
Abstract
FOF1-ATP synthase is a highly conserved enzyme of eukaryotic or bacterial cells. This enzyme contains eight species of various subunits in bacteria. The F1 sector contains subunits α3, β3, γ, δ, and ε, and the FO sector contains subunits a, b2, and c(10–15). According to modern nomenclature, the rotor of FOF1-ATF synthase consists of the γ, ε, and c subunits; the stator of the enzyme molecule includes the α3, β3, δ, a, and b2 subunits. The rotation of the complex relative to the stator subunits leads to the synthesis or hydrolysis of ATP with the translocation of protons through the a subunit and c ring of the FO sector. The most famous ATP synthase inhibitor is oligomycin A. Oligomycin A inhibits proton translocation in the FOF1-ATP synthase complexes, which leads to an impaired energy metabolism in cells. Oligomycin has a cytotoxic effect against a number of pathogenic bacteria and a high antitumor activity due to the inactivation of FOF1-ATF synthase, a promising biological target for modern drugs. Since FOF1-ATP synthase is highly conserved and ATP synthesis is one of the central processes necessary for the vital functions of cells, this enzyme is a promising biotarget for new antibacterial drugs synthesized based on oligomycin A. This article describes the latest data on the functioning of ATP synthase in bacterial and eukaryotic cells, as well as recent work on the development of new antibacterial drugs based on oligomycin A and its derivatives.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
The membrane-bound FОF1-ATP synthase (ЕС 3.6.3.14) is a unique enzyme complex with a bifunctional catalytic mechanism of ATP synthesis and hydrolysis. Detailed research on the FОF1-ATP synthase structure and function remains an important challenge for modern biochemistry. Functional impairment of ATP synthase is associated with different diseases (Ahmad et al., 2013). The enzyme consists of two structurally and functionally different parts: a membrane-integrated ion-translocating FО sector and a peripheral F1 sector that contains the catalytic sites for ATP synthesis and hydrolysis (Walker, 2013). Although the structures of FOF1-ATP synthases differ in different living organisms, this enzyme is highly conservative in both bacteria and humans. At present, the function of FОF1-ATP synthase is considered in the context of a single complex with other enzymes contained in the eukaryotic mitochondria and bacterial cell walls. The FОF1-ATP synthase complex is a promising molecular target for the treatment of infections and cancer. The best known FОF1-ATP synthase inhibitor is oligomycin A. Oligomycin A binds to the C subunit and selectively suppresses proton translocation in FОF1-ATP synthase of the eukaryotic mitochondria and the cytoplasmic complex of bacterial FОF1-ATP synthase, which leads to impairment of energy metabolism (Shchepina et al., 2002; Cole and Alzari, 2005).
Detailed research on the FОF1-ATP synthase structure and function remains an important challenge for modern biochemistry.
MACROLIDE ANTIBIOTICS: CLASSIFICATION AND MECHANISMS OF ACTION
The term “macrolide” is used to describe agents with a chemical structure represented by a macrocyclic lactone ring with 12 or more members (Mazzei et al., 1993). This class of compounds comprises a lot of biologically active substances, including antibiotics, antifungal drugs, and prokinetic and immunosuppressive agents. The 14-, 15- and 16-membered macrolides are a widely used family of antibiotics. They excellently penetrate tissues and have a high antimicrobial activity, mostly against gram-positive cocci and atypical pathogens (Bearden and Rodvold, 1999). Erythromycin A, a 14-membered macrolide, was isolated more than 50 years ago from Streptomyces cultures; it was the first macrolide antibiotic used in clinical practice (Mazzei et al., 1993). The class of macrolide antibiotics includes chemical agents such as azithromycin, clarithromycin, telithtomycin, erythromycin, carbomycin, oleandomycin, spiramycin, tylosin, roxithromycin, oligomycin A, etc. The mechanism of action of macrolide antibiotics is based on the inhibition of protein synthesis in cells. Macrolide antibiotics reversibly bind to the P site of the bacterial 50S ribosomal subunit. It is believed that inhibition occurs when the incorporation of the next amino acid in the growing peptide attached to the tRNA by peptidyl transferase is prevented (Tenson et al., 2003) (chloramphenicol has a similar mechanism of action (Drainas et al., 1987)) and when the translation process is inhibited. Another potential mechanism is the untimely dissociation of peptidyl-tRNA from the ribosome (Tenson et al., 2003).
Mechanisms of Emergence of Resistance to Macrolide Antibiotics
Since the first widespread application of antibacterial agents in the 1940s, bacterial pathogens have begun to acquire resistance to existing drugs, especially in case of their excessive use. Today, there are three major mechanisms of antibiotic resistance in bacteria:
(1) The presence of antibiotic-inactivating enzymes (e.g., β-lactamase hydrolyzing the β-lactam ring of penicillins);
(2) Target-site mutations in the receptor, enzyme, or ribosomal subunit that prevent the binding of the antibiotic to its target;
(3) The modification or overexpression of transport proteins, upon which antibiotics cannot enter or are released from the cells (Tenover, 2006).
These mechanisms have been found in strains producing macrolide antibiotics; at the same time, cells often have several mechanisms of protection from the antimicrobial effects of antibiotics produced by these cells. When macrolide antibiotics are used to treat diseases caused by pathogenic microorganisms, one should take into consideration the level of microbial sensitivity to these antibiotics. This particularly concerns the ratio between the activity of agents and their toxicity for humans. Single-drug therapy, especially in the case of nonobservance related to treatment length, can lead to the emergence of drug-resistant forms of pathogenic microorganisms; therefore, complex therapies should be used for diseases caused by the latter. For example, modifications in the ribosome induce resistance to a broad range of antibiotics that use it as a target (antibiotics can belong to different chemical classes), while the release of antibiotic from the cell and its inactivation can cause resistance to only one antibiotic (or to structurally similar antibiotics), since it affects the molecule per se.
Thus, there is an urgent need to study the development of novel strategies to detect and obtain effective antibiotics in order to overcome the widespread and growing antibiotic resistance. One potential strategy is to search for fundamentally new bacterial proteins that can become targets for novel classes of antibiotics. Of particular interest is the development of antibiotics that would affect not only on a single biotarget within a cell but several targets simultaneously.
