
Abstract
The effect of peroxide-induced oxidation of fibrinogen on modification of its primary structure and functional properties was investigated. The oxidation sites were shown to be Met, Trp, and His residues. Using the DLS method, it was found that the oxidative modification of fibrinogen results in the change of microrheological characteristics of fibrin network. The fibrinogen oxidation diminishes its tolerance to plasmin hydrolysis and deteriorates the factor XIIIa ability to stabilize the fibrin gel.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Fibrinogen (FG) is a plasma protein most susceptible to oxidative modification. FG plays an important role in blood clotting, fibrinolysis, cell and matrix interactions, inflammation, wound healing, and neoplasia.
Oxidation of FG damages its structure and affects the function of the protein. It is known that oxidative modification of human FG molecules causes changes in the fibrin self-assembly, which eventually leads to abnormal clots. They are characterized by thin individual fibrin fibers, low porosity and low permeability. It is believed that oxidation affects the self-assembly of fibrin primarily by inhibiting the lateral association of protofibrils [1].
Previously, we investigated the oxidative modification of the molecule after its ozone- and hypochlorite-induced oxidation [2, 3]. The data obtained demonstrated the ability of the FG structure to preserve the functionally important amino acid residues during oxidation, which, in our opinion, is the results of the presence of a large number of methionine residues in proteins that are considered as ROS scavengers [4]. This allowed us to conclude that the FG structure is adapted to the action of ROS. To additionally confirm this hypothesis, in this study we investigated the oxidative modification of FG by hydrogen peroxide (H2O2), which is one of the most important oxidants produced in the human body.
FG was isolated from citrate donor blood plasma by glycine precipitation [5]. Oxidation of FG was induced by adding H2O2 solution (Sigma-Aldrich, United States) at a concentration of 150, 300, 600, and 1000 μM. After FG oxidation, the reduced samples of the protein as well as its polypeptide chains covalently linked under the influence of activated coagulation factor XIII (FXIIIa) were analyzed by electrophoresis. The oxidative damage of FG molecules was also assessed by the changes in the accumulation of plasmin hydrolysis degradation products [6].
The oxidation sites were identified by liquid chromatography-tandem mass spectrometry analysis (HPLC-MS/MS) in a system consisting of an Agilent 1100 chromatograph with automatic sampling (Agilent Technologies Inc., Santa Clara, United States) and a 7T LTQ-FT Ultra tandem mass spectrometer (Thermo, Bremen, Germany) [7]. To prepare samples for analysis, they were treated with dithiothreitol (DTT) to reduce the disulfide bonds with subsequent alkylation with iodoacetamide and hydrolysis with trypsin (Promega, United States). The tryptic peptides were identified using the PEAKS Studio software (V. 8.5, Bioinformatics Solutions Inc., Waterloo, On, Canada). All experiments were performed in triplicate. When comparing the results, we took into account the amino acids that were not oxidized in the control as well as those whose oxidation level increased by more than 1% compared to the control.
The microrheological characteristics of fibrin gels formed under the action of thrombin from the native and oxidized FG were studied by dynamic light scattering (DLS) in a Malvern Zetasizer Nano S instrument. Correlation functions (KF) of g(2)(τ) fluctuations of the radiation intensity at 633 nm, scattered by the fibrin gel at an angle of 173° in the range of the delay time τ = 0.5–4 × 106 μs were recorded using the Zetasizer software in the form [g(2)(τ) – 1].
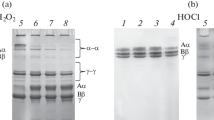
Figure 1a shows the results of electrophoresis of the polypeptide chains of native and oxidized FG. Irrespective of the oxidant concentrations used, neither fragmentation of the protein nor the formation of covalent cross-links of its chains were observed.

