
Abstract
ATP synthases are unique rotatory molecular machines that supply biochemical reactions with adenosine triphosphate (ATP)—the universal “currency”, which cells use for synthesis of vital molecules and sustaining life. ATP synthases of F-type (FOF1) are found embedded in bacterial cellular membrane, in thylakoid membranes of chloroplasts, and in mitochondrial inner membranes in eukaryotes. The main functions of ATP synthases are control of the ATP synthesis and transmembrane potential. Although the key subunits of the enzyme remain highly conserved, subunit composition and structural organization of ATP synthases and their assemblies are significantly different. In addition, there are hypotheses that the enzyme might be involved in the formation of the mitochondrial permeability transition pore and play a role in regulation of the cell death processes. Dysfunctions of this enzyme lead to numerous severe disorders with high fatality levels. In our review, we focus on FOF1-structure-based approach towards development of new therapies by using FOF1 structural features inherited by the representatives of this enzyme family from different taxonomy groups. We analyzed and systematized the most relevant information about the structural organization of FOF1 to discuss how this approach might help in the development of new therapies targeting ATP synthases and design tools for cellular bioenergetics control.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cellular bioenergetics is essentially based on the catalysis of the energy storage molecules using conformational changes of protein complexes involved in the oxidative phosphorylation (OXPHOS) or photophosphorylation processes. The actual synthesis of these small molecules, which are vital for most organisms, takes place at the interfaces between different subunits of these protein complexes.
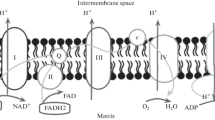
ATP synthase is a key enzyme in the OXPHOS and photophosphorylation processes. It produces adenosine triphosphate (ATP)—a small energy storage molecule powering vital biochemical reactions in living cells. F-type ATP synthase consists of F1 and FO multisubunit complexes (water-soluble and membrane integral, respectively), which are connected by central and peripheral stalks. The central stalk rotates with the c-ring as a rigid body, triggering conformational changes and rotation of the catalytic part α3β3 (Fig. 1). ATP is synthesised at the interaction interface of the α/β catalytic subunits of the ATP synthase by chemo-mechanical coupling via the rotation of the c-ring subunit complex in the membrane [1, 2].

The figure is reprinted from [4]
An overall view of the cFOF1 ATP synthase (most of the bFOF1 comprise similar subunits). FO part consists of abb’cN subunits, F1 part consists of α3β3γɛδ subunits, bb’ is a peripheral stalk. Inside the cN-ring pore, there are molecules that interact with its inner surface. The PDB structure 6FKF of cFOF1 from the spinach chloroplasts is shown [3].
ATP synthase is extremely wide-spread and is present in almost all organisms from bacteria and archaea to eukaryotes. F-type ATP synthase was found in the bacterial cellular membranes, thylakoid membranes of chloroplasts, and cristae of mitochondria in eukaryotes. Various types of structural oligomeric organization of ATP synthases were found in different species. Bacterial ATP synthases (bFOF1) are considered to be monomers [5,6,7]. Chloroplast ATP synthases (cFOF1) are also found to be mostly monomers [3, 8]. However, some evidence showed dimerization of cFOF1, which is probably connected with the regulation processes of ATP synthesis in chloroplasts [9]. Mitochondrial F-ATP synthases (mtFOF1) are arranged as dimers, tetramers, and, in some cases, hexamers [10], thereby determining the topology of mitochondrial cristae in eukaryotes [5]. Intriguingly, slight differences in the peripheral subunit composition may lead to the significantly different assemblies of ATP synthases among organisms, even if the assembly of the mtFOF1 dimer (type of dimer) remains the same [10, 11].
A vital importance of ATP synthase for cellular bioenergetics implies that dysfunctions of this enzyme result in a number of severe diseases with high fatality. Severe metabolic disorders, such as malfunctioning of tissues and organs and diabetes, are only a part of the problems that can be caused by improper functioning of ATP synthase [12, 13]. In addition, there is a hypothesis that ATP synthase might be involved in the formation of the mitochondrial permeability transition pore (mPTP) [14,15,16], also known as the mitochondrial mega-channel (MMC) [17,18,19,20,21,22], which represents a non-selective mega-channel in the inner mitochondrial membranes. The permeability transition phenomenon of acute swelling and uncoupling of mitochondria when exposed to high calcium concentrations and phosphate, oxidative stress, or other conditions leads to the dissipation of the proton motive force (PMF, promoting movement of protons across membranes downhill the electrochemical potential) and, consequently, to the cell death.
The membrane rotary part of the ATP synthases—c-ring and, in particular, its inner pore—might play a crucial role in proper functioning of the whole enzyme. Furthermore, a hypothesis suggesting that the c-ring itself might act as a mega-channel in the mPTP was proposed [14]. In our previous reports [4, 23], we hypothesised that the isoprenoid quinones might be present inside the inner pore of the c-rings in almost all organisms, especially in bacterial and chloroplast ATP synthases. They might form a complex dynamic interaction interface with the inner α-helices of the c-ring, and this interface is extremely important in stabilization of the c-ring, preventing ion leakage, and influencing the rate of the ATP synthesis.
The gaps in the knowledge of the molecular mechanism of the ATP synthase action can be filled mainly via structural studies of the enzyme. However, obtaining high-resolution structures of the protein complex is not sufficient. To clearly understand molecular mechanisms of stabilization of the ATP synthase during two synchronized rotation processes (α3β3 catalytic part and the c-ring in the membrane), it is necessary to track the conformational changes or obtain a series of high-resolution structures at different stages of the enzyme action [7]. Nevertheless, static high-resolution structures of ATP synthases are also extremely useful for the development of new therapies of various diseases by FOF1-structure-based drug design. One of the successful examples of using FOF1-structure-based approach was understanding the molecular mechanisms of a novel anti-tuberculosis (TB) drug bedaquiline (BDQ), based on the high-resolution structures of a c-ring [24] and the complete bFOF1 complex [25] from mycobacteria.
In this review, we highlight recent advances in structural biology of ATP synthases, systematize high-resolution structural data, and describe FOF1-structure-based approach to design new therapies. We demonstrate the similarities in structural organization of various ATP synthases found in the representatives of different phylogenetic groups. We discuss how to use these structural features for developing ligands capable of binding to the corresponding FOF1 and other possible ways of modulating FOF1 functioning. We emphasise such an approach involves information on high-resolution structural data of different families of FOF1.
Similarity and diversity of ATP synthases
In different organisms, ATP synthases demonstrate unexpectedly high diversity of their subunit composition. It is intriguing that almost all bFOF1 and cFOF1 have minimal differences in the subunit composition of the enzyme (α3β3γεδbb’can—9 types of subunits, except Mycobacteria, which lack the δ-subunit—it is an extension of the b-subunit of a peripheral stalk [25]). Furthermore, all these enzymes are mostly monomers, although subtomographic studies of chloroplasts showed that the population of the cFOF1 represents a distribution of oligomeric forms containing 85% of monomers, 12% of “pairs”/dimers, and 3% of higher oligomers of cFOF1 [8]. On the other hand, cryo-EM studies of spinach cFOF1 revealed that the enzyme is characterized by a monomeric structure that can be resolved to about 3 Å [3]. However, the sample preparation included reconstitution of the ATP synthase into nanodiscs with the scaffold protein MSP2N2. This means that the maximum diameter of these nanodiscs was about 145 Å [26], which is able to fit only a monomeric form of the cFOF1 (Fig. 1).
In contrast to bacteria, eukaryotes have significantly different subunit composition of ATP synthases, which are incorporated into the inner membranes of their mitochondria. Mitochondria themselves are thought to descend from alphaproteobacteria (a class of bacteria in the Proteobacteria phylum), and the bacterial intracellular membranes that were used to harness energy are thought to become the mitochondrial cristae [27]. These alphaproteobacteria had a monomeric ATP synthase, which further should have been modified to create dimers in the mitochondrial cristae. Therefore, the machinery, which determines the topology of the cristae [28], should have occurred simultaneously with the advent of the ATP synthase dimers. The transition from the monomeric bFOF1 to the mtFOF1 dimers added functionality to the enzyme. In fact, the determination of the cristae topology and, therefore, the morphology of the mitochondria in different species was the direct consequence of that transition [29].
mtFOF1
Interestingly, the core subunits of ATP synthases (α3β3γε, a-subunit and the membrane part of a c-ring) are present with slight modifications in different groups of the Eukaryota superkingdom. At the same time the peripheral subunits are significantly different, so that mtFOF1 has four types of dimers (Fig. 2).

The figure was slightly modified from ref. [4]
Phylogenetic tree of species with available structural data on F-type ATP synthases. The phylogenetic tree includes the species from two superkingdoms (Bacteria and Eukaryota), and eukaryotes from Viridiplantae, Opisthokonta, Sar, and Discoba clades (the representatives of the superkingdom Archaea have A-type ATP synthases and are not discussed in this review). The type of ATP synthase oligomeric state is determined for cellular membrane bFOF1 in the case of bacteria and for mtFOF1 in the case of eukaryotes. Some eukaryotes which have chloroplasts, and consequently cFOF1, are highlighted green. The letter “F” means the reported functional activity of purified and isolated FOF1. Structures of ATP synthases are marked with the corresponding PDB IDs. Unknown structures of ATP synthases are painted as a faded grey sketch with a question mark.
Currently Eukaryota superkingdom contains seventeen groups of different rank according to the NCBI taxonomy classification [30]. The structures of complete ATP synthases are available for representatives of only four of them: Opisthokonta, Sar and Discoba clades, and Viridiplantae. Interesting fact is that mtFOF1 dimers have differences in their subunit composition and consequently are characterized by different shape only between representatives of different groups [5].
The catalytic activity of purified and isolated ATP synthase was demonstrated only for limited cases [3, 25, 31, 32]. The rest of the papers reported the high-resolution structures of FOF1 do not report about validation of ATP synthesis activity.
Inside each group, mtFOF1 dimers belong to the same type and show minimal differences according to available information. For example, Opisthokonta clade representatives have V-shape mtFOF1 dimers of type I, which produce ~ 86° of membrane curvature (Fig. 3A, E). High-resolution structures are available for mtFOF1 from yeast [33] and porcine heart [34]. The mtFOF1 tetramer was described as the largest assembly for this group (Fig. 3A). On the other hand, the Viridiplantae representatives have V-shape mtFOF1 dimers of type II, which results in the less membrane curvature ~ 55°–56° (Fig. 3B, F) than that of the I type. Viridiplantae also have chloroplasts with the monomeric cFOF1, which can assemble to oligomers under specific conditions [9, 35, 36]. High-resolution structures are available for the mtFOF1 dimer from unicellular alga Polytomella sp. [37] and for the cFOF1 monomer from spinach leaves [3]. Dimeric form is the highest oligomeric state of the ATP synthases from this group (Fig. 3B). Sar clade representatives have mtFOF1 dimers of type III, which almost do not curve the membrane and have a U-shape structure (Fig. 3C, G). In this group, the representatives of Ciliophora phylum (Alveolata clade) have tetramers of mtFOF1 (two U-shape dimers), which determine the cristae morphology, as it was shown by solving the high-resolution structure of the mtFOF1 tetramer from Tetrahymena thermophila [11]. Interestingly, the representatives of the Apicomplexa phylum (Alveolata clade) have six ATP synthases assembling in hexamers that form bipentagonal pyramids with icosahedric symmetry, which determines the cristae morphology (high-resolution cryo-EM structure is available for the mtFOF1 hexamer from Toxoplasma gondii [10]).