Macrolide Antibiotics of the Class of Oligomycins
Macrolides are antibiotics of the class of oligomycins that contain a 26-membered α,β-unsaturated lactone with conjugated dienes condensed with a bicyclic, spiroketal ring system. Oligomycins A, B, and C were isolated from Streptomyces diastatochromogenes in 1954 (Smith et al., 1954). The major products (A, B, C oligomycins) are synthesized by cells in different proportions depending on the strain and the cultivation and isolation conditions (Kim et al., 1999).
At present, there are many different, known antibiotics with similar structures belonging to the class of oligomycins (A, B, C, E, F, D, rutamycin A, B, etc.) (Fig. 1). The mechanism of action of oligomycin A includes the selective suppression of proton translocation in the FОF1-ATP synthase of eukaryotic mitochondria and in the cytoplasmic complex of the FОF1-ATP synthase of actinobacteria, which leads to impairment of the energy metabolism. Oligomycin A has a cytotoxic effect on some pathogenic bacteria of the genus Actinobacterium due to the inactivation of FОF1-ATP synthase, a promising biotarget for modern drugs (Ahmad et al., 2013). According to the available data, oligomycin A has several unstudied biotargets in bacterial cells different from FOF1-ATP synthases (Wender et al., 2006). Oligomycin A and its analogs also demonstrate a stable antitumor activity (Kobayashi et al., 1987; Yamazaki et al., 1992; Kim et al., 1997). However, the use of oligomycin A in chemotherapy for infectious diseases is limited by its high toxicity.

Chemical structure of the family of oligomycins (A, B, C, E, F, rutamycin A, B and 44-homooligomycin A, B) (according to Nakata et al.,1995, with modifications).
ATP SYNTHASE: A BIOTARGET FOR OLIGOMYCIN A IN EUKARYOTES
ATP synthase is the main complex for the production of cellular energy in all animals and plants and in almost all microorganisms. Adenosine triphosphate (ATP) is a universal energy source. It is synthesized by ATP synthase via oxidation or phosphorylation in bacterial membranes, mitochondria, and chloroplasts. The overall reaction sequence can be put down as follows:
ATP synthesis requires mechanical rotation in which the ATP synthase subunits rotate at a velocity of ~100 revolutions per second in order to synthesize energy via oxidation reactions. ATP synthase (ЕС 3.6.3.14) is a generic term for the enzyme capable of ATP synthesis from ADP (adenosine diphosphate) and inorganic phosphate (Pi). ATP synthase is one of the smallest known biological nanomotors in living organisms. An average person under normal life conditions can roughly generate about 2.0 million kg of ATP from ADP and Pi by the age of 75 years, and the daily approximate amount of ATP utilized by a human being per day can be up to 40 kg. It may be supposed that each ADP and ATP molecule in an organism is phosphorylated and dephosphorylated, respectively, 1000 times per day on average (Senior et al., 2002; Ahmad and Laughlin, 2010; Ahmad et al., 2011). The structural and functional activities of ATP synthase are essentially similar in all prokaryotes and eukaryotes (Boyer, 1997; García et al., 2000; Kabaleeswaran et al., 2006, 2009; Dibrova et al., 2010).
The total number of protons necessary for the synthesis of a single ATP molecule in different organisms varies from three to four; moreover, this number in a cell can vary depending on the physiological requirements (van Walraven et al., 1996; Schemidt et al., 1998; Yoshida et al., 2001).
ATP synthase is one of the most conservative enzymes. Consequently, ATP synthases from the inner mitochondrial membrane and the thylakoid membrane of chloroplasts have identical structural and functional properties relative to bacterial ATP synthase; thus, eukaryotic and bacterial ATP synthases are very similar (Kucharczyk et al., 2009).
Classification of Different ATP Synthases
Before discussing the detailed structure of FОF1-ATP synthase, it would be reasonable to describe in brief other types of ATPases. The F-type (phosphorylation Factor) ATP synthases (also known as FОF1-ATPases and H+-transporting ATPases) are extremely conservative proteins and are the main enzymes performing ATP synthesis in living systems. These enzymes are localized in the plasma membranes of bacteria, the thylakoid membranes of chloroplasts, and the inner mitochondrial membranes. Some bacteria also have Na+-transporting F-ATP synthases.
V-type (Vacuole) ATP synthases are found in the eukaryotic endomembrane systems, e.g., vacuoles, the Golgi apparatus, endosome, lysosome, the plasma membrane of prokaryotes, and some specialized eukaryotic cells. V-ATPases are able to hydrolyze ATP for proton pump function but cannot operate in the reverse direction; for ATP synthesis (Gogarten et al., 1992; Nelson et al., 2000), they occur only in archaea, and perform the same function as F-ATPases. The A‑type ATPases probably appeared as an adaptation to different needs of cells under the more extreme environmental conditions encountered by some archaeal species.
The P-type ATP synthases (P-ATPases, also known as E1-E2-ATP synthases) are found in bacteria and some eukaryotic plasma membranes and organelles. The function of P-ATPase is the transport of various compounds, including ions and phospholipids, across the membrane, with ATP hydrolysis as an energy source. There are many different classes of P‑ATPases, each of which is involved in the transport of particular types of ions: H+, Na+, K+, Mg2+, Ca2+, Ag+ and Ag2+, Zn2+, Со2+, Pb2+, Ni2+, Cd2+, Cu+ and Cu2+.
The E-type (Extracellular) ATP synthases are bound to cell surface membranes and have a broad substrate specificity. Their function is to hydrolyze other nucleoside triphosphates in addition to ATP, as well as intracellular ATP (Wilms et al., 1996; Müller et al., 1999; Toyoshima et al., 2000).
FОF1-ATP Synthase of Eukaryotic and Prokaryotic Organisms
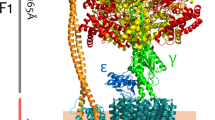
FОF1-ATP synthase has a long history of research. The F1 particle was isolated for the first time by Efraim Racker’s team in 1961. The FО particle was so named due to its inhibition by oligomycin in the membrane ATP synthase. The FОF1-ATP synthase of E. coli has the simplest structure. In prokaryotes, it contains eight types of different subunits (in mammals, the number of subunits varies from 16 to 18): α3, β3, γ, ẟ, ε, a, b2, and c10. The total molecular mass is ~530 kDa (varying from 550 to 650 kDa). F1 contains the subunits α3, β3, γ, δ, and ε, and FО contains the subunits a, b2, and c10 at a stoichiometric ratio of 1 : 2 : 10–14 (Fig. 2).

