sea cucumber Stichopus monotuberculatus. Fish Shellfish Immun. 2014;38(1):265–74.
Ren CH, Chen T, Jiang X, Wang YH, Hu CQ. The first characterization of gene structure and biological function for echinoderm translationally controlled tumor protein (TCTP). Fish Shellfish Immun. 2014;41(2):137–46.
Yan AF, Ren CH, Chen T, Huo D, Jiang X, Sun HY, Hu CQ. A novel caspase-6 from sea cucumber Holothuria leucospilota: Molecular characterization, expression analysis and apoptosis detection. Fish Shellfish Immun. 2018;80:232–40.
García-Arrarás JE, Dolmatov IY. Echinoderms: potential model systems for studies on muscle regeneration. Curr Pharm Des. 2010;16(8):942–55.
Sun LN, Lin CG, Li XN, Xing L, Huo DC, Sun JC, Zhang LB, Yang HS. Comparative Phospho- and Acetyl Proteomics Analysis of Posttranslational Modifications Regulating Intestine Regeneration in Sea Cucumbers. Front Physiol. 2018;9:836.
Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30.
Kanehisa M, Furumichi M, Sato Y, Kawashima M, Ishiguro-Watanabe M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 2023;51(D1):D587-d592.
Funding
This work was supported by the Innovation Team Project of Guangdong Universities (No. 2022KCXTD017), the National Natural Science Foundation of China (42176132), the National Key R & D Program of China (2022YFD2401301), the Key Deployment Project of Centre for Ocean Mega-Research of Science, Chinese Academy of Sciences (COMS2020Q03), and the Guangdong International High-end Talents Exchange Project (Overseas famous experts, YKHZZ 2021 No. 1480).
Author information
Authors and Affiliations
Contributions
Conceptualization: Lihong Yuan, Ting Chen and Aifen Yan; Investigation: Renhui Liu, Xinyue Ren, Junyan Wang, Xinyu Sun, Jiasheng Huang, Zhengyan Guo and Ling Luo; Methodology: Ting Chen, Tiehao Lin, Chunhua Ren, Xudong Cao, Aifen Yan and Lihong Yuan; Formal analysis: Renhui Liu, Xinyue Ren and Ting Chen; Writing - original draft preparation: Renhui Liu, Xinyue Ren, Junyan Wang and Ting Chen; Writing - review and editing: Aifen Yan and Lihong Yuan; Funding acquisition: Lihong Yuan, Ting Chen, Chunhua Ren, Peng Luo and Chaoqun Hu; Resources: Lihong Yuan, Chunhua Ren, Peng Luo, Chaoqun Hu and Aifen Yan; Supervision: Lihong Yuan, Ting Chen and Aifen Yan.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liu, R., Ren, X., Wang, J. et al. Transcriptomic analysis reveals the early body wall regeneration mechanism of the sea cucumber Holothuria leucospilota after artificially induced transverse fission. BMC Genomics 24, 766 (2023). https://doi.org/10.1186/s12864-023-09808-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-023-09808-1