Four types of mtFOF1 dimers. A–D Show cryo-EM structures of mtFOF1 and E, F—their arrangement in 3D volume cristae. A, E—the tetramers of mtFOF1 from animals & fungi (dimers of type I). B, F—the dimers of mtFOF1 from unicellular green algae (type II). C, G—the tetramers of mtFOF1 from ciliates and hexamers of mtFOF1 from Apicomplexa (dimers of type III). D, H—the dimers of mtFOF1 from Euglena and trypanosomes (dimers of type IV). E, F Were reprinted from ref. [38]. C, G were reprinted from refs. [10, 11]. H Was reprinted from ref. [39]
This is a new form of mtFOF1 organization in comparison with other known species, where the mtFOF1 complexes have a tendency to arrange into rows. Finally, representatives of Discoba clade have V-shape mtFOF1 dimers of type IV, producing approximately the same membrane curvature as that generated by the type II of mtFOF1 dimers (~ 55°–56°) (Fig. 3D, H). These organisms also have chloroplasts with monomeric cFOF1. High-resolution structure is available for mtFOF1 dimer from Euglena gracilis [40], and no structural data are currently available for the cFOF1 from any representative of the Discoba clade. Considering subunit composition of ATP synthases, in bacteria and chloroplasts, bFOF1 and cFOF1 are similar and consist of 9 or 8 types of subunits. In mitochondria of eukaryotes, the mtFOF1 form dimers of type I, II, III, and IV, and have 19, 18, 27, and 28 different types of subunits, respectively (Table 1) according to the high-resolution structures of the corresponding mtFOF1 complexes.
The interesting fact is that there might be different mtFOF1 assemblies from the same type of the mtFOF1 dimer defining different morphology of the mitochondria cristae. These assemblies are occasionally defined by slight changes in the peripheral subunit machinery. This fact is also important in terms of the roles of different cofactors and small-molecule ligands that participate in the assembly of oligomeric mtFOF1 complexes, especially the FO parts of ATP synthases (see details in the “Small-molecule cofactors of the c-ring”).
Interestingly, representatives of the Viridiplantae kingdom and the Discoba clade have both mitochondria and chloroplasts. Therefore, they carry two different ATP synthase species (mtFOF1 and cFOF1) in their proteomes. It might be very intriguing to learn how the ATP regulation is organized in these species in terms of the coordination of the biochemical processes as well as the structures of ATP synthases. However, no structural information about mtFOF1 and cFOF1 from one organism is currently available. Although there is a high-resolution structure of the cFOF1 from thylakoid membranes of spinach, there is no structural data about its mtFOF1. A vice versa situation is observed for alga (Polytomella sp.), where a high-resolution structure of the mtFOF1 dimer is available [37] but there is no structural information about the corresponding cFOF1. Therefore, the capability of the spinach mtFOF1 to form dimers of type II is only a hypothesis that needs to be directly confirmed by the high-resolution structural data.
cFOF1
Although the chloroplast ATP synthases were primarily found as monomers, in some cases they were actually reported as dimers [8, 35, 36], and biochemical evidence of their cFOF1 dimerization were shown. The existence of different cFOF1 (e.g. via F1–F1 interface) dimerization mechanisms is claimed, which might depend on the phosphate concentration. Structural data obtained by small-angle X-ray scattering for cFOF1 from spinach chloroplasts were reported [41]. However, the lack of high-resolution structural data of cFOF1 dimer does not allow understanding whether such dimers are functional or non-functional protein complexes.
bFOF1
Concerning bacterial ATP synthases it is necessary to mention that although there is no significant differences between different clades in the form-factors of their enzymes, representatives included in one clade show structural differences between their bFOF1. For example, bFOF1 from Mycobacterium smegmatis [25] and Bacillus sp. [32] belonging respectively to the Actinobacteria and Firmicutes phyla of the Terrabacteria group clade have 8 and 9 types of subunits and are characterized by different intersubunit interaction interfaces (less contacts within b/a interface, 35 a.a. extension of α-subunits, etc.) [25].
In addition, it would be interesting to investigate structures of ATP synthases from various bacterial and archaeal phyla from the point of view of their diversity and evolution. For example bFOF1 from Chloroflexus aurantiacus, a representative of Chloroflexi phylum, contains four copies of b-subunit per complex instead of usual two and the connection between FO and F1 moieties by the peripheral stalk of b- and δ-subunits is designed differently [42, 43]. Structural comparison of proton and sodium ATP synthases seems very perspective, as well as comparative structural analysis of bacterial and archaeal ATP synthases, seem very promising approaches.
Therefore, while highlighting the structural features of the bFOF1, one should pay closer attention to the phylum than to the clades for the corresponding organisms.
High-resolution structural studies
Since the first high-resolution structure (2.8 Å) of the F1 part of mtFOF1 from the bovine mitochondria was solved by X-ray diffraction (XRD) crystallography technique [44] many attempts were made to get the high-resolution structure of the whole complex. However, crystallization of the whole FOF1 ATP synthase is still a challenge. As a result, since 1999, there were only six successful attempts, which resulted in XRD high-resolution structures of ATP synthase from different organisms [45,46,47,48,49,50] (Table 2). These structures generated data crucial for understanding the molecular mechanisms of action of the important enzyme.
Firstly, the mechanism of the transmission of c-ring rotation to the conformational changes in the α3β3 domain powering ATP synthesis became clear. For example, the structure of monomeric yeast mtFOF1 showed ten c-subunits in the c-ring and close contacts between the c-ring and γδ subunits [45]. This allowed the authors to suggest an ensemble rotation of cγδ as a rigid body. In ref. [46], the structure of Mg-ADP-inhibited state of the yeast ATP synthase was described. The contacts between the c-ring/γδ were found to be formed by electrostatic forces, and Glu59 in the active center of the c-ring appeared not to be involved in the hydrogen bonding between c-subunits, therefore indicating that this residue does not determine a c-ring stoichiometry.
The paradigm of structural organization of ATP synthases changed when the c-rings with a stoichiometry that is not multiple of three were discovered. Besides observing this in the first structures of yeast mtFOF1, for spinach chloroplast c-ring it was also shown that it has 14 c-subunits instead of 12 as assumed before [51], and a c-ring with eight subunits was later discovered in the bovine mtFOF1 [47].
The tilt between the F1 central stalk and the c-ring [48] provided understanding of a flexibility of ATP synthase during its functioning. The work reported in ref. [6] demonstrated twelve c-subunits in bacterial ATP synthase from Paracoccus denitrificans, described the structure of peripheral stalk bb’ subunits, and a-subunit in FO part of bFOF1. This was one of the first studies, where a close interaction interface of subunit a with the c-ring was shown, and a mechanism of proton transfer through the a/c-ring interface via two half-channels formed by the a-subunit was suggested. This provided a better understanding of the molecular mechanisms of converting proton gradient electrochemical energy to ATP synthesis. The studies of α3β3 active center pointed to a native regulation by Mg-ATP and Mg-ADP inhibitors of ATP synthases [50].
The first Cryo-EM structure of FOF1 (resolution 20 Å) was obtained in 2003 [56]. Due to the significant progress in cryo-EM technique, more high-resolution structural data of ATP synthases became available. The resolution of cryo-EM-based structures of the enzyme was improved from 20 Å in 2003 to 2.5 Å in 2020, and an increasing number of structures of the complete FOF1 was collected during the recent 5 years [3, 7, 10, 11, 29, 31, 32, 34, 37, 40, 50, 52,53,54,55, 57]. These studies revealed the structural and functional organization of ATP synthases and their assemblies, helped understanding the chemo-mechanical coupling and transmembrane proton transport mechanisms, and revealed how organisation of the mtFOF1 influences the cristae topology and defines a morphology of mitochondria.
The fact that mtFOF1 can assemble into dimers, which determine the topology of cristae in mitochondria of mammals, plants, and fungi was demonstrated in refs. [8, 58,59,60] However, for a long time, the mechanisms determining the form of ensembles of mtFOF1 dimers remained hidden. The understanding that structural elements of FO are responsible for dimerization and larger assemblies was developed by subsequent studies [29]. The structural organization of the a-subunit was established [52] for the bovine mtFOF1, and the following studies [53] discovered that four α-helices of the a-subunit arrange parallel to the membrane plane. This arrangement creates the invaginations in FO called half-channels which establish proton transport via a/c-ring interaction interface.
Revealing the existence of the different conformational states of ATP synthases allowed modelling the whole rotational cycle of the enzyme [31, 53]. The participation of FO subunits in the ATP synthase functioning revealed that ATP8 subunit provides an additional brace between the peripheral stalk and the a-subunit in yeast mtFOF1, and flexibility of the peripheral stalk bdh subunits allows a-subunit to adjust lateral movements during the activity of F1 part [54]. The involvement of FO subunits in the ATP synthase dimerization process revealed that the f-subunit establishes FO-FO contacts, whereas e- and g-subunits determine a membrane curvature of ∼100° forming so-called V-shape dimer of yeast mtFOF1 [50]. Further improvement of the yeast mtFOF1 V-shape dimer model showed [57] that subunits f,8,b determine the base of a peripheral stalk with d-subunit, which acts as a clip. Subunits a, i/j are also responsible for the dimerization and the b-, e-, g-, and k-subunits define a membrane curvature for the mtFOF1 V-shape dimer.
High-resolution structures of entire ATP synthases and their parts opened a possibility of designing small-molecule cofactors capable of binding to ATP synthases and modulating or inhibiting their activity. For example, the detailed structure of the FO part of the yeast mtFOF1 allows suggesting that the ATP synthase inhibitor oligomycin binds to the c-ring residing in the lipid surrounding [55]. More details of functioning of cFOF1 from spinach chloroplasts were summarized in ref. [3], and the adaptation mechanism of chloroplast ATP synthase redox potential to the day-night cycle was uncovered.
Structural studies of mtFOF1 from green algae Polytomella sp. demonstrated flexibility of the a-subunit during rotation of the c10-ring and discrete ATP synthesis [37]. In addition, ten subunits composing a rigid peripheral stalk established dimerization contacts between two enzymes and participated in the formation of an mtFOF1 dimer of type II (see the “Similarity and diversity of ATP synthases”). This type of mtFOF1 dimers was demonstrated to have an ion (presumably Zn2+) coordinated by the His-248 and His-252 of aH5 of a-subunit, and it possibly synchronizes protonation of the c-ring active center with its rotation.
Another type of dimeric organization (type IV) of mtFOF1 was reported in ref. [5] The work showed a number of peripheral subunits, associated small-molecule cofactors, such as cardiolipins and other phospholipids, and unusual structural organization of a c-ring, where the extension of the c-subunits form a β-barrel that have contacts with ATPEG1-subunit [40].
Very recently, high-resolution structures of ATP synthases revealed more complex assemblies of these enzymes for dimeric types I, III, and IV (corresponding oligomerization states are 4, 4 and 6, respectively). The structure of the mtFOF1 tetramer from mammalian mitochondria revealed that subunits b, k, g, and inhibitory factor 1 (IF1) are responsible for connecting two V-shape mtFOF1 dimers and form a H-shape tetramer [34]. The authors also suggested that a 6.8PL subunit is inside the c-ring pore. The authors proposed three different mechanisms of mtFOF1 inhibition: by IF1, by Mg-ADP and by e-subunit establishing contacts with the 6.8PL subunit.
The structure of the mtFOF1 tetramer from ciliates was recently resolved [11]. This mtFOF1 forms U-shaped dimers of a type III. The peripheral subunit machinery establishing dimerization was determined including a subunit ATPTT2 which occurred to be a single copy in the mtFOF1 dimer. Two U-shape dimers create a tetramer that determines an angle of membrane curvature of ~ 11°. Interestingly, small-molecule cofactors, such as cardiolipins, NAD, and CoQ-8, were found associated with the subunits in FO and their function remains unknown.
The structure of the mtFOF1 (V-shape dimer of a type IV) from apicomplexan parasite revealed hexameric cyclic assembly of the enzyme that forms pentagonal pyramids [10]. The subunit ATPTG11 was found responsible for assembling the hexamers, and its knock-out resulted in the disruption of the pentagonal nets of the mtFOF1 hexamers and altered cristae morphology.
Besides mtFOF1, bacterial ATP synthases were also studied by cryo-EM method. The structure of recombinant bFOF1 helped clarify the mechanisms of enzyme inhibition by the ε-subunit and the proton pathway through the membrane [32].
It is worth noting that successful crystallization trials of ATP synthases were done by vapour diffusion (VD) method, which however, has limitations, such as leading to rapid kinetics of molecules in solution and destabilization of membrane proteins. Therefore, the VD method is commonly used for crystallization of water-soluble proteins and is not optimal for membrane protein complexes [61]. This might also explain the fact of the existence of only partial crystallization of ATP synthases (separate domains or subunits) by VD method even when using the whole protein complex as a starting crystallization material [62].
Crystallization in lipid mesophases (in meso method) is a promising technique to obtain crystals of complete membrane protein complexes, such as FOF1 because it uses membrane-mimicking systems which provide more native conditions for membrane proteins than VD method does [63, 64]. However, there are limitations on size of membrane systems when using in meso-crystallization method. A lipid cubic phase (LCP) forms two non-intersecting nets of water channels. The size of the channel diameter is limited by the physico-chemical properties of a matrix lipid that is used to prepare LCP and the composition of a precipitate, where the LCP with the membrane protein is embedded (Fig. 4A, B). F1 part of ATP synthase is approximately 160 Å, which is about 3–4 times larger than the average diameters of the water channel of common LCPs (Fig. 4C) [65].