Schematic representation of FOF1-ATP synthase (according to Gao et al., 2005, with modifications).
In mammals, additional subunits are localized mainly in the F1 region. At the same time, according to the literature data, the FОF1-ATP synthase of yeast is the most complex enzyme; it consists of 20 types of different subunits (Kabaleeswaran et al., 2009). In plants, ATP synthase is localized in chloroplasts (CFОF1-ATP synthase). The enzyme is incorporated into the thylakoid membrane; the CF1 part is incorporated into the stroma and is an integral structure in the nocturnal photosynthesis reactions (the Calvin cycle) and ATP synthesis. In chloroplasts, FОF1-ATP synthase has an identical structure, except that it has two isoforms. In mitochondria, there are seven to nine additional subunits depending on the species, but their masses are too low relative to the whole enzyme and they perform regulatory functions (Senior, 1988; Karrasch and Walker, 1999; Devenish et al., 2000). The synthesis and hydrolysis of ATP occur at three catalytic sites in the F1 sector, while proton transfer occurs via the FO sector incorporated in the membrane. The γ subunit forms a helical coil of alpha-helices, which is localized in the central space of the α3β3 hexagon. The proton gradient triggers a clockwise rotation (when observed from the outer membrane) of the γ subunit, which results in ATP synthesis, while the counterclockwise rotation of the γ subunit results in ATP hydrolysis. Three ATP molecules are synthesized at each rotation of the γ subunit by 360°.
Mechanism of ATP Synthesis and Hydrolysis
When an ATP molecule is bound to one catalytic region, the interaction is highly stable, but ATP hydrolysis is very slow. An excess of ATP leads to binding at all three catalytic centers but with a much lower substrate affinity at the second and third catalytic centers. The Kd value for ATP binding is <1 nM at the first catalytic center but ~1 and 30 µM at the second and third catalytic centers, respectively. When the third catalytic center is bound to ATP, the intensity of ATP hydrolysis increases by 104–105 times (Weber and Senior, 2000). Thus, the F1 sector consists of three α and three β subunits (αβ tandem subunits form a structure characterized by a third-order symmetry axis with a rotation angle of 120°), which show the strict negative cooperativity of substrate binding and, at the same time, the strict positive cooperativity of enzyme activity. In the literature, it is referred to as the binding site–modification hypothesis (Boyer, 1993). The key peculiarity of this hypothesis is that each of the three catalytic sites (three pairs of αβ subunits containing these sites) has a different conformation at any moment of time. One of them is open and ready to bind ATP (or ADP + Pi), whereas the second and third sites are partially open and closed, respectively. ATP binding, which results in closure of the site, initiates conformational changes such that the closed site becomes partially open and the partially open site becomes completely open. Thus, each site switches between three states, both in ATP hydrolysis and in ATP synthesis, but in a different direction (Fig. 3).

Binding site–modification hypothesis and rotation of the catalytic binding site by the γ subunit (according to Capaldi et al., 2002, with modifications).
Each catalytic site goes through three states during the cycle: T, L, and O. ATP binds to the O site (open and empty) in order to transform it into the T site (bound to ATP). After bond is broken, the T site transforms into an L site (bound to ADP + Pi), while ADP + Pi is detached from the O site. At any given time, each of the three catalytic sites is in the O, T, and L state, respectively. The switching between these states at each of the sites leads to hydrolysis (or synthesis) on one ATP molecule and rotation of the γ rotor by 120°. Figure 3 shows the stages of hydrolysis or synthesis of one ATP molecule at three bindings sites (Capaldi et al., 2002). According to the published data, the rotor consists of the subunits γ, ε, c, and the stator consists of the α3, β3, δ, a, and b2 subunits (Diez et al., 2004; Itoh et al., 2004; Weber, 2006). The function of the stator is to prevent joint rotation of the catalytic centers and the rotor.
There are altogether six nucleotide binding sites in the F1 sector of ATP synthase. Three catalytic sites are present mostly in the β subunit, while three noncatalytic binding sites are present mostly in the λ subunit. The three catalytic sites are designated as βTP, PDP, and βE; they were found via X-ray crystallography based on ATP, ADP, and Pi binding, respectively (Leslie and Walker, 2000; Menz et al., 2001). Pi originally binds to βE (the empty site) for ATP synthesis. The synthesis reaction occurs at the three catalytic sites interdependently and successively. Each catalytic site undergoes conformational changes resulting in the following reaction: substrate (ADP + Pi) binding → ATP synthesis → ATP release. In the FО sector, the mechanical rotation of the rotor shaft leads to conformational changes in the catalytic domains in F1 for ATP synthesis. The reverse reaction of ATP hydrolysis causes reverse conformational changes in the FО sector and consequently changes the direction of rotation of the rotor shaft. These conformational changes in the catalytic centers are associated with rotation of the γ subunit. A deeper understanding of the FОF1 structure and function could highlight the potential pathways for the development of ATP synthase as a target for molecular medicinal agents and its application in nanotechnologies and nanomedicine (Whitesides, 2003; Khataee, H. and Khataee, A., 2009).
Consequently, it is extremely important to understand the structure and catalytic function of ATP synthase, in particular, the way in which Pi binding leads to ATP formation, for an understanding of the processes of inhibition of enzyme function when it binds to medicinal agents (Bullough et al., 1989; Gledhill and Walker, 2005; Hong and Pedersen, 2008).
Mechanism of Oligomycin-A Binding to the C Subunit of FОF1-ATP Synthase
The study of the oligomycin-binding site is of great interest due to the wide use of oligomycin-like inhibitors and the existing potential to detect novel medicinal agents based on this mechanism (inhibition of the C subunit of FОF1-ATP synthase).
The C subunit of ATP synthase is a membrane-integrated protein consisting of two helices, 1 and 2, that penetrate the inner mitochondrial membrane (Fig. 3a). The C subunit is a ring consisting of different numbers of subunits depending on the type. For example, this complex comprises ten subunits in the yeast ATP synthase and eight subunits in the bovine ATP synthase (Stock et al., 1999; Watt et al., 2010).