Electrophoresis of various samples of FG. (a) SDS-PAGE of FG polypeptide chains (5% stacking gel, 12% separating gel): 1—unoxidized FG, 2—FG oxidized with 150 μM H2O2, 3—FG oxidized with 300 μM H2O2, 4—FG oxidized with 600 μM H2O2, 5—FG oxidized with 1000 μM H2O2. (b) SDS-PAGE of the products of FG crosslinking by FXIIIa (4% stacking gel, 8% separating gel): (1) products of FXIIIa reaction with the unoxidized FG, and FG oxidized with 150 μM H2O2 (2), 300 μM H2O2 (3), 600 μM H2O2 (4), and 1000 μM H2O2 (5). (c) SDS-PAGE of hydrolysis products of FG molecule by plasmin (4% stacking gel, 8% separating gel): 1 —nonhydrolyzed FG; hydrolysis products of the unoxidized GF (2), FG oxidized with 150 μM H2O2 (3), FG oxidized with 300 μM H2O2 (4), FG oxidized with 600 μM H2O2 (5), and FG oxidized with 1000 μM H2O2 (6).
In the presence of FXIIIa, the polypeptide chains of fibrin were involved in covalent cross-linking, which was manifested in the formation of γ–γ dimers and α–α polymers [8]. With increasing oxidant concentration, the amount of produced α–α polymers and γ–γ dimers is reduced, as evidenced as an increase in the content source Aα- and γ chains (Fig. 1b). Obviously, this is a consequence of oxidative modification of the FG molecule structure.
When assessing the susceptibility of the FG molecule to plasmin hydrolysis during oxidation, it is clearly seen that even at a minimum amount of oxidant (150 μM), the amount of degradation products significantly increased (Fig. 1c).
Samples of unoxidized FG and FG treated with 300 μM H2O2 were analyzed by mass spectrometry. Under the influence of H2O2, methionine, tryptophan, and histidine residues in the FG molecule were most susceptible to oxidation. The modifications found in these amino acid residues included the cases of formation of methionine sulfoxide, 2-oxohistidine, and hydroxytryptophan as a result of attachment of one oxygen atom to the side chain (+15.99), oxidation of tryptophan to kynurenine (+3.99,) and methanethiol cleavage from the side chain of Met (–48.00). As can be seen in Fig. 2, the amino acid residues modified as a result of the induced oxidation were found in all three polypeptide chains and in all structural regions of the FG molecule, except the E region.

Primary structure (amino acid residue sequence corresponds to UniProt P02671 (FIBA _HUMAN), P02675 (FIBB _HUMAN), and P02679(FIBG _HUMAN), http://www.uniprot.org) and the sequences of peptide fragments of the FG polypeptide chains that were obtained experimentally by HPLC–MS/MS. Double line underlines the polypeptide chains regions detected in both the control and the oxidized (300 μM H2O2) samples, single line below underlies the regions detected only in the oxidized sample (300 μM H2O2), and single line above shows the regions detected only in the control sample. The modification sites are marked by black triangles on top. The amino acid residues at the NH2- terminal regions of the polypeptide chains are shown in gray and are signal peptides.
In the unoxidized sample, oxidative modification of the following amino acid residues were also found: AαMet91, AαMet207, AαMet240, AαTrp276, AαMet476, AαMet517, AαMet584, BβMet118, BβMet190, BβMet305, BβMet314, BβMet367, γMet78, and γTrp227. This can be explained by the baseline oxidation of the FG molecule in blood plasma as well as by additional oxidation during its preparative isolation, storage, and sample injection. Almost all amino acid residues modified in the control showed a moderate degree of oxidation, which significantly increased at the induced oxidation.
The amino acid residues localized in the E region that were involved in thrombin binding (AαTrp33, AαPhe35, AαAsp38, AαGlu39, BβAla68, BβAsp69, γAsp27, and γSer30 [9]) were not affected by oxidative modification, which indicated the preservation of the thrombin-binding sites in FG during oxidation. The greatest number of oxidation sites was detected in the αC region (AαMet240, AαTrp276, AαTrp341, AαTrp391, AαMet476, AαMet517, AαHis544, AαHis545, and AαMet584), which is consistent with our previous data [2, 3] and confirms the hypothesis that this region may serve as a trap for ROS molecules [10].