The figure was taken from ref. [4]
A Schematic representation of a lipid cubic phase (LCP) of Im3m type of symmetry. The lattice parameter (LP) and the diameter of water channels (dw) are shown. B A schematic reconstruction of one ATP synthase molecule in the LCP. Subunits colored in red do not fit in the unit cell of the LCP. C Required dw and LP and completeness of the ATP synthase complex that can be reconstituted in corresponding LCPs of different types of symmetries are shown. The PDB model 6FKF of ATP synthase from spinach chloroplasts was used for illustration [3].
It might be also useful to swell lipid mesophases by using charged glycerophospholipids (1,2-Distearoyl-sn-glycero-3-phosphoglycerol) (DSPG) [66]. However, this method works well only if a low-salt precipitant (not higher than 150 mM NaCl of ionic strength) is used. Precipitants with high salinity will shrink the LCP and decrease the size of water channel diameters.
Cryo-EM techniques are commonly working successfully with proteins larger than 100 kDa, which is a suitable case for studying ATP synthases (Mw > 500 kDa), and they have recently allowed improving a resolution up to 2.5–3.0 Å in the case of the F1 part. Unfortunately, the resolution of the FO part hardly reaches 3.0–3.1 Å. The lack of the structural information makes it difficult to clearly understand the molecular mechanisms of intersubunit interactions, which play key roles in functioning of the enzyme.
A possible way for obtaining a high-resolution structure of ATP synthase uses complementary XRD and cryo-EM techniques. In this approach, XRD can allow obtaining high-resolution structures of separate membrane subunits, which can be crystallized in meso, and the cryo-EM could provide high-resolution structures of F1 part of ATP synthase. Then the mosaic model can be constructed as it was done in ref. [50] (see Table 2).
Intrinsically disordered regions in ATP synthases
High-resolution structural studies of ATP synthases is still a challenge, despite great recent advances that were achieved with new developments of X-ray crystallography or cryo-EM studies of membrane proteins. There are several reasons for that in the case of complex structural organization and transmembrane nature of such proteins. Another important factor that is commonly hindering the attempts of the crystallographic analysis of proteins is the presence of intrinsically disordered regions [67]. Among well-known examples of disordered proteins recalcitrant to crystallization is a myelin basic protein (MBP) [68]. In fact, due to the exhaustive series of attempts to crystallize MBP for X-ray diffraction, where 4600 different crystallization conditions failed to induce crystallization of MBP, this protein was included to the category of “uncrystallizable” proteins [69]. Even proteins with solved crystal structures can possess noticeable levels of intrinsic disorder, since very significant fraction of proteins in PDB contains regions with missing electron density, which are potentially intrinsically disordered [70, 71]. In line with these considerations, analysis of the PDB structure 6FKF of cFOF1 from the spinach chloroplasts revealed that each subunit of this machine contains variable regions of missing electron density: residues 1–7 and 504–507 in chain α; residues 1–17 and 495–498 in chain β; residues 1–42 and 364 in chain γ; residues 1–70 and 250–257 in chain δ; residues 132–134 in chain ε; residues 1–21 and 245–247 in chain a; residues 1–22 and 183–184 in chain b; residues 1–77 and 221–222 in chain b’; and residues 1–2 in chain c. Therefore, very significant parts of each cFOF1 subunit are expected to be disordered even within the assembled complex. Figure 5 further illustrates this concept by showing per-residue disorder profiles generated for each cFOF1 subunit.

Per-residue intrinsic disorder predisposition of the subunits of cFOF1 from the spinach chloroplasts. Intrinsic disorder profiles were generated based on the outputs of six commonly used disorder predictors: PONDR® VLXT [72], PONDR® VL3 [73], PONDR® VLS2 [74], PONDR® FIT [75], and IUPred2 (Short) and IUPred2 (Long) [76]. The outputs of the evaluation of the per-residue disorder propensity by these tools are represented as real numbers between 1 (ideal prediction of disorder) and 0 (ideal prediction of order). A threshold of 0.5 is used to identify disordered residues and regions in query proteins. Residues with the disorder scores (DS) 0.25 ≤ DS < 0.5 are considered as moderately disordered, whereas residues with 0.15 ≤ DS < 0.25 are flexible. The figure clearly shows that all subunits contain noticeable levels of intrinsic disorder, with some of them being rather disordered
The corresponding intrinsically disordered regions of ATP synthases might help to understand the mechanisms of regulation of the enzymes. For example, intrinsically disordered proteins (IDPs) were found to regulate activity of ATP synthases from bacteria (E. coli, Paracoccus denitrificans) (Fig. 6A, B) as well as from mitochondria of eukaryotes (S. cerevisiae) (Fig. 6C) [77]. The activity regulation of chloroplast ATP synthases occurs via a redox switch on its γ-subunit [78]. Figure 5C shows the presence of intrinsically disordered regions in γ-subunit (residues 1–42 and 364), however, the structural changes of the redox switch are in another region of γ-subunit (residues 238–282) [78]. Therefore, additional experiments are required to reveal how the mechanisms of activity regulation of ATP synthases can be investigated in terms of IDPs.

The figure is from [77]
The structures of inhibited ATP synthases by intrinsically disordered regions of their subunits (colored in red). A bFOF1 from E. coli inhibited by ε-subunit, B bFOF1 from Paracoccus denitrificans inhibited by ζ-subunit, C mtFOF1 from S. cerevisiae inhibited by IF1 subunit.
However, even when high-resolution structures of query proteins are not available, there is always a possibility to try different organisms belonging to the same phylogenetic group as the organism of interest (e.g. pathogen), if one can properly account for the structural features of their FOF1, which are described in this review. There are a number of hints on which type of structural features might be used to affect or modulate the activity of ATP synthases. In the case of mitochondrial [11] and chloroplast [23] ATP synthases, the presence of isoprenoid quinones as the FO cofactors might be a strong indicator that different derivatives of ubiquinones (UQ) (mitochondria), plastoquinones (PQ) (chloroplasts) and menaquinones (MK) (bacteria) might influence the functioning of ATP synthases and can become the tools of cellular bioenergetic control. We discuss here a generalized approach based on the relevant structural information on the ATP synthases, which might be helpful in the further studies, development of new therapies, and structure-based drug design.
Validation of ATP synthase functionality
Structural information about ATP synthase is commonly validated via the functional activity assays of the investigated enzyme. Typically, functional studies are done by incorporating ATP synthases into liposomes and then checking the activity by the luciferin-luciferase assay [79,80,81,82].
A typical protocol includes preparation of the unilamellar vesicles (ULVs) of phosphatidyl choline lipids (PC) and reconstitution of the ATP synthase there. Here, ULVs are the spherical vesicles bounded by a single lipid bilayer. ATP synthase is initially solubilized in detergent (DDM, tPCC-α-M, digitonin, etc.). Thereafter, Bio-Beads or Amberlite XAD-2 resin is used to remove detergent and trigger incorporation of the enzyme into ULV bilayers (Fig. 7A). Detailed description of the protocol can be found in ref. [62].