The C subunit is a significant component of the proton turbine of ATP synthase, which converts electrochemical energy into mechanical rotation and vice versa. An indispensable part of the C subunit is the glutamic acid (Glu59) in helix 2; its function is direct proton movement from the cytosol to the mitochondrial matrix during ATP synthesis. The side chain of the carboxyl group of Glu59 is almost in the middle of helix 2 and is located in the lipid bilayer. It is involved in protonation due to the closed conformation. Subunit A forms two proton half-channels through which a proton is transferred and bound to the carboxyl group of Glu59 in the open conformation, which makes the protonation/deprotonation process possible (Pogoryelov et al., 2010; Fillingame and Steed, 2014). Oligomycin A binds to the two neighboring subunits of the c ring positioned at the proton-access channel and blocks the major carboxyl groups, which leads to inhibited proton translocation. The molecule of oligomycin A binds to amino acid residue Glu59 and prevents the binding of the side chain of the carboxyl group to the water molecule. The side chain of the carboxyl group of amino acid Glu59 forms a hydrogen bond with the water molecule, which, in turn, forms a hydrogen bond with the oxygen of the carbonyl group of Leu57 and the oxygen of the carbonyl group of oligomycin (O36). Thus, Glu59 forms a hydrogen bond with oligomycin via the link with the water molecule. Other interactions between oligomycin and the C subunit primarily occur due to van der Waals forces (Fig. 4). The interatomic distances between the c subunit and oligomycin atoms are 3.7–4.9 Å. Amino-acid residue Phe64 forms a series of interactions with six carbon atoms of oligomycin, including C2 and C3, which have a double bond with oligomycin A (Palmer and Potter, 2008) and thus can also interact via π–π stacking. Each of the two amino acid residues, Ala56 and Ala60, interacts with the opposite ends of oligomycin molecule via bonding between oligomycin and the C ring (the N and O chains). Other amino acid residues interact with oligomycin via bonding to only one of the C subunits.

Interaction between C-subunit atoms and oligomycin A. (a) Atoms are shown as spheres, and residues are shown as rods within a range of 5 Å from the atoms in the oligomycin molecule. Dotted lines indicate atomic distances in the range of 2.5–2.8 Å for three hydrogen bonds and 3.7–4.8 Å for hypothetical van der Waals forces; (b) the interactions between oligomycin A and the C ring. The residues for which the atom is within the range of 4.8 Å are shown by dotted lines (possible interaction) (according to Symersky et al., 2012, with modifications).
The study of the binding mechanism of the recently studied agent R207910 (TMC207), which inhibits ATP synthase and is highly effective against tuberculosis mycobacteria, showed that strains resistant to this agent contain three independent mutations in the gene encoding the C subunit (Asp26Val, Ala61Pro, Ile64Met) (Andries et al., 2005; Koul et al., 2008).
In the model of the C subunit of yeast, amino-acid residue Asp26 is localized in helix 1 next to Gly25, which imparts resistance to oligomycin and cross resistance to related drugs in the case of mutation (Nagley et al., 1986; Galanis et al., 1989). In yeasts, Ala61 is localized next to Ala60, one of the two residues interacting with oligomycin in both molecules of the C subunit that form a binding site.
The Ala(61)Pro mutation causes a kink in the alpha helix and destroys the binding site for medicinal agents. Amino-acid residue Ile64 corresponds to Phe64 in yeasts, which forms critical contacts with oligomycin. Though the structure of R207910 is highly different from that of oligomycin, this chemical compound has some similar chemical properties, which confirms the similarity between the binding site of oligomycin A and the C subunit of mitochondria and bacteria. In addition, this binding site for oligomycin A in the C subunit can be also used by other inhibitors of bacterial c ring and probably by analogous inhibitors binding to VО (the FО analog) of the vacuolar-type ATPase (Dröse and Altendorf, 1997), which can yield one more potential target agent.
Thus, the study of biotargets for oligomycin A and its derivatives is one of the key trends in the development of novel drugs on their basis.
ANTIBIOTIC OLIGOMYCIN A AND ITS DERIVATIVES; THEIR MECHANISMS OF ACTION IN ACTINOBACTERIA
Oligomycin-like antibiotics belonging to the group of macrolides are produced by many actinobacteria, including Streptomyces sp. WK-6150 (Enomoto et al., 2001), S. virginiae 17 (Danilenko et al., 2012), Streptomyces sp. MCI-2225 (Kobayashi et al., 1987), S. libani (Kim et al., 1999), S. diastatochromogenes, S. diastaticus (Yang et al., 2010), Actinoalloteichus sp. NPS702 (Sato et al., 2012), etc. These strains are resistant to oligomycin-like antibiotics, because they produce antibiotics of this class. At present, the mechanisms of sensitivity in different strains susceptible to oligomycin-like antibiotics remain unstudied. S. fradiae (xinghaiensis) ATCC 19609, which is supersensitive to oligomycin A (<0.001 nmol/mL or 0.0005 nmol/disc), seems to be one such strain (Alekseeva et al., 2009). The available data demonstrate the existence of several targets for oligomycin A in the cells (Wender et al., 2006). It can be supposed that the mechanisms of sensitivity of actinobacterial strains to oligomycin A are determined by several biotargets in the cells, the antibiotic capacity to penetrate the cell, the absence of a mechanism of antibiotic release from the cell, the absence of cell repair systems, etc. Novel semisynthetic derivatives of oligomycin A were synthesized at the Gauze Scientific Research Institute of New Antibiotics to determine the sensitivity mechanisms and to detect additional biotargets and to establish in detail the mechanism of interaction between oligomycin A and the latter, including FОF1-ATP synthase. The derivatives were selectively modified at the fragments of antibiotic molecule (position 33 of the side chain) involved in the binding to the C subunit of FОF1-ATP synthase (Fig. 5).

Scheme of the synthesis of 33-O-mesyl oligomycin A (2), (33S)-azido-33-deoxyoligomycin (3), and 33-deoxy-33-(1,2,3-triazol-1-yl) oligomycin A (4a–d) from oligomycin A (1) (according to Lysenkova et al., 2013, with modifications).