For the oxidized protein, deceleration of accumulation of α–α polymers and γ–γ dimers was observed, as also evidenced by the increase in the amount of the initial Aα and γ chains (Fig. 1b). FXIIIa formed covalent crosslinks between γGln398/399 and γLys406 [11], producing γ–γ dimers and, at a lower rate, α–α polymers between the Aα chains at several sites: AαGln328, AαGln336, AαLys508, AαLys556, and AαLys562 [12]. All the detected amino acid residues (γGln398/399 and γLys406, AαGln336, and AαLys508) during oxidation remained in the native form. This is additional evidence that the inhibition of covalent crosslinking of FG chains during oxidation is not the result of disruption of the structure of functional sites but is due to the conformational rearrangements in the oxidized protein, making these functional sites less accessible to FXIIIa. Among the regions of the molecule that were susceptible to plasmin hydrolysis, oxidative modification sites were also not found. This fact confirms the earlier assumption that the oxidized proteins, due to their increased hydrophobicity, become more susceptible to proteolytic degradation [13].
The obtained data on the FG peroxidation are consistent with the results of the previous study of ozone- and hypochlorite-induced oxidation of the FG molecule [2, 3, 10]. Hypochlorite and ozone caused significantly greater damage of the FG molecule, facilitating modification of a wider range of amino acid residues and increasing the diversity of oxidative modification types [2, 3] as compared to H2O2; however, in general, they did not affect the functional key sites in FG. Out of the 24 identified modified amino acid residues, 18 are methionine residues, 12 of which were oxidized even in the control sample. The residues include AαMet476, the oxidation of which is regarded a number of studies [1, 5] as the main factor responsible for the disturbed ability of αC-regions to participate in the lateral aggregation of protofibrils. However, the preferential oxidation of AaMet476 (manifested even in the control sample as a result of autoxidation of FG molecule) indicates that this residue is apparently exposed on the surface of the molecule. It is known that the exposed methionine residues in proteins are easily oxidized, have antioxidant function, and have no effect on the biological activity [4].
Oxidation of native FG changes the geometrical and mechanical characteristics of the formed fibers and, ultimately, the structure and mechanical properties of fibrin clots. In the context of the oxidative processes in the body, the study of these changes is of fundamental medical and biological importance. Figure 3 shows the normalized dependences [g(2)(τ) – 1] for the gels formed by native and oxidized FG.

Normalized correlograms of dynamic light scattering in gels of native (solid line) and oxidized FG: dash-and-dash line, 300 μM H2O2 per 1 μM protein; dash-and-dot line, 500 μM H2O2 per 1 μM protein.
Recording was performed 43, 73, and 22 min after the preparation of the gel. The shape of the correlation functions represents the dynamics of overdamped oscillations of elastic fibrin network of the gel, which is characterized, to a first approximation, by the decay time τeff ∝ f/G, where f and G are effective values of the friction coefficient of fibers in the surrounding liquid and the elastic modulus of fibrin network, correspondingly [14]. The τeff value can be estimated integrally by the formula: \({{\tau }_{{\operatorname{eff} }}} = \int {{{{[{{g}^{{(2)}}}(\tau ) - 1]}}^{{1/2}}}d\tau } \). This gives values τeff = 8.5, 7.8, and 10.5 ms for the gels from the native FG and FG oxidized with 300 and 500 μM of H2O2 per 1 μM protein, correspondingly. The shape of the recorded correlation functions and the obtained estimates of τeff suggest that the oxidation of FG with hydrogen peroxide at doses of approximately 500 μM H2O2 per 1 μM protein markedly decreases the stiffness of the fibrin network of a freshly formed gel. As it was shown previously [15], this may be associated with the structural changes due to formation of local aggregates of thin fibers .