Quantitative analysis of ATP synthase activity by ATP synthesis luciferin-luciferase test. A Schematic model of a proteoliposome with reconstituted ATP synthases is shown with the imposed ΔpH and Δμ(K+) gradients to energize ATP synthesis. B Schematic and C experimental data of the luminescence signal. C is from ref. [62] and the figure is from ref. [4]
The luciferin-luciferase assay is based on the capability of luciferase to hydrolyse ATP to AMP and luciferin to oxyluciferin emitting light during this chemical reaction, which can be detected by spectroscopy (Fig. 7B, C). Based on these data, the efficiency of ATP synthesis (ATP/sec/enzyme) is calculated in comparison with the ATP standard added (Fig. 7B, C).
The method for reconstitution of FOF1 into liposomes, energisation of the liposome membrane by an acid base transition and measurement of ATP synthesis (using radioactive phosphate as well as luciferin/luciferase) was developed for optimisation of the isolation of the FOF1 from chloroplasts. It resulted in isolated cFOF1 with all subunits showing activities of about half of the activity observed in thylakoid membranes. Reconstitution and ATP synthesis activity of the cFOF1 was reported first in ref. [83].
Reconstitution in liposomes with full ATP synthesis activity of a mtFOF1 driven by an acid–base transition was reported first in ref. [84]. H+/ATP ratio of proton transport-coupled ATP synthesis have been first described in ref. [85]. In addition, mtFOF1 reconstituted into liposomes together with a photosensitive membrane protein bacteriorhodopsin showed light-induced ∆pH-driven ATP synthesis [86].
In earlier work the luciferin/luciferase was used to determine the amount of ATP synthesized in an acid base transition using a separate assay. This allows one to optimize the conditions for energisation with an acid base transition and, in the separate assay, the optimal conditions for the detection of ATP with luciferin/luciferase can be used [83].
These experiments revealed that different ATP synthases have different rates of ATP synthesis and even the same ATP synthase can be characterized by noticeable difference in its ATP synthesis rate. For example, cFOF1 from Spinacia Oleracea has a measured efficiency ranging from 40 to 200 ATP/FOF1/sec in similar functional studies conducted in different groups [62, 87, 88].
Bacterial FOF1 was reported to be reconstituted into liposomes and its ATP synthesis activity was measured firstly by Fischer et al. in 1994 [79]. Bacterial ATP synthases were reported to have lower ATP synthesis rate, e.g. ~ 11–15 ATP/sec/enzyme for bFOF1 from Ilyobacter tartaricus [89].
Surprisingly, functional activity measurements were reported only for several FOF1 from Fig. 2 and only for monomers of FOF1. It could be interesting to measure the functional activity of dimeric FOF1 and compare it with one of monomeric FOF1.
ATP-dependent luciferases from firefly or others are used for in vivo functional studies of ATP synthases [90, 91], for purified mitochondria or chloroplasts [92, 93], and for two-hybrid systems to evaluate the interaction of cofactors with ATP synthases [94]. A perspective approach might be the usage of combined sensory systems of eukaryotic (ATP-dependent) and bacterial (FMN—NADH-dependent) luciferases [95, 96] for functional studies of respiratory chain proteins and ATP synthases both in vivo and in vitro [97], as well as ATP-dependent processes such as refolding of thermoinactivated proteins [98].
Vinkler et al. [93] investigated the ATP synthesizing system of chloroplasts with electric field pulses and for ATP detection with the luciferin/luciferase included in the reaction medium. However, the results described in this work mainly seem to be artefacts resulting from electrode reactions and from the effect of the temperature jump generated by the voltage pulse in combination with the adenylate kinase omnipresent in chloroplasts.
Mitochondrial permeability transition pore (mPTP)
One of the most intriguing phenomena in mitochondria is the existence of the mitochondrial permeability transition pore (mPTP), also known as the mitochondrial mega-channel (MMC). Although this phenomenon has been intensively discussed during the last several decades, structural information about its molecular mechanisms is still missing. Importance of the mPTP is defined by the fact that activation of this pore triggers cell death (apoptosis and necrosis). PTP is defined as a Ca2+-dependent non-selective channel permeable to ions and solutes with a molecular mass below 1500 Da. Although its maximal conductivity reaches 1.5 nS, for a number of substates the lower conductivity is observed [15].
In mammals, the PTP is positively modulated by binding of cyclophilin D (CyPD) and inhibited by cyclosporin A (CsA). Phenomenologically, the mPTP activation is characterized by high ion conductivity, rapid membrane depolarization, and cellular death. According to the recent discussions, mPTP is an assembly that might be either adenine nucleotide translocase (ANT) or/and ATP synthase [15, 99]. This model is based on the observations that the purified ANT, as well as purified ATP synthase were reconstituted into liposomes and showed electrophysiological characteristics similar to mPTP measured by patch clamp technique (conductivity of 300–600 pS for ANT [100] and 1–1.3 nS for ATP synthase [101], respectively). However, the mPTP phenomenon was shown to persist after the knockdown of ANT [102] and the c-subunit of ATP synthase expression [103].
Below we briefly describe the dominating hypotheses of the pore formation via ATP synthase and discuss how the oligomeric assemblies of the enzyme might form a huge non-selective channel in the mitochondrial inner membranes.
The c-ring hypothesis
The c-ring hypothesis suggests that the c-rings under the physiological conditions are capable of non-selective conductivity (Fig. 8A). The most notable evidence was described in ref. [104], demonstrating that the purified c-rings show biophysical characteristics similar to mPTP, but are not sensitive to Ca2+ and are not regulated by CsA. Therefore, a hypothesis was proposed that the regulatory elements are located in the F1 part, and the pore itself consists of c-rings. F1 participates in the inhibition of mPTP, and during the formation of a pore, F1 dissociates from FO. It was shown by fluorescence microscopy applying the bipartite tetracysteine display (FlAsh) method, which indicated that the structures of open and closed pores are different.

Different hypotheses of ATP synthase involvement in mPTP phenomenon. A The c-ring hypothesis. B ATP synthase oligomer hypothesis suggesting that mPTP might occur between of FO parts of ATP synthase monomers. C A “death finger” hypothesis suggesting that conformational changes induced by Ca2+ and CyPD lead to removal of e-subunit and of a lipid plug by conformational changes via b-subunit. A, B were adapted from ref. [15]; C from ref. [113]
The theory of the mPTP formation via c-rings was often criticized [105,106,107]. Briefly, the group of Walker [107] showed that the channel inside the c-ring is hydrophobic and has the shape of a sand clock, narrowing from 14.2 to 10.4 Å in the middle. Based on these structural data, they suggested that the formation of a pore with conductivity up to 1.5 nS from c-rings is not possible. Along the same vein, Bernardi [105] claimed that (1) dissociation of F1 from FO occurs under extremely specific conditions (2 M urea), and when returning to physiological conditions, reverse assembly occurs; (2) the assumption that the β-subunit can close the FO channel is quite controversial, since normally the β-subunit has no affinity for c-rings, and the presence of a free β-subunit itself is rather unlikely; (3) the induction of Ca2+ mPTP in intact/complete ATP synthase can be suppressed by the addition of CsA, and it is not clear how this can happen if F1 has already completely dissociated from FO.
In addition, based on the atomic modeling [106] it was shown that (1) the hydrated c-ring does not exhibit the properties of mPTP; (2) under physiological conditions, the c-ring lumen part is not hydrated and is plugged with lipids. All this was confirmed for the cases of the c10-ring from Saccharomyces cerevisiae, and for the c13-ring from B. pseudofirmus. The models without two c-subunit monomers also showed that the c-ring remained clogged with lipid molecules. Therefore, the formation of a pore by the ATP synthase c-rings in their physiological conditions is still questionable.
The oligomer hypothesis
Another hypothesis suggests the formation of a pore between two monomers of the ATP synthase dimer or between four monomers of the ATP synthase tetramer (Fig. 8B). This model [101] is based on the observations that dimers, but not monomers of the ATP synthase, after blue native gel purification and reconstitution into the lipid bilayers in the presence of Ca2+, can open a channel with currents that were typical of the mitochondrial mega channel. The channel opening was inhibited by the ATP synthase inhibitor AMP-PNP and Mg2+/ADP. Based on the additional considerations [108,109,110], it was suggested that the pore is formed at the dimeric interface.
The main refutation of this hypothesis was presented in [111], where the authors showed that mPTP requires the dissociation of ATP synthase dimers. Furthermore, the authors of other study [112] showed that the ATP synthase monomer was sufficient for the formation of a pore and criticized previous publications [101, 110], where the oligomeric state of the ATP synthase reconstituted in liposomes was not verified unambiguously.
In the beginning of 2020, Walker et al. claimed [16] that based on the knockout data of various ATP synthase subunits, it can be argued that mPTP is not necessarily associated with the ATP synthase dimer.
In conclusion [16], it seems that the final answer on the possibility of the mPTP formation via the ATP synthase dimer will be obtained via determining the high-resolution structure of the ATP synthase dimer. In 2020, the dimer structure was obtained by the J. Walker group using Cryo-EM technique [107], and it was shown that the c-rings cannot form a pore under native conditions. However, there was no statement about the model of the pore between the dimeric FO parts of ATP synthase.
The “death-finger” hypothesis
Despite the evidence against the hypothesis of pore formation via c-rings, research towards this hypothesis was continued, and new evidence supporting this model was recently obtained [112,113,114]. As a result, currently, the modified model of the mPTP formation via c-rings is suggested. This modified model of the mPTP formation via a c-ring is as follows. High Ca2+ levels lead to the displacement of Mg2+ from the pocket in the β-subunit and causes conformational changes in F1, which eventually lead to the separation of the e-subunit from the c-ring and the stretching of the lipid plug by conformational changes via the b-subunit (Fig. 8C). At the initial stage, these changes are reversible, but eventually they lead to the separation of the F1 part and the increase of the pores of c-rings to 2–3 nm [113]. It is quite possible that this increasing in the diameter of the c-ring pores occurs similarly to the formation of non-specific pores by amyloidogenic proteins [114]. Next (or simultaneously) this leads to the destruction of the oligomeric interaction interfaces and the dissociation of the dimers and tetramers of ATP synthase [111]. It is assumed [112] that the formation of mPTP occurs at the level of the ATP synthase monomer, and the decay of the oligomers is a consequence, but not the cause of this phenomenon. In this study, not only patch clamp measurements were performed with ATP synthase reconstituted in small unilamellar vesicles (SUVs), which showed that ATP synthase forms a voltage-gated and Ca2+—dependent channel, but also the monomeric state of ATP synthase was confirmed by the Cryo-EM.
Thus, there are different hypotheses on the nature of mPTP and additional experiments are required to reveal a real mechanism of the transition.
C-ring
An intriguing fact is the diversity of the molecular mechanisms utilized for stabilization of ATP synthases in their rotary part—c-ring. Different organisms have different contexts of interaction interfaces of their c-subunits. In fact, their ATP synthases can be found in completely different surroundings, show different subunit stoichiometry of the c-ring (Fig. 9), possess variability of other subunits of the ATP synthase that are interacting with the c-ring, and sometimes even contain the extended polypeptide chain of the c1-subunit [40].

Phylogenetic tree showing the c-ring stoichiometry of different organisms. The histogram illustrates the number of the c-subunits. The stoichiometry of eukaryotic H+ mitochondrial c-rings is colored red and H+ chloroplast c-rings—green; bacterial H+ c-rings—purple and Na+ c-rings—orange. Acetobacterium woodii is colored pink due to a hybrid F/V type Na+ c-ring with a stoichiometry of 9F/1V. Red and green question marks indicate unknown stoichiometry of mtFOF1 and cFOF1 c-rings for the corresponding species, respectively.
The figure was reprinted from ref. [4] with minor modifications
The stoichiometry of c-rings from different organisms is different and ranges from 8 to 17 subunits (Fig. 9). In the case of eukaryotes, the most common numbers of mitochondrial c-subunits are 8 and 10. Bacteria show more diverged c-ring stoichiometry, which is ranging from 9 to 17, with high variability being sometimes observed within the closely related organisms (Fig. 9). Some bacterial species have a hybrid F/V type Na+ c-ring with 9F/1V stoichiometry, though the form factor of this c-ring is similar to a c-ring with 11F protomers [115].
One of the important details here is that the c-ring might be involved in the mPTP formation. However, as we have discussed, determination of the exact molecular mechanisms of this phenomenon is still a big challenge. Based on the diversity of the stoichiometries of c-rings in different organisms (Fig. 9) and different context of interaction interfaces between a c-ring and surrounding subunits of ATP synthase (Fig. 10), it seems that the different types of mtFOF1 dimers will undergo different pathways of disassembly, therefore presenting diversified mechanisms of the processes of mPTP formation in eukaryotes. There are also interesting questions on the existence in bacteria or chloroplasts of the processes similar to mPTP in mitochondria and molecular mechanisms that could underlie these processes.