Nitron-oligomycin was synthesized via the interaction between oligomycin A and hydroxylamine hydrochloride in pyridine in the presence of sodium acetate (Lysenkova et al., 2009); (33S)-azido-33-deoxyoligomycin was synthesized via the interaction between 33‑O-mesyl oligomycin A and sodium azide in dimethylformamide at 60–65°С; 33-O-mesyloligomycin A was synthesized via the interaction between methane sulfochloride and oligomycin A in pyridine (Lysenkova et al., 2013); in addition, a new method of chemical modification of oligomycin A in the side chain was developed (Fig. 6). The interaction between 33-O-mesyloligomycin and potassium thiocyanate was used to synthesize (33S)-33-deoxy-33-thiocyanate oligomycin (Fig. 6). (33S)-33-deoxy-33-thiocyanate oligomycin showed a slightly lower activity against S. fradiae as compared to the initial oligomycin A (Lysenkova et al., 2015).

Synthesis of (33S)-33-deoxy-33-thiocyanate oligomycin (3). Oligomycin A (1). 33-O-mesylate oligomycin A (2).
The activity of newly synthesized derivatives of oligomycin A was monitored with a test system based on the bacterial strain S. fradiae (xinghaiensis) ATCC 19609, which is supersensitive to oligomycin A. All derivatives showed lower activity against S. fradiae ATCC 19609; moreover, some of them were less toxic for oligomycin A.
For the further development of a new generation of chemotherapeutic agents, it will be necessary to detect all potential intracellular targets and to study the mechanisms of resistance of bacterial cells to oligomycin A and its derivatives.
Sensitivity and Resistance to Oligomycin A in Bacteria, Including Actinobacteria
Most bacteria are sensitive to various antibiotics of different chemical classes (Jorgensen et al., 2009). The sensitivity or resistance of a strain to any antibiotic is determined by the following parameters: the strain is considered sensitive if MIC in vitro (1–10 µg/mL) makes therapy successful; the strain is considered naturally resistant if MIC in vitro (more than 100 µg/mL) makes successful therapy impossible (Rodloff et al., 2008). Eukaryotic strains, including Saccharomyces cerevisiae, are sensitive to oligomycin A at a level of 25 µg/mL (Park et al., 2011), while gram-negative bacteria are naturally resistant to oligomycin A and its derivatives (Kim et al., 1997). Some gram-positive microorganisms are sensitive to oligomycin A and its derivatives; they belong to the class of actinobacteria. Bacteria of the genus Streptomyces have different levels of sensitivity to oligomycin A. The MIC of oligomycin A for the strain S. albus ATCC 21132 is 0.01 nm/disc (Lysenkova et al., 2015); the level of sensitivity is slightly less than in S. lividans 66 (MIC 1 nm/disc (Alekseeva et al., 2009)). The strain S. fradiae (xinghaiensis) ATCC 19609 is supersensitive to oligomycin A: MIC < 0.001 nmol/mL (Lysenkova et al., 2015).
CONCLUSIONS
S. fradiae (xinghaiensis) ATCC 19609 is supersensitive to oligomycin A and is used as a model object for the study of FОF1-ATP synthase. FОF1-ATP synthase is a unique, universal machine of pro- and eukaryotic organisms that supplies them with energy as a result of ATP synthesis under aerobic conditions and the generation of a transmembrane proton gradient under anaerobic conditions. FОF1-ATP synthase is a rather well-studied nanomachine and, hence, it is possible to start to develop on its basis a natural-like biotechnological construct for proton transport in order to create a novel energy source. Oligomycin A and its derivatives are classical chemical molecules that have been used for many decades to study the function of FОF1-ATP synthase. Earlier, we found the strain S. fradiae (xinghaiensis) ATCC 19609, which is sensitive to oligomycin A in nanoscale quantities (0.001 µg/mL). In addition, it was shown that the function of FОF1-ATP synthase probably depends on the phosphorylation by serine-threonine protein kinases. Tens of new semisynthetic derivatives of oligomycin A with different effects on FОF1-ATP synthase have been synthesized. It was shown that, in addition to FОF1-ATP synthase, oligomycin A can affect DNA repair and replication systems (Bekker et al., 2019). All of these findings make it possible to proceed to a new level of research on FОF1-ATP synthase as a nanomachine for ATP synthesis and hydrolysis and its application as a biotarget for new drugs. Deeper study of the mechanisms of supersensitivity to oligomycin A and its derivatives in S. fradiae (xinghaiensis) ATCC 19609 will make it possible to use the discovered new mechanisms for the detection of new biotargets in bacterial cells and the creation of novel antibacterial agents.
The importance of ATP synthase as a promising biotarget for drug development is also indicated by the fact that many antibiotics, such as efrapeptin, aurovertin, and oligomycin, inhibit precisely ATP synthase. Efrapeptin and aurovertin inhibit ATP synthesis and hydrolysis (van Raaij et al., 1996). Oligomycin is a powerful inhibitor of ATP synthase via binding to the FО complex, which blocks proton conductivity. The literature data show that oligomycin induces an apoptotic response in cultured human lymphoblastoid cells and other mammalian cells within 12–18 h but, at the same time, mitochondrial inhibitors do not cause apoptosis in cells without the mitochondrial respiratory chain (Wolvetang et al., 1994). Another such study showed that oligomycin interacts with separate mitochondrial components, which, in turn, can result in the apoptosis of individual cells due to enhanced expression of СD14 (Mills et al., 1999). Thus, it is quite possible that the interactions between ATP synthase and other inhibitors can play a significant role in apoptotic mechanisms in mitochondria.
REFERENCES
Ahmad, Z. and Laughlin, T.F., Medicinal chemistry of ATP synthase: a potential drug target of dietary polyphenols and amphibian antimicrobial peptides, Curr. Med. Chem., 2010, vol. 17, no. 25, pp. 2822–2836.
Ahmad, Z., Okafor, F., and Laughlin, T.F., Role of charged residues in the catalytic sites of Escherichia coli ATP synthase, J. Amino Acids., 2011, vol., 2011, art. ID 785741.