REFERENCES
Weigandt, K.M., White, N., Chung, D., Ellingson, E., Wang, Y., Fu, X., and Pozzo, D.C., Biophys. J., 2012, vol. 103, no. 11, pp. 2399–2407.
Yurina, L.I., Vasilyeva, A.D., Bugrova, A.E., Indeykina, M.I., Kononikhin, A.S., Nikolaev, E.N., and Rosenfeld, M.A., Dokl. Biochem. Biophys., 2019, vol. 484, pp. 37–41.
Yurina, L., Vasilyeva, A., Indeykina, M., Bugrova, A., Biryukova, M., Kononikhin, A., Nikolaev, E., and Rosenfeld, M., Free Radic. Res., 2019, vol. 53, no. 4, pp. 430–455.
Luo, S. and Levine, R.L., FASEB J., 2009, vol. 23, no. 2, pp. 464–472.
White, N.J., Wang, Y., Fu, X., Cardenas, J.C., Martin, E.J., Brophy, D.F., Wade, C.E., Wang, X., St, John, A.E., Lim, E.B., Stern, S.A., Ward, K.R., Lopez, A., and Chung, D., Free Radic. Biol. Med., 2016, vol. 96, pp. 181–189.
Rosenfeld, M.A., Shchegolikhin, A.N., Bychkova, A.V., Leonova, V.B., Biryukova, M.I., and Kostanova, E.A., Free Radic. Biol. Med., 2014, vol. 77, pp. 106–120.
Galetskiy, D., Lohscheider, J.N., Kononikhin, A.S., Popov, I.A., Nikolaev, E.N., and Adamska, I., Rapid Commun. Mass Spectrom., 2011, vol. 25, no. 1, pp. 184–190.
Lorand, L. and Chenoweth, D., Proc. Natl. Acad. Sci. U. S. A., 1969, vol. 63, no. 4, pp. 1247–1252.
Pechik, I., Madrazo, J., Mosesson, M.W., Hernandez, I., Gilliland, G.L., and Medved, L., Proc. Natl. Acad. Sci. U. S. A., 2004, vol. 101, no. 1, pp. 2718–2723.
Rosenfeld, M.A., Vasilyeva, A.D., Yurina, L.V., and Bychkova, A.V., Free Radic. Res., 2018, vol. 52, no. 1, pp. 14–38.
Chen, R. and Doolittle, R.F., Biochemistry, 1971, vol. 10, no. 24, pp. 4487–4491.
Sobel, J.H. and Gawinowicz, M.A., J. Biol. Chem., 1996, vol. 271, no. 32, pp. 19288–19297.
Davies, M.J., Biochem. J., 2016, vol. 473, no. 7, pp. 805–825.
Tanaka, T., Hocker, L.O., and Benedek, G.B., J. Chem. Phys., 1973, vol. 59, no. 9, pp. 5151–5159.
Wang, L., Li, L., Wang, H., and Liu, J., Biochem. J., 2016, vol. 473, no. 23, pp. 4373–4384.
ACKNOWLEDGMENTS
In this study, we used the equipment of the Core Facility of the Emanuel Institute of Biochemical Physics, Russian Academy of Sciences.
Funding
The study was supported by the Russian Foundation for Basic Research (project no. 18-04-01313). Mass spectrometric data were obtained with the support of the Russian Science Foundation (project no. 14-24-00114).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest. The authors declare that they have no conflict of interest.
Statement of compliance with standards of research involving humans as subjects. All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. Informed consent was obtained from all individual participants involved in the study.
Additional information
Translated by M. Batrukova
Rights and permissions
About this article

Cite this article
Yurina, L.V., Vasilyeva, A.D., Kononenko, V.L. et al. The Structural–Functional Damage of Fibrinogen Oxidized by Hydrogen Peroxide. Dokl Biochem Biophys 492, 130–134 (2020). https://doi.org/10.1134/S1607672920020167
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1607672920020167