The figure was reprinted from ref. [4] with modifications
Interaction interfaces for different types of mtFOF1 dimers with a focus on the c-ring. A mtFOF1 dimer of type I has a PL6.8 subunit inside the inner pore of the c-ring. B mtFOF1 dimer of type II has a large peripheral stalk stabilizing the c-ring. C mtFOF1 dimer of type III has an ATPTT6 subunit with a cardiolipin inside the inner pore of the c-ring. D mtFOF1 dimer of type IV has an extended polypeptide chain at the N-terminus, which forms a β-barrel establishing sliding contacts with ATPEG-1 subunit.
C-rings differ not only by the number of c1-subunit copies, but also by the secondary structure of the polypeptide chains. Typically, in most known cases, the c1-subunit consists of two α-helices connected by a conserved loop, which establishes contacts between a c-ring and a central stalk of an ATP synthase. Two transmembrane α-helices create a so-called hairpin, so that the N- and C-termini of the c1-subunit are on the same side and are very close to the membrane surface. However, the representatives from the Discoba clade have an extension at the N-terminus of the c1 polypeptide chain, which forms a β-barrel serving as an interface, which connects the c-ring with the ATPEG-1 subunit via sliding contacts (Fig. 10D).
Small-molecule cofactors of the c-ring
The question on what compounds can be found inside the inner pore of the c-rings of ATP synthases from different organisms was risen 2 decades ago while investigating the c11-ring of bFOF1 from Ilyobacter tartaricus [116] and the c14-ring of cFOF1 from spinach chloroplasts [51, 117]. Some authors claimed the lipidic nature of the so-called “plug” inside the central pore of c-rings and showed AFM data [116] or results of computer modelling [118, 119], whereas other authors claimed the proteinaceous nature of the “plug” [117]. Nowadays, due to the existence of the high-resolution structural information for ATP synthases from different species one can observe that the nature of the “plug” of the c-rings from mtFOF1 can be either lipid or protein or protein/lipid. In case of mtFOF1 dimers of type I, it was shown that a subunit PL6.8 is inserted inside the inner pore of the c8-ring of mtFOF1 from the porcine heart [34]. In case of mtFOF1 dimers of type III, a cardiolipin was found inside the pore of the c10-ring [11]. The other types of mtFOF1 dimers (II and IV) might have a lipid “plug” preventing ion leakage through the corresponding c-rings [37, 40].
Concerning the chloroplast ATP synthases the purified samples of the c-ring from the ATP synthase of spinach chloroplasts were firstly published in 1987 [120]. Later, the similar samples were reported to contain small-molecule supplements despite the harsh conditions of sample purification (1% w/v N-lauryl sarcosine (NLS), 65 °C) [87]. The structural evidence of the possible presence of small-molecule supplements inside the c-ring inner pore was presented in refs. [23, 121] based on the analysis of high-resolution crystallographic data (Fig. 11A–D). Another evidence of the presence of small molecules, such as beta-carotene or chlorophyll a, is also indicated by the corresponding color of the c-ring crystals [62, 87] (Fig. 11E–J), studies with UV–Vis spectroscopy (Fig. 11K) and reversed-phase HPLC (Fig. 11L) [87].

The figure was reprinted from ref. [4] with modifications
A, B [121], C, D [23] Crystallographic data of the c14-ring from spinach chloroplasts demonstrate additional electron densities inside the inner pore of the c-ring. E–J A gallery of the crystals of the c14-ring from spinach chloroplasts [23, 62, 87]. K UV–Vis spectra of dissolved c14-ring crystals and L the reversed-phase HPLC diagram pointing to beta-carotene and chlorophyll a at 480 and 665 nm, respectively [87].
Additional experimental evidence of possible presence of small molecules in c-rings was reported in our previous work [23] (Fig. 12), where the purified samples of the c14-ring from spinach chloroplasts were investigated by UV–Vis differential spectroscopy before and after a qualitative reaction with NaBH4. The difference spectra between an oxidative and a reduced states of the samples showed the similar fingerprint as for a quinonic compound—trimethyl benzoquinone (TMBQ), which is a polar moiety of a plastoquinone-9 (PQ-9) (Fig. 12A, D, E).

The figure was reprinted from ref. [4]
Differential UV–Vis spectroscopy of the purified c14-ring from spinach chloroplasts dissolved in EtOH. A “Ox-redox” differential spectra of the purified c-ring and TMBQ (colored in red and black, respectively). B UV–Vis spectra of the c-ring samples before (ox) and after addition of NaBH4 (redox) (black and red lines, respectively). C A control experiment with pure EtOH before and after addition of NaBH4 (black and red lines, respectively). Schemes of reducing D TMBQ to TMBQH2 and E PQ9 to PQ9H2.
In line with these observations, additional examples of the interaction of different isoprenoid quinones with ATP synthase were presented [11]. The authors demonstrated a direct interaction of UQ-8 with subunits in FO part of a tetrameric mtFOF1 from Tetrahymena thermophila.
Initially, hypothesis that isoprenoid quinones might be cofactors of ATP synthases from different organisms was proposed in [4, 23]. The authors showed the presence of additional positive electron densities inside the c-ring from spinach chloroplasts and found similar densities in the available high-resolution structures of c-rings from bacteria and mitochondria of eukaryotes. This hypothesis gains stronger evidence if one calculates the distances between the polar/apolar interfaces inside the c-rings from different organisms and compares them with the corresponding isoprenoid quinones that are natural components of their membranes (Table 3) [4].
Since 2019, a boom of cryo-EM studies on the ATP synthases resulted in the emergence of high-resolution structures of mtFOF1 from different species, where the electron densities observed inside the pore of the corresponding c-rings were annotated as a cardiolipin molecule [11] (mtFOF1 dimer of type III), or α-helix of PL6.8 subunit [34] (mtFOF1 dimer of type I). Intriguingly, if there are isoprenoid quinones inside the c-rings of mtFOF1, there should be complex interaction interfaces between a lipidic mixture or a protein-lipid mixture and the inner surface of the c-ring (Fig. 13B) [4].

The figure was reprinted from ref. [4]
Schematic picture of possible interaction interfaces of c-rings from different organisms. Isoprenoid quinones are shown in orange. A The c14-ring from Spinacia oleracea (6TQJ) [23] and PQ-9 molecules. B The c10-ring from Saccharomyces cerevisiae (4F4S) [122] and the possible UQ-6/6.8PL subunit interface. C The c9-ring from Mycobacterium phlei (4V1H) [123] and MK-9 molecules. D The c13-ring from Bacillus pseudofirmus (4CBK) [123] and MK-7 molecules. E The c15-ring from Arthrospira platensis (2XQU) [124] and PQ-7 molecules.
In case of bacterial and chloroplast ATP synthases the densities were not annotated in the literature due to low resolution of the corresponding structures. In addition to what is known, there is no large peripheral machinery in bFOF1 and cFOF1 therefore it is highly likely that the plug inside corresponding c-rings might consist of isoprenoid quinones or at least a mixture of lipids and quinone molecules (Fig. 13A, C, D, E) [4].
ATP synthases as potential therapeutic targets
Recent advances in the structural biology of ATP synthases directly demonstrated that representatives of different taxonomy groups carry unique structural features of this enzyme, thereby representing potential targets for drug design and gene therapies, or in more general context, open new possibilities towards the development of promising tools for cellular bioenergetics control. For example, it was already shown [10, 11, 29, 125] that a specific architecture of the mitochondria cristae is formed by different mtFOF1 oligomers (see Fig. 3), and their disassembly to mtFOF1 monomers leads to the loss of cristae shape and swelling.
World Health Organization (WHO) presented a list of the diseases caused by the different pathogens that require new antibiotics for treatment (Table 4) [126]. We added to this list a taxonomy classification of the corresponding pathogens to highlight, which structural features of their ATP synthases might be used for therapies.
A successful case of drug design based on the structural features of ATP synthases is bedaquiline (BDQ), a drug developed against tuberculosis (Fig. 14). The pathogen Mycobacterium tuberculosis carries the same structural features of bFOF1 as Mycobacterium smegmatis, for which the high-resolution ATP synthase structure was solved by the cryo-EM and demonstrated an inhibition of the enzyme by BDQ via binding of this molecule to the c9-ring active centers of the bFOF1 from mycobacteria [25]. In 2015, the molecular mechanism of BDQ binding was elucidated by X-ray crystallographic analysis of a crystal of the c9-ring [24]. Nowadays, the studies of BDQ efficiency showed high culture conversion rate (65–100%) and a satisfactory treatment outcomes [128].