Ahmad, Z., Okafor, F., Azim, S., et al., ATP synthase: a molecular therapeutic drug target for antimicrobial and antitumor peptides, Curr. Med. Chem., 2013, vol. 20, no. 15, pp. 1956–1973.
Alekseeva, M.G., Elizarov, S.M., Bekker, O.B., et al., F0F1 ATP synthase of Streptomycetes: modulation of activity and oligomycin resistance by protein Ser/Thr kinases, Biochemistry (Moscow)Suppl. Ser. A: Membr. Cell Biol., 2009, vol. 3, no. 1, pp. 16–23.
Andries, K., Verhasselt, P., Guillemont, J., et al., A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis,Science, 2005, vol. 307, no. 5707, pp. 223–227.
Antimicrobial Therapy in Veterinary Medicine, Giguère, S., Prescott, J.F., Baggot, J.D., et al., Eds., Chichester: Wiley, 2006.
Bearden, D.T. and Rodvold, K.A., Penetration of macrolides into pulmonary sites of infection, Infect. Med., 1999, vol. 16, no. 7, pp. 480–484.
Bekker, O.B., Vatlin, A.A., Zakharevich, N.V., et al., Draft genome sequence of Streptomyces xinghaiensis (fradiae) OlgR, a strain resistant to oligomycin A, Microbiol. Res. Ann., 2019, vol. 8, no. 2, pp. e01531-18.
Boyer, P.D., The binding change mechanism for ATP synthase—some probabilities and possibilities, Biochim. Biophys. Acta,Bioenerg., 1993, vol. 1140, no. 3, pp. 215–250.
Boyer, P.D., The ATP synthase—a splendid molecular machine, Annu. Rev. Biochem., 1997, vol. 66, pp. 717–749.
Bullough, D.A., Ceccarelli, E.A., Roise, D., and Allison, W.S., Inhibition of the bovine-heart mitochondrial F1-ATPase by cationic dyes and amphipathic peptides, Biochim. Biophys. Acta,Bioenerg., 1989, vol. 975, no. 3, pp. 377–383.
Capaldi, R.A., Aggeler, R., Moser, T.L., et al., Mechanism of the F1F0-type ATP synthase, a biological rotary motor, Trends Biochem. Sci., 2002, vol. 27, no. 3, pp. 154–160.
Cole, S.T. and Alzari, P.M., Microbiology. TB—a new target, a new drug, Science, 2005, vol. 307, no. 5707, pp. 214–215.
Danilenko, A.N., Bibikova, M.V., Spiridonova, I.A., et al., Physicochemical properties and structure of oligomycin SC-II, produced by Streptomyces virginiae 17, Antibiot. Khimioter., 2012, vol. 57, nos. 11–12, pp. 3–7.
Devenish, R.J., Prescott, M., Roucou, X., et al., Insights into ATP synthase assembly and function through the molecular genetic manipulation of subunits of the yeast mitochondrial enzyme complex, Biochim. Biophys. Acta,Bioenerg., 2000, vol. 1458, nos. 2–3, pp. 428–442.
Dibrova, D.V., Galperin, M.Y., and Mulkidjanian, A.Y., Characterization of the N-ATPase, a distinct, laterally transferred Na+-translocating form of the bacterial F-type membrane ATPase, Bioinformatics, 2010, vol. 26, no. 12, pp. 1473–1476.
Diez, M., Zimmermann, B., Börsch, M., et al., Proton-powered subunit rotation in single membrane-bound F0F1-ATP synthase, Nat. Struct. Mol. Biol., 2004, vol. 11, no. 2, pp. 135–141.
Drainas, D., Kalpaxis, D.L., and Coutsogeorgopoulos, C., Inhibition of ribosomal peptidyltransferase by chloramphenicol. Kinetic studies, Eur. J. Biochem., 1987, vol. 164, no. 1, pp. 53–58.
Dröse, S. and Altendorf, K., Bafilomycins and concanamycins as inhibitors of V-ATPases and P-ATPases, J. Exp. Biol., 1997, vol. 200, no. 1, pp. 1–8.
Enomoto, Y., Shiomi, K., Matsumoto, A., et al., Isolation of a new antibiotic oligomycin G produced by Streptomyces sp. WK-6150, J. Antibiot. (Tokyo), 2001, vol. 54, no. 3, pp. 308–313.
Fillingame, R.H. and Steed, P.R., Half channels mediating H+ transport and the mechanism of gating in the F0 sector of Escherichia coli F1F0 ATP synthase, Biochim. Biophys. Acta,Bioenerg., 2014, vol. 1837, no. 7, pp. 1063–1068.
Galanis, M., Mattoon, J.R., and Nagley, P., Amino acid substitutions in mitochondrial ATP synthase subunit 9 of Saccharomyces cerevisiae leading to venturicidin or ossamycin resistance, FEBS Lett., 1989, vol. 249, no. 2, pp. 333–336.
Gao, Y.Q., Yang, W., and Karplus, M., A structure-based model for the synthesis and hydrolysis of ATP by F1-ATPase, Cell., 2005, vol. 123, no. 2, pp. 195–205.
García, J.J., Ogilvie, I., Robinson, B.H., et al., Structure, functioning, and assembly of the ATP synthase in cells from patients with the T8993G mitochondrial DNA mutation. Comparison with the enzyme in Rho(0) cells completely lacking mtDNA, J. Biol. Chem., 2000, vol. 275, no. 15, pp. 11075–11081.
Gledhill, J.R. and Walker, J.E., Inhibition sites in F1-ATPase from bovine heart mitochondria, Biochem. J., 2005, vol. 386, no. 3, pp. 591–598.
Gogarten, J.P., Starke, T., Kibak, H., et al., Evolution and isoforms of V-ATPase subunits, J. Exp. Biol., 1992, vol. 172, pp. 137–147.
Hong, S. and Pedersen, P.L., ATP synthase and the actions of inhibitors utilized to study its roles in human health, disease, and other scientific areas, Microbiol. Mol. Biol. Rev., 2008, vol. 72, no. 4, pp. 590–641.
Itoh, H., Takahashi, A., Adachi, K., et al., Mechanically driven ATP synthesis by F1-ATPase, Nature, 2004, vol. 427, no. 6973, pp. 465–468.
Jorgensen, J.H. and Ferraro, M.J., Antimicrobial susceptibility testing: a review of general principles and contemporary practices, Clin. Infect. Dis., 2009, vol. 49, no. 11, pp. 1749–1755.
Kabaleeswaran, V., Puri, N., Walker, J.E., Leslie, A.G.W., et al., Novel features of the rotary catalytic mechanism revealed in the structure of yeast F1 ATPase, EMBO J., 2006, vol. 25, no. 22, pp. 5433–5442.
Kabaleeswaran, V., Shen, H., Symersky, J., et al., Asymmetric structure of the yeast F1 ATPase in the absence of bound nucleotides, J. Biol. Chem., 2009, vol. 284, no. 16, pp. 10546–10551.
Karrasch, S. and Walker, J.E., Novel features in the structure of bovine ATP synthase, J. Mol. Biol., 1999, vol. 290, no. 2, pp. 379–384.
Khataee, H.R. and Khataee, A.R., Advances in F0F1-ATP synthase biological protein nanomotor: from mechanisms and strategies to potential applications, Nano, 2009, vol. 4, no. 2, pp. 55–67.
Kim, B.S., Moon, S.S., and Hwang, B.K., Isolation, identification, and antifungal activity of a macrolide antibiotic, oligomycin A, produced by Streptomyces libani,Can. J. Bot., 1999, vol. 77, pp. 850–858.
Kim, H.S., Band, H.J., Lee, S.Y., et al., 44-Homooligomycin E, a new cytotoxic macrolide antibiotic from Streptomyces ostreogriseus,Biosci., Biotechnol. Biochem., 1997, vol. 61, no. 2, pp. 378–380.
Kobayashi, K., Nishino, C., Ohya, J., et al., Oligomycin E, a new antitumor antibiotic produced by Streptomyces sp. MCI-2225, J. Antibiot. (Tokyo), 1987, vol. 40, no. 7, pp. 1053–1057.
Koul, A., Vranckx, L., Dendouga, N., et al., Diarylquinolines are bactericidal for dormant mycobacteria as a result of disturbed ATP homeostasis, J. Biol. Chem., 2008, vol. 283, no. 37, pp. 25273–25280.
Kucharczyk, R., Zick, M., Bietenhader, M., et al., Mitochondrial ATP synthase disorders: molecular mechanisms and the quest for curative therapeutic approaches, Biochim. Biophys. Acta, Mol.Cell Res., 2009, vol. 1793, no. 1, pp. 186–199.
Leslie, A.G. and Walker, J.E., Structural model of F1-ATPase and the implications for rotary catalysis, Philos. Trans. R. Soc., B, 2000, vol. 355, no. 1396, pp. 465–471.
Lysenkova, L.N., Turchin, K.F., Danilenko, V.N., et al., The first examples of chemical modification of oligomycin A, J. Antibiot. (Tokyo), 2009, vol. 63, pp. 17–22.
Lysenkova, L.N., Turchin, K.F., Korolev, A.M., et al., Synthesis and cytotoxicity of oligomycin A derivatives modified in the side chain, Bioorg. Med. Chem., 2013, vol. 21, pp. 2918–2924.
Lysenkova, L.N., Godovikov, I.A., Korolev, A.M., et al., Synthesis and anti-actinomycotic activity of the oligomycin A thiocyanato derivative modified at 2-oxypropyl side chain, Macroheterocycles, 2015, vol. 8, no. 4, pp. 424–428.
Mazzei, T., Mini, E., and Novelli, A.P.P., Chemistry and mode of action of macrolides, J. Antimicrob. Chemother., 1993, vol. 31, suppl., pp. 1–9.
Menz, R.I., Walker, J.E., and Leslie, A.G., Structure of bovine mitochondrial F1-ATPase with nucleotide bound to all three catalytic sites: implications for the mechanism of rotary catalysis, Cell, 2001, vol. 106, no. 3, pp. 331–341.
Mills, K.I., Woodgate, L.J., Gilkes, A F., et al., Inhibition of mitochondrial function in HL60 cells is associated with an increased apoptosis and expression of CD14, Biochem. Biophys. Res. Commun., 1999, vol. 263, no. 2, pp. 294–300.
Müller, V., Ruppert, C., and Lemker, T., Structure and function of the A1A0-ATPases from methanogenic archaea, J. Bioenerg. Biomembr., 1999, vol. 31, no. 1, pp. 15–27.
Nagley, P., Hall, R.M., and Ooi, B.G., Amino acid substitutions in mitochondrial ATPase subunit 9 of Saccharomyces cerevisiae leading to oligomycin or venturicidin resistance, FEBS Lett., 1986, vol. 195, nos. 1–2, pp. 159–163.
Nakata, M., Ishiyama, T., Akamatsu, S., et al., Synthetic studies on oligomycins. Synthesis of the oligomycin B spiroketal and polypropionate portions, Bull. Chem. Soc. Jpn., 1995, vol. 68, no. 3, pp. 967–989.
Nelson, N., Perzov, N., Cohen, A., et al., The cellular biology of proton-motive force generation by V-ATPases, J. Exp. Biol., 2000, vol. 203, no. 1, pp. 89–95.
Palmer, R.A. and Potter, B.S., X-ray structures and absolute configurations of the antibiotics oligomycins A, B and C: inhibitors of ATP synthase, J. Chem. Crystallogr., 2008, vol. 38, no. 4, pp. 243–253.
Park, J.W., Park, S.R., Han, A.R., et al., Generation of reduced macrolide analogs by regio-specific biotransformation, J. Antibiot. (Tokyo), 2011, vol. 64, no. 1, pp. 155–157.
Pogoryelov, D., Krah, A., Langer, J.D., et al., Microscopic rotary mechanism of ion translocation in the F0 complex of ATP synthases, Nat. Chem. Biol., 2010, vol. 6, no. 12, pp. 891–899.
Rodloff, A., Bauer, T., Ewig, S., et al., Susceptible, intermediate, and resistant—the intensity of antibiotic action, Dtsch. Arztebl. Int., 2008, vol. 105, no. 39, pp. 657–662.
Sato, S., Iwata, F., Yamada, S., and Katayama, M., Neomaclafungins A–I: oligomycin-class macrolides from a marine-derived actinomycete, J. Nat. Prod., 2012, vol. 75, no. 11, pp. 1974–1982.