The figure was modified from [25]
Among human pathogens, a significant part is represented by protists. Their ATP synthases can become a potential target for new antibiotics and inhibitors. Since the mtFOF1 from different species within the same group are structurally similar, it makes them prospective targets for ligands and small-molecule drugs or other therapies targeting ATP synthases. To date, high-resolution structures are available only for four groups of eukaryotes (see Fig. 2).
Existing structure of mtFOF1 from Euglena gracilis [40] reveals structural features of the mtFOF1 (type IV dimer) belonging to the Discoba clade. This clade also includes pathogens from the genus Leishmania [129] that cause leishmaniasis. In addition to Leishmania, this group includes Trypanosoma brucei [130], the causative agent of African Trypanosomiasis. WHO classifies both of these diseases as so-called “forgotten” diseases. Although the diseases from this list (Table 4) do not attract public attention, as they are dangerous mainly for residents of developing countries, nevertheless, they take about half a million lives annually.
One more example of the potential targeting is the mtFOF1 dimers type III from Tetrahymena thermophila [11] and Toxoplasma gondii [10] belonging to the Ciliophora and Apicomplexa phylum, respectively, from the Alveolata clade of the Sar clade. As it was mentioned in the “Similarity and diversity of ATP synthases”, the high-resolution structures showed similar U-shape form-factors of the mtFOF1 dimers from these organisms. However, these dimers mtFOF1 showed completely different macroscale organization in mitochondria cristae. In the case of Ciliophora phylum, the tubular mitochondria cristae topology is formed by a tetrameric array of mtFOF1 (see Fig. 3C, G the left side). In the case of Apicomplexa phylum, the mitochondrial cristae are formed by the pentagonal bypiramids formed by the mtFOF1 hexamers (see Fig. 3C, G the right side). Though different oligomeric organization of mtFOF1 is present in representatives of the Sar clade, they carry similar structural features in their dimeric organization, except for the peripheral distant subunits in the FO part of the enzymes. The Sar clade includes a large number of pathogens, such as Babesia spp. (Apicomplexa phylum) [131], Toxoplasma gondii (Ciliophora phylum) [132], Balantidium coli (Ciliophora phylum) [133], and Cryptosporidium spp. (Apicomplexa phylum) [134]. In addition, the causative agent of malaria Plasmodium spp. (Apicomplexa phylum) [135] also belongs to Alveolata clade from the Sar clade.
However, there are pathogens belonging to the groups, for which there is still no mtFOF1 structural data (representatives of Amoebozoa and Metamonada clades). For example, the pathogen Entamoeba histolytica [136, 137], belonging to Amoebozoa clade, causes amoebiasis (also belongs to the forgotten diseases as well as trypanosomiasis and leishmaniasis). The Metamonada clade includes pathogens, such as Giardia lamblia [138], which causes giardiasis, and Trichomonas vaginalis [139], which causes trichomoniasis, a sexually transmitted disease. Obtaining high-resolution structures of the corresponding ATP synthases could help in the development of new drugs against these diseases.
Outlook
Despite recent advances in the structural biology of ATP synthases, these enzymes are still a focus of modern research due to their vital importance for living organisms, structural complexity, and diverse subunit composition in distant lineages. The main divergence of the ATP synthases generally follows the NCBI taxonomy [30]. For example, Bacteria superkingdom and chloroplasts carry ATP synthase monomers. Although in some cases, the cFOF1 dimers were reported, there is neither structural data showing the presence of cFOF1 functional dimers nor biochemical evidence of the subunit machinery establishing these dimers.
ATP synthases from the Eukaryota superkingdom representatives reside within the mitochondria and chloroplasts. In mitochondria, ATP synthases tend to assemble into arrays, which determine the topology of their cristae. The next level of divergence of mtFOF1 subunit machinery occurs at the level of clades of Eukaryota superkingdom. For example, the representatives of Opisthokonta, Sar and Discoba clades, and Viridiplantae kingdom have significantly different types of dimeric assemblies (V- and U-shape dimers of four types). Before 2021, one could make the conclusion that a type of a dimer exactly defines the morphology of mitochondria. However, a recent study [10] showed that Toxoplasma gondii—a representative of Apicomplexa phylum from Alveolata clade belonging to Sar clade—has completely different assembly of mtFOF1 arrays (hexamers of mtFOF1 which form pentagonal bipyramids, with each hexamer consisting of three mtFOF1 dimers of type III) than Tetrahymena thermophile—a representative of a Ciliophora phylum, which also belongs to Alveolata clade (its mtFOF1 dimers of type III form arrays of tetramers creating tubular cristae) [11]. Unexpectedly, the difference in the organization of the mtFOF1 assembly is established by minimal changes in the peripheral subunit machinery of dimers of the enzyme. In particular, the hexameric net of the mtFOF1 is established by a single subunit ATPTG11, making it an attractive target for therapies and other various applications.
The structural features of bacterial ATP synthases can also be utilized for the development of drugs. One of the promising cases is a development of the anti-tuberculosis drug BDQ, where the ATP synthase-structure-based approach revealed the molecular mechanisms of inhibiting the enzyme [24, 25], which might help improving the drug efficiency in the future.
We summarize here a possible systematic strategy for the development of new therapies based on the FOF1-structure approach (Fig. 15). Briefly, the approach starts from the classification of pathogens (see Table 4). If a pathogen belongs to a phylogenetic group, for which a high-resolution structure of the corresponding FOF1 is available for one of the representatives, one can use it for searching for the specific structural features carried by the ATP synthase of these organisms. Usually, model systems chosen for studies by means of structural biology are non-pathogenic. However, they might accurately reflect the behavior of the corresponding pathogens belonging to the same phylogenetic group.

Promising FOF1-structure-based drug-design approach. If a target pathogen belongs to a phylogenetic group for which a corresponding high-resolution structure of FOF1 is unavailable, one might choose a model system carrying the same structural features of FOF1 for expression or purification of intact FOF1 from it. When the high-resolution structure of the FOF1 is obtained one can search for structural features that can be utilized for FOF1-structure-based drug-design with following standard procedures of functional testing, preclinical and clinical trials
If a pathogen belongs to a phylogenetic group, for which there are no representatives with available high-resolution structure of FOF1, then one has to choose an appropriate model system from the same phylogenetic group, preferentially at the phylum level, but at least at level of clades.
Then, one has to obtain a recombinant or intact ATP synthase, depending on the model system that was chosen, and after that obtain a high-resolution structure of FOF1 by means of structural biology according to modern protocols of cryo-EM studies (see Table 2) and probably using protein crystallization and X-ray diffraction as a complementary method to refine the high-resolution structure especially in a membrane FO region, where commonly the resolution of cryo-EM technique is lower than for the water-soluble part F1.
Finally, when the high-resolution structure of corresponding FOF1 is obtained one can search for unique structural features belonging to the exact phylogenetic group but missing in other groups. Typically, a host organism and an infecting pathogen belong to the distant lineages and therefore carry significant differences in the FOF1 organization. These structural differences become the target for small-molecule ligands that can partially or completely inhibit pathogenic FOF1 without affecting the activity of the host enzyme. Further functional tests, preclinical and clinical trials following standard procedures are finalizing the development of new FOF1 targeting drugs against the pathogenic infections.
The useful support on the way to new therapies targeted to FOF1 may also come from the state-of-the-art machine learning method such as AlphaFold [140, 141] and RoseTTAFold [142]. Despite revolutionary advances for protein structure predictions, the use for drug development is still elusive [143]. However, when structural information for parts of FOF1 remains unavailable, predicted models may be a source of therapeutic ideas which can later be validated in functional tests.
The scheme in Fig. 15 is not the only way of how the approach using structural information on the FOF1 from different organisms can be used. FOF1-structure-based approach is much wider and might open new perspectives for the gene therapies, drug design, and development of tools for the control of cellular bioenergetics. One of the intriguing perspectives is the possible role of mtFOF1 in the mPTP phenomenon, which is, however, still unknown in detail. The possibility of using mPTP as a target for therapies is actively discussed in the literature [144]. A complementary use of the FOF1-structure-based approach might significantly help in the development of the mPTP-based therapies.
Regarding bacterial and chloroplast ATP synthases, the question of the small-molecule compounds inside the inner pore of their c-rings is still not yet settled. The possible presence of isoprenoid quinones inside the c-rings of corresponding FOF1 [23] is intriguing both in terms of understanding the mechanisms of the enzyme functioning and development of the powerful small-molecule ligands that can modulate the bFOF1 and cFOF1 efficiencies.
Availability of data and materials
Not applicable.
References
Walker JE (2013) The ATP synthase: the understood, the uncertain and the unknown. Biochem Soc Trans 41:1–16
Junge W, Nelson N (2015) ATP synthase. Annu Rev Biochem 84:631–657
Hahn A, Vonck J, Mills DJ, Meier T, Kühlbrandt W (2018) Structure, mechanism, and regulation of the chloroplast ATP synthase. Science 360:eaat4318
Vlasov AV (2021) New structural insights in chloroplast F1FO-ATP synthases—RWTH Publications. RWTH Aachen University. https://doi.org/10.18154/RWTH-2021-00849
Kühlbrandt W (2019) Structure and mechanisms of F-Type ATP synthases. Annu Rev Biochem 88:515–549
Morales-Rios E, Montgomery MG, Leslie AGW, Walker JE (2015) Structure of ATP synthase from Paracoccus denitrificans determined by X-ray crystallography at 4.0 Å resolution. Proc Natl Acad Sci USA 112:13231–13236
Sobti M et al (2020) Cryo-EM structures provide insight into how E. coli F1Fo ATP synthase accommodates symmetry mismatch. Nat Commun 11:1–10
Daum B, Nicastro D, Austin J, Richard MJ, Kühlbrandt W (2010) Arrangement of photosystem II and ATP synthase in chloroplast membranes of spinach and pea. Plant Cell 22:1299–1312
Seelert H, Dencher NA (2011) ATP synthase superassemblies in animals and plants: two or more are better. Biochim Biophys Acta Bioenerg 1807:1185–1197
Mühleip A et al (2021) ATP synthase hexamer assemblies shape cristae of Toxoplasma mitochondria. Nat Commun 12:1–13
Flygaard RK, Mühleip A, Tobiasson V, Amunts A (2020) Type III ATP synthase is a symmetry-deviated dimer that induces membrane curvature through tetramerization. Nat Commun 11:1–11
Nuskova H et al (2019) Biochemical thresholds for pathological presentation of ATP synthase deficiencies. Biochem Biophys Res Commun. https://doi.org/10.1016/j.bbrc.2019.11.033
Neupane P, Bhuju S, Thapa N, Bhattarai HK (2019) ATP synthase: structure, function and inhibition. Biomol Concepts 10:1–10
Gerle C (2016) On the structural possibility of pore-forming mitochondrial FoF1 ATP synthase. Biochim Biophys Acta Bioenerg 1857:1191–1196
Carraro M, Carrer A, Urbani A, Bernardi P (2020) Molecular nature and regulation of the mitochondrial permeability transition pore(s), drug target(s) in cardioprotection. J Mol Cell Cardiol 144:76–86
Walker JE, Carroll J, He J (2020) Reply to Bernardi: the mitochondrial permeability transition pore and the ATP synthase. Proc Natl Acad Sci 117:2745–2746
Petronilli V, Szabò I, Zoratti M (1989) The inner mitochondrial membrane contains ion-conducting channels similar to those found in bacteria. FEBS Lett 259:137–143
Szabo I, Zoratti M (1991) The giant channel of the inner mitochondrial membrane is inhibited by cyclosporin A. J Biol Chem 266:3376–3379
Szabó I, Zoratti M (1992) The mitochondrial megachannel is the permeability transition pore. J Bioenerg Biomembr 24:111–117
Szabo I, Bernardi P, Zoratti M (1992) Modulation of the mitochondrial megachannel by divalent cations and protons. J Biol Chem 267:2940–2946
Kinnally KW, Zorov DYA, Perini S (1991) Calcium modulation of mitochondrial inner membrane channel activity. Biochem Biophys Res Commun 176:1183–1188
Bernardi P et al (1992) Modulation of the mitochondrial permeability transition pore. Effect of protons and divalent cations. J Biol Chem 267:2934–2939
Vlasov AV et al (2019) Unusual features of the c-ring of F1FO ATP synthases. Sci Rep 9:18547
Preiss L et al (2015) Structure of the mycobacterial ATP synthase Fo rotor ring in complex with the anti-TB drug bedaquiline. Sci Adv 1:e1500106–e1500106
Guo H et al (2021) Structure of mycobacterial ATP synthase bound to the tuberculosis drug bedaquiline. Nature 589:143–147
Grinkova YV, Denisov IG, Sligar SG (2010) Engineering extended membrane scaffold proteins for self-assembly of soluble nanoscale lipid bilayers. Protein Eng Des Sel 23:843–848
Muñoz-Gómez SA, Wideman JG, Roger AJ, Slamovits CH (2017) The origin of mitochondrial cristae from alphaproteobacteria. Mol Biol Evol 34:943–956
Eydt K, Davies KM, Behrendt C, Wittig I, Reichert AS (2017) Cristae architecture is determined by an interplay of the MICOS complex and the F1Fo ATP synthase via Mic27 and Mic10. Microb Cell 4:259–272
Davies KM, Anselmi C, Wittig I, Faraldo-Gómez JD, Kühlbrandt W (2012) Structure of the yeast F1Fo-ATP synthase dimer and its role in shaping the mitochondrial cristae. Proc Natl Acad Sci USA 109:13602–13607
Schoch CL et al (2020) NCBI taxonomy: a comprehensive update on curation, resources and tools. Database 2020: baaa062. PubMed: 32761142 PMC: PMC7408187
Sobti M et al (2016) Cryo-EM structures of the autoinhibited E. coli ATP synthase in three rotational states. Elife 5:e21598
Guo H, Suzuki T, Rubinstein JL (2019) Structure of a bacterial ATP synthase. Elife 8:e43128
Guo H, Bueler SA, Rubinstein JL (2017) Atomic model for the dimeric FOregion of mitochondrial ATP synthase. Science 358:936–940
Gu J et al (2019) Cryo-EM structure of the mammalian ATP synthase tetramer bound with inhibitory protein IF1. Science 364:1068–1075
Rexroth S et al (2004) Dimeric H+-ATP synthase in the chloroplast of Chlamydomonas reinhardtii. Biochim Biophys Acta Bioenerg 1658:202–211
Schwaßmann HJ, Rexroth S, Seelert H, Dencher NA (2007) Metabolism controls dimerization of the chloroplast FoF1 ATP synthase in Chlamydomonas reinhardtii. FEBS Lett 581:1391–1396
Murphy BJ et al (2019) Rotary substates of mitochondrial ATP synthase reveal the basis of flexible F1–Fo coupling. Science 364:eaaw9128
Blum TB, Hahn A, Meier T, Davies KM, Kühlbrandt W (2019) Dimers of mitochondrial ATP synthase induce membrane curvature and self-assemble into rows. Proc Natl Acad Sci USA 116:4250–4255
Mühleip AW, Dewar CE, Schnaufer A, Kühlbrandt W, Davies KM (2017) In situ structure of trypanosomal ATP synthase dimer reveals a unique arrangement of catalytic subunits. Proc Natl Acad Sci 114:992–997
Mühleip A, McComas SE, Amunts A (2019) Structure of a mitochondrial ATP synthase with bound native cardiolipin. Elife 8:e51179
Vlasov AV, Ryzhykau YL, Gordeliy VI, Kuklin AI (2017) Spinach ATP-synthases form dimers in nanodiscs. Small-angle X-ray and neutron scattering investigations. FEBS J 284:87
Yanyushin MF (1993) Subunit structure of ATP synthase from chloroflexus aurantiacus. FEBS Lett 335:85–88
Yanyushin MF (1997) Determination of subunit composition of the F 1 and F 0 moieties of ATP synthase from Chloroflexus aurantiacus. Biochem 62:285–288
Abrahams JP, Leslie AGW, Lutter R, Walker JE (1994) Structure at 28 Â resolution of F1-ATPase from bovine heart mitochondria. Nature 370:621–628
Stock D, Leslie AGW, Walker JE (1999) Molecular architecture of the rotary motor in ATP synthase. Science 286:1700–1705
Dautant A, Velours J, Giraud MF (2010) Crystal structure of the Mg·ADP-inhibited state of the yeast F 1c10-ATP synthase. J Biol Chem 285:29502–29510
Watt IN, Montgomery MG, Runswick MJ, Leslie AGW, Walker JE (2010) Bioenergetic cost of making an adenosine triphosphate molecule in animal mitochondria. Proc Natl Acad Sci USA 107:16823–16827
Giraud MF et al (2012) Rotor architecture in the yeast and bovine F 1-c-ring complexes of F-ATP synthase. J Struct Biol 177:490–497
Morales-Rios E et al (2015) Purification, characterization and crystallization of the F-ATPase from Paracoccus denitrificans. Open Biol 5:150119
Hahn A et al (2016) Structure of a complete ATP synthase dimer reveals the molecular basis of inner mitochondrial membrane morphology. Mol Cell 63:445–456
Seelert H et al (2000) Proton-powered turbine of a plant motor. Nature 405:418–419
Zhou A et al (2015) Structure and conformational states of the bovine mitochondrial ATP synthase by cryo-EM. Elife 4:e10180
Allegretti M et al (2015) Horizontal membrane-intrinsic α-helices in the stator a-subunit of an F-type ATP synthase. Nature 521:237–240
Vinothkumar KR, Montgomery MG, Liu S, Walker JE (2016) Structure of the mitochondrial ATP synthase from Pichia angusta determined by electron cryo-microscopy. Proc Natl Acad Sci USA 113:12709–12714
Srivastava AP et al (2018) High-resolution cryo-EM analysis of the yeast ATP synthase in a lipid membrane. Science 360:6389
Mellwig C, Böttcher B (2003) A unique resting position of the ATP-synthase from Chloroplasts*. J Biol Chem 278:18544–18549
Guo H, Bueler SA, Rubinstein JL (2017) Atomic model for the dimeric FO region of mitochondrial ATP synthase. Science 358:936–940
Allen RD, Schroeder CC, Fok AK (1989) An investigation of mitochondrial inner membranes by rapid-freeze deep-etch techniques. J Cell Biol 108:2233–2240
Nicastro D, Frangakis AS, Typke D, Baumeister W (2000) Cryo-electron tomography of neurospora mitochondria. J Struct Biol 129:48–56
Giraud MF et al (2002) Is there a relationship between the supramolecular organization of the mitochondrial ATP synthase and the formation of cristae? Biochim Biophys Acta Bioenerg 1555:174–180
Benvenuti M, Mangani S (2007) Crystallization of soluble proteins in vapor diffusion for X-ray crystallography. Nat Protoc 2:1633–1651
Balakrishna AM, Seelert H, Marx S-H, Dencher NA, Grüber G (2014) Crystallographic structure of the turbine C-ring from spinach chloroplast F-ATP synthase. Biosci Rep 34:e00102
Caffrey M, Cherezov V (2009) Crystallizing membrane proteins using lipidic mesophases. Nat Protoc 4:706–731
Li D, Shah STA, Caffrey M (2013) Host lipid and temperature as important screening variables for crystallizing integral membrane proteins in lipidic mesophases. Trials with diacylglycerol kinase. Cryst Growth Des 13:2846–2857
Ishchenko A et al (2017) Chemically stable lipids for membrane protein crystallization. Cryst Growth Des 17:3502–3511
Zabara A et al (2018) Design of ultra-swollen lipidic mesophases for the crystallization of membrane proteins with large extracellular domains. Nat Commun 9:1–9
Johnson DE et al (2012) High-throughput characterization of intrinsic disorder in proteins from the protein structure initiative. J Struct Biol 180:201–215
Sedzik J, Kirschner DA (1992) Is myelin basic protein crystallizable? Neurochem Res 17:157–166
Harauz G et al (2004) Myelin basic protein-diverse conformational states of an intrinsically unstructured protein and its roles in myelin assembly and multiple sclerosis. Micron 35:503–542
Le Gall T, Romero PR, Cortese MS, Uversky VN, Dunker AK (2007) Intrinsic disorder in the protein data bank. J Biomol Struct Dyn 24:325–341
DeForte S, Uversky VN (2016) Resolving the ambiguity: making sense of intrinsic disorder when PDB structures disagree. Protein Sci 25:676–688