Schemidt, R.A., Qu, J., Williams, J.R., and Brusilow, W.S., Effects of carbon source on expression of F0 genes and on the stoichiometry of the c subunit in the F1F0 ATPase of Escherichia coli,J. Bacteriol., 1998, vol. 180, no. 12, pp. 3205–3208.
Senior, A.E., ATP synthesis by oxidative phosphorylation, Physiol. Rev., 1988, vol. 68, no. 1, pp. 177–231.
Senior, A.E., Nadanaciva, S., and Weber, J., The molecular mechanism of ATP synthesis by F1F0-ATP synthase, Biochim. Biophys. Acta,Bioenerg., 2002, vol. 1553, no. 3, pp. 188–211.
Shchepina, L.A., Pletjushkina, O., Avetisyan, A., et al., Oligomycin, inhibitor of the F0 part of H+-ATP-synthase, suppresses the TNF-induced apoptosis, Oncogene, 2002, vol. 21, no. 53, pp. 8149–8157.
Smith, R.M., Peterson, W.H., and McCoy, E., Oligomycin, a new antifungal antibiotic, Antibiot. Chemother. (Northfield), 1954, vol. 4, no. 9, pp. 962–970.
Stock, D., Leslie, A.G., and Walker, J.E., Molecular architecture of the rotary motor in ATP synthase, Science, 1999, vol. 286, no. 5445, pp. 1700–1705.
Symersky, J., Osowski, D., Walters, D.E., and Mueller, D.M., Oligomycin frames a common drug-binding site in the ATP synthase, Proc. Natl. Acad. Sci. U.S.A., 2012, vol. 109, no. 35, pp. 13961–13965.
Tenover, F.C., Mechanisms of antimicrobial resistance in bacteria, Am. J. Infect. Control, 2006, vol. 34, no. 5, pp. 3–10.
Tenson, T., Lovmar, M., and Ehrenberg, M., The mechanism of action of macrolides, lincosamides and streptogramin B reveals the nascent peptide exit path in the ribosome, J. Mol. Biol., 2003, vol. 330, no. 5, pp. 1005–1014.
Toyoshima, C., Nomura, H., and Tsuda, T., Crystal structure of the calcium pump of sarcoplasmic reticulum at 2.6 A resolution, Nature, 2000, vol. 405, no. 6787, pp. 647–655.
van Raaij, M.J., Abrahams, J.P., Leslie, A.G., and Walker, J.E., The structure of bovine F1-ATPase complexed with the antibiotic inhibitor aurovertin B, Proc. Natl. Acad. Sci. U.S.A., 1996, vol. 93, no. 14, pp. 6913–6917.
van Walraven, H.S., Strotmann, H., Schwarz, O., and Rumberg, B., The H+/ATP coupling ratio of the ATP synthase from thiol-modulated chloroplasts and two cyanobacterial strains is four, FEBS Lett., 1996, vol. 379, no. 3, pp. 309–313.
Walker, J.E., The ATP synthase: the understood, the uncertain and the unknown, Biochem. Soc. Trans., 2013, vol. 41, no. 1, pp. 1–16.
Watt, I.N., Montgomery, M.G., Runswick, M.J., et al., Bioenergetic cost of making an adenosine triphosphate molecule in animal mitochondria, Proc. Natl. Acad. Sci. U.S.A., 2010, vol. 107, no. 39, pp. 16823–16827.
Weber, J., ATP synthase: subunit-subunit interactions in the stator stalk, Biochim. Biophys. Acta,Bioenerg., 2006, vol. 1757, nos. 9–10, pp. 1162–1170.
Weber, J. and Senior, A.E., ATP synthase: what we know about ATP hydrolysis and what we do not know about ATP synthesis, Biochim. Biophys. Acta,Bioenerg., 2000, vol. 1458, nos. 2–3, pp. 300–309.
Wender, P.A., Jankowski, O.D., Longcore, K., et al., Correlation of F0F1-ATPase inhibition and antiproliferative activity of apoptolidin analogues, Org. Lett., 2006, vol. 8, no. 4, pp. 589–592.
Whitesides, G.M., The “right” size in nanobiotechnology, Nat. Biotechnol., 2003, vol. 21, no. 10, pp. 1161–1165.
Wilms, R., Freiberg, C., Wegerle, E., et al., Subunit structure and organization of the genes of the A1A0 ATPase from the archaeon Methanosarcina mazei Go1, J. Biol. Chem., 1996, vol. 271, no. 31, pp. 18843–18852.
Wolvetang, E.J., Johnson, K.L., Krauer, K., et al., Mitochondrial respiratory chain inhibitors induce apoptosis, FEBS Lett., 1994, vol. 339, nos. 1–2, pp. 40–44.
Yamazaki, M., Yamashita, T., Harada, T., et al., 44-Homooligomycins A and B, new antitumor antibiotics from Streptomyces bottropensis. Producing organism, fermentation, isolation, structure elucidation and biological properties, J. Antibiot. (Tokyo), 1992, vol. 45, no. 2, pp. 171–179.
Yang, P.W., Li, M.G., Zhao, J.Y., et al., Oligomycins A and C, major secondary metabolites isolated from the newly isolated strain Streptomyces diastaticus, Folia Microbiol. (Praha), 2010, vol. 55, no. 1, pp. 10–16.
Yoshida, M., Muneyuki, E., and Hisabori, T., ATP synthase—a marvelous rotary engine of the cell, Nat. Rev. Mol. Cell Biol., 2001, vol. 2, no. 9, pp. 669–677.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interests. The authors declare that they have no conflicts of interest.
Statement on the welfare of humans or animals. This article does not contain any studies involving animals performed by any of the authors.
Additional information
Translated by E. Makeeva
Rights and permissions
About this article

Cite this article
Vatlin, A.A., Danilenko, V.N. Bacterial FOF1 ATP: Nanomotor for ATP Synthesis and Hydrolysis and Mechanism of Interaction with the Macrolide Antibiotic Oligomycin A. Biol Bull Rev 10, 483–494 (2020). https://doi.org/10.1134/S2079086420060067
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S2079086420060067