Romero P et al (2000) Sequence complexity of disordered protein. Proteins Struct Funct Bioinform 42:38–48
Peng K, Radivojac P, Vucetic S, Dunker AK, Obradovic Z (2006) Length-dependent prediction of protein intrinsic disorder. BMC Bioinform 7:208–212
Peng K et al (2005) Optimizing long intrinsic disorder predictors with protein evolutionary information. J Bioinform Comput Biol 3:35–60
Xue B, Dunbrack RL, Williams RW, Dunker AK, Uversky VN (2010) PONDR-FIT: a meta-predictor of intrinsically disordered amino acids. Biochim Biophys Acta 1804:996–1010
Mészáros B, Erdos G, Dosztányi Z (2018) IUPred2A: context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res 46:W329–W337
Mendoza-Hoffmann F, Zarco-Zavala M, Ortega R, García-Trejo JJ (2018) Control of rotation of the F1FO-ATP synthase nanomotor by an inhibitory α-helix from unfolded ε or intrinsically disordered ζ and IF1 proteins. J Bioenerg Biomembr 50:403–424
Yang J-H, Williams D, Kandiah E, Fromme P, Chiu P-L (2020) Structural basis of redox modulation on chloroplast ATP synthase. Commun Biol 3:1–12
Fischer S et al (1994) ATP synthesis catalyzed by the ATP synthase of Escherichia coli reconstituted into liposomes. Eur J Biochem 225:167–172
Poetsch A et al (2003) Characterisation of subunit III and its oligomer from spinach chloroplast ATP synthase. Biochim Biophys Acta Biomembr 1618:59–66
Suhai T, Dencher NA, Poetsch A, Seelert H (2008) Remarkable stability of the proton translocating F1FO-ATP synthase from the thermophilic cyanobacterium Thermosynechococcus elongatus BP-1. Biochim Biophys Acta Biomembr 1778:1131–1140
Suhai T, Heidrich NG, Dencher NA, Seelert H (2009) Highly sensitive detection of ATPase activity in native gels. Electrophoresis 30:3622–3625
Schmidt G, Gräber P (1985) The rate of ATP synthesis by reconstituted CF0F1 liposomes. Biochim Biophys Acta Bioenerg 808:46–51
Förster K et al (2010) Proton transport coupled ATP synthesis by the purified yeast H+-ATP synthase in proteoliposomes. Biochim Biophys Acta Bioenerg 1797:1828–1837
Turina P, Samoray D, Gräber P (2003) H+/ATP ratio of proton transport-coupled ATP synthesis and hydrolysis catalysed by CF0F1-liposomes. EMBO J. https://doi.org/10.1093/emboj/cdg073
Groth G, Walker JE (1996) ATP synthase from bovine heart mitochondria: reconstitution into unilamellar phospholipid vesicles of the pure enzyme in a functional state. Biochem J 318:351–357
Varco-Merth B, Fromme R, Wang M, Fromme P (2008) Crystallization of the c14-rotor of the chloroplast ATP synthase reveals that it contains pigments. Biochim Biophys Acta Bioenerg 1777:605–612
Grotjohann I, Gräber P (2002) The H+-ATPase from chloroplasts: effect of different reconstitution procedures on ATP synthesis activity and on phosphate dependence of ATP synthesis. Biochim Biophys Acta Bioenerg 1556:208–216
Pogoryelov D et al (2012) Engineering rotor ring stoichiometries in the ATP synthase. Proc Natl Acad Sci USA 109:E1599–E1608
Luz AL, Lagido C, Hirschey MD, Meyer JN (2016) In vivo determination of mitochondrial function using luciferase-expressing caenorhabditis elegans: contribution of oxidative phosphorylation, glycolysis, and fatty acid oxidation to toxicant-induced dysfunction. Curr Protoc Toxicol 69:2581–25822
Boerries M et al (2007) Ca2+-dependent interaction of S100A1 with F1-ATPase leads to an increased ATP content in cardiomyocytes. Mol Cell Biol 27:4365–4373
Drew B, Leeuwenburgh C (2003) Method for measuring ATP production in isolated mitochondria: ATP production in brain and liver mitochondria of Fischer-344 rats with age and caloric restriction. Am J Physiol Regul Integr Comp Physiol 285:1259–1267
Vinkler C, Korenstein R (1982) Characterization of external electric field-driven ATP synthesis in chloroplasts. Proc Natl Acad Sci 79:3183–3187
Sun T et al (2000) Ralstonia solanacearum elicitor RipX induces defense reaction by suppressing the mitochondrial atpA gene in host plant. Int J Mol Sci 2020:21
Meighen EA (1991) Molecular biology of bacterial bioluminescence. Microbiol Rev 55:123–142
Nijvipakul S et al (2008) LuxG is a functioning flavin reductase for bacterial luminescence. J Bacteriol 190:1531–1538
Kalyabina VP, Esimbekova EN, Torgashina IG, Kopylova KV, Kratasyuk VA (2019) Principles for construction of bioluminescent enzyme biotests for analysis of complex media. Dokl Biochem Biophys 485:107–110
Zavilgelsky GB, Kotova VY, Mazhul’ MM, Manukhov IV (2002) Role of Hsp70 (DnaK–DnaJ–GrpE) and Hsp100 (ClpA and ClpB) chaperones in refolding and increased thermal stability of bacterial luciferases in Escherichia coli cells. Biochem 67:986–992
Bernardi P (2020) Mechanisms for Ca2+-dependent permeability transition in mitochondria. Proc Natl Acad Sci USA 117:2743–2744
Neginskaya MA et al (2019) ATP synthase c-subunit-deficient mitochondria have a small cyclosporine a-sensitive channel, but lack the permeability transition pore. Cell Rep 26:11-17.e2
Giorgio V et al (2013) Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc Natl Acad Sci 110:5887–5892
Kokoszka JE et al (2004) The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature 427:461–465
He J et al (2017) Persistence of the mitochondrial permeability transition in the absence of subunit c of human ATP synthase. Proc Natl Acad Sci 114:3409–3414
Alavian KN et al (2014) An uncoupling channel within the c-subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. Proc Natl Acad Sci 111:10580–10585
Bernardi P, Di Lisa F (2015) The mitochondrial permeability transition pore: molecular nature and role as a target in cardioprotection. J Mol Cell Cardiol 78:100–106
Zhou W, Marinelli F, Nief C, Faraldo-Gómez JD (2017) Atomistic simulations indicate the c-subunit ring of the F1Fo ATP synthase is not the mitochondrial permeability transition pore. Elife 6:e23781
Spikes TE, Montgomery MG, Walker JE (2020) Structure of the dimeric ATP synthase from bovine mitochondria. Proc Natl Acad Sci 117:23519–23526
Carraro M et al (2014) Channel formation by yeast F-ATP synthase and the role of dimerization in the mitochondrial permeability transition. J Biol Chem 289:15980–15985
Carraro M et al (2018) High-conductance channel formation in yeast mitochondria is mediated by F-ATP synthase e and g subunits. Cell Physiol Biochem 50:1840–1855
Urbani A et al (2019) Purified F-ATP synthase forms a Ca2+-dependent high-conductance channel matching the mitochondrial permeability transition pore. Nat Commun 10:1–11
Bonora M et al (2017) Mitochondrial permeability transition involves dissociation of F1FO ATP synthase dimers and C-ring conformation. EMBO Rep 18:1077–1089
Mnatsakanyan N et al (2019) A mitochondrial megachannel resides in monomeric F1FO ATP synthase. Nat Commun 10:1–11
Pinke G, Zhou L, Sazanov LA (2020) Cryo-EM structure of the entire mammalian F-type ATP synthase. Nat Struct Mol Biol 27:1077–1085
Amodeo GF et al (2021) C subunit of the ATP synthase is an amyloidogenic calcium dependent channel-forming peptide with possible implications in mitochondrial permeability transition. Sci Rep 11:1–10
Matthies D et al (2014) High-resolution structure and mechanism of an F/V-hybrid rotor ring in a Na+-coupled ATP synthase. Nat Commun 5:5286
Meier T, Matthey U, Henzen F, Dimroth P, Müller DJ (2001) The central plug in the reconstituted undecameric c cylinder of a bacterial ATP synthase consists of phospholipids. FEBS Lett 505:353–356
Seelert H, Dencher NA, Müller DJ (2003) Fourteen protomers compose the oligomer III of the proton-rotor in spinach chloroplast ATP synthase. J Mol Biol 333:337–344
Novitskaia O, Buslaev P, Gushchin I (2019) Assembly of spinach chloroplast ATP synthase rotor ring protein–lipid complex. Front Mol Biosci 6:135
Meier T et al (2009) Complete ion-coordination structure in the rotor ring of Na+-dependent F-ATP synthases. J Mol Biol 391:498–507
Fromme P, Gräber P, Boekema EJ (1987) Isolation and characterization of a supramolecular complex of subunit III of the ATP-synthase from chloroplasts. Zeitschrift fur Naturforsch Sect C J Biosci 42:1239–1245
Vollmar M, Schlieper D, Winn M, Büchner C, Groth G (2009) Structure of the c14 rotor ring of the proton translocating chloroplast ATP synthase. J Biol Chem 284:18228–18235
Symersky J et al (2012) Structure of the c10 ring of the yeast mitochondrial ATP synthase in the open conformation. Nat Struct Mol Biol 19:485–491
Preiss L et al (2014) The c-ring ion binding site of the ATP synthase from Bacillus pseudofirmus OF4 is adapted to alkaliphilic lifestyle. Mol Microbiol 92:973–984
Pogoryelov D et al (2010) Microscopic rotary mechanism of ion translocation in the Fo complex of ATP synthases. Nat Chem Biol 6:891–899
Spikes TE, Montgomery MG, Walker JE (2021) Interface mobility between monomers in dimeric bovine ATP synthase participates in the ultrastructure of inner mitochondrial membranes. Proc Natl Acad Sci 118
Tacconelli E et al (2018) Discovery, research, and development of new antibiotics: the WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect Dis 18:318–327
Gao W, Howden BP, Stinear TP (2018) Evolution of virulence in Enterococcus faecium, a hospital-adapted opportunistic pathogen. Curr Opin Microbiol 41:76–82
Li Y, Sun F, Zhang W (2019) Bedaquiline and delamanid in the treatment of multidrug-resistant tuberculosis: promising but challenging. Drug Dev Res 80:98–105
Kevric I, Cappel MA, Keeling JH (2015) New world and old world leishmania infections: a practical review. Dermatol Clin 33:579–593
Migchelsen SJ, Büscher P, Hoepelman AIM, Schallig HDFH, Adams ER (2011) Human African trypanosomiasis: a review of non-endemic cases in the past 20 years. Int J Infect Dis 15:e517–e524
Young KM et al (2019) Zoonotic Babesia: a scoping review of the global evidence. PLoS One 14:e0226781
Saadatnia G, Golkar M (2012) A review on human toxoplasmosis. Scand J Infect Dis. https://doi.org/10.3109/00365548.2012.693197
Schuster FL, Ramirez-Avila L (2008) Current world status of Balantidium coli. Clin Microbiol Rev 21:626–638
Fayer R, Ungar BLP (1986) Cryptosporidium spp. and Cryptosporidiosis. Microbiol Rev 50:458–483
Tuteja R (2007) Malaria—an overview. FEBS J 274:4670–4679
Clark C, Espinosa Cantellano M, Bhattacharya A (2000) Entamoeba Histolytica: an overview of the biology of the organism. Amebiasis. https://doi.org/10.1142/9781848160583_0001
El-Dib NA (2017) Entamoeba histolytica: an overview. Curr Trop Med Rep 4:11–20
Visvesvara GS (1983) Giardiasis: an overview. IMJ Ill Med J 164:34–39
Docampo R, Ryan CM, de Miguel N, Johnson PJ (2011) Trichomonas vaginalis: current understanding of host-parasite interactions. Essays Biochem 51:161–175
Jumper J et al (2021) Highly accurate protein structure prediction with AlphaFold. Nature 596:583–589
Tunyasuvunakool K et al (2021) Highly accurate protein structure prediction for the human proteome. Nature 596:590–596
Baek M et al (2021) Accurate prediction of protein structures and interactions using a three-track neural network. Science 373:871–876
Mullard A (2021) What does AlphaFold mean for drug discovery? Nat Rev Drug Discov 20:725–727
Cui Y et al (2020) Recent progress in the use of mitochondrial membrane permeability transition pore in mitochondrial dysfunction-related disease therapies. Mol Cell Biochem 476:493–506
Acknowledgements
AVV greatly acknowledges the Council for Grants of the President of the Russian Federation for state support of young Russian scientists and for state support of leading scientific schools of the Russian Federation (Scholarship of the President of the Russian Federation, Order of the Ministry of Education and Science of the Russian Federation of January 26, 2021 No. 54 on the appointment of a scholarship for 2021-2023). SDO acknowledges funding from the Foundation of Promoting Innovations for financial support in the framework of the program UMNIK. VIG acknowledges funding from Frankfurt: Cluster of Excellence Frankfurt Macromolecular Complexes (to E.B.) by the Max Planck Society (to E.B.) and by the Commissariat à l’Energie Atomique et aux Energies Alternatives (Institut de Biologie Structurale) – Helmholtz-Gemeinschaft Deutscher Forschungszentren (Forschungszentrum Jülich) Special Terms and Conditions 5.1 specific agreement.
Funding
The work is supported by RFBR 19-29-12022. The work is supported by the Ministry of Science and Higher Education of the Russian Federation (075-00337-20-03/FSMG-2020-0003; 075–00958-21–05, project # 730000F.99.1.BV10AA00006).
Author information
Authors and Affiliations
Contributions
AVV designed, conceived and wrote the manuscript. SDO strongly contributed to the section “ATP synthases as potential therapeutic targets”, edited the text of the manuscript. NAB strongly contributed to the section “Mitochondrial permeability transition pore (mPTP)”. VNU strongly contributed to the section “Intrinsically disordered regions in ATP synthases”, contributed to all sections and edited the text of the manuscript. VIB contributed to the section “Outlook”, edited the text of the manuscript. MFY contributed to the section “Similarity and diversity of ATP synthases”. IVM contributed to the section “Validation of ATP synthase functionality”, edited the text of the manuscript. AVR organized funding acquisition, edited the text of the manuscript. ADV contributed to the section “Small-molecule cofactors of the c-ring”, edited the text of the manuscript. NSI contributed to the section “Intrinsically disordered regions in ATP synthases”, edited the text of the manuscript. AIK contributed to the section “High-resolution structural studies”. NAD contributed to all sections and edited the text of the manuscript. VIG supervised the project, strongly contributed to all sections and edited the text of the manuscript. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interests
The authors declare that they have no conflict of interest.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Vlasov, A.V., Osipov, S.D., Bondarev, N.A. et al. ATP synthase FOF1 structure, function, and structure-based drug design. Cell. Mol. Life Sci. 79, 179 (2022). https://doi.org/10.1007/s00018-022-04153-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00018-022-04153-0