
Abstract
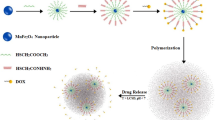
A novel pH-responsive magnetic nanosystem was designed and developed for cancer therapy. For this purpose, a well-defined terpolymer was synthesized through the polymerization of 2-hydroxyethyl methacrylate (HEMA) monomer using reversible addition fragmentation chain transfer (RAFT) technique and simultaneous grafting of ε-caprolactone (ε-CL) monomer via ring-opening polymerization (ROP) approach followed by block copolymerization of acrylic acid (AA) monomer via RAFT polymerization method, in order to produce a poly(acrylic acid)-block-[poly(2-oxyethyl methacrylate)-grafted-poly(ε-caprolactone)] [PAA-b-(POEMA-g-PCL)]. Afterward, magnetite nanoparticles (Fe3O4 NPs) were synthesized through chemical co-precipitation method, and then incorporated into the developed terpolymer through physical interactions (e.g., electrostatic and hydrogen bonding) via solution mixing approach. The doxorubicin hydrochloride (Dox), as an anticancer model drug, loading (LE) and encapsulation (EE) efficiencies of the fabricated magnetic nanosystem were calculated to be 96 ± 5 and 9.65 ± 0.5%, respectively. The drug release study results revealed that the developed drug delivery system (DDS) could be efficiently control the delivery of Dox in response to pH stimuli.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
INTRODUCTION
Based on cancer statistics, this disease will soon become the most important healthcare problem. Conventional cancer treatment approaches (e.g., chemo- and radiation-therapies) affects both cancerous and healthy tissues that lead to numerous side effects, including fatigue, hair loss, nausea and vomiting, easy bruising and bleeding [1‒4]. Thus, development of more efficient treatment approaches is necessary for reducing the side effects of current cancer therapy methods. In this context, the advent of nanotechnology has been sensible role in the development of novel and efficient approach for cancer therapy. Among the various nanomaterials, magnetic nanoparticles (Fe3O4 NPs) have been attracted great deal of attention mainly due to facile fabrication process and low cost raw materials, high safety, and excellent magnetic property [5‒9].
The most important biomedical applications of Fe3O4 NPs can be listed as enzyme immobilization, bioanalytical fields, magnetic resonance imaging (MRI), magnetic fluid hyperthermia therapy, as well as drug and gene delivery [10‒13]. Among these, the development of drug delivery systems (DDSs) based on Fe3O4 NPs is particular of interest due to some inherent features of magnetite DDSs, including rapid and easy isolation at the targeted area using an external magnetic field, diagnosis via MRI technique, and possibility for magnetic fluid hyperthermia therapy. However, for in vivo applications the surface modification of Fe3O4 NPs is necessary in order to protect them reticuloendothelial system as well as increase the stability and bio-availability [13, 14]. In this context, the surface modification of Fe3O4 NPs with macromolecules, especially stimuli-responsive polymers can be led to “smart” DDSs.
The stimuli-responsive systems can be self-assembled into one or more forms of micellar aggregates in aqueous solutions or supramolecular gels in response to external stimuli triggers [13, 15]. Among the numerous stimuli, pH-responsive DDSs are more fascinating mainly due to diversity of pH in cancerous site (4.0 and 5.4 are the average pH values of cancer cell lysosomal and endosomal, respectively) and blood or normal tissues (pH = 7.4) that lead to “smart” drug release at the target site [16‒18]. In general, the pH-responsive polymers possess acidic (e.g., carboxyl) or basic (e.g., ammonium salt) pendent groups in own main chains, which either release or accept protons in response to pH changes [19‒20]. Among the various pH-responsive polymers, poly(acrylic acid) (PAA) with carboxyl pendent group have received more attention mainly due to its inherent physicochemical and biological features such as water solubility, good pH responsibility, excellent and instantaneous mucoadhesive properties, as well as good biocompatibility and biodegradability [13, 20].
On the other hand, the synthesis of stimuli-responsive polymeric DDSs using reversible deactivation radical polymerization (RDRP; otherwise known as controlled or “living” radical polymerization) have received great deal of interest mainly due to its powerful capacity for the synthesis of polymeric materials with well-defined structures with narrow dispersity [13, 20, 21]. Three main approaches in this polymerization technique are atom transfer radical polymerization (ATRP) [22], reversible addition–fragmentation chain transfer (RAFT) polymerization [23], and nitroxide-mediated polymerization (NMP) [24]. Amongst, RAFT polymerization approach is more interesting in the case of biomedical purposes mainly due to its easy and cheap experimental set-up, applicability to numerous types of vinyl monomers, as well as there is no metal contamination in this approach against ATRP method [25‒27].
According to inherent physicochemical as well as biological features of pH-responsive polymers and Fe3O4 NPs that discussed above, the association of them can be led to “smart” DDS in the case of cancer treatment. Therefore, the main purpose of the present study was introduction of a facile strategy to synthesize a novel pH-responsive magnetite DDS for simultaneous cancer diagnosis and therapy (known as theranostic nanomedicines). For this purpose, a well-defined terpolymer, namely PAA-b-(POEMA-g-PCL), was synthesized through the combination of ROP and RAFT polymerization techniques. In another experimental section, Fe3O4 NPs were synthesized using chemical co-precipitation approach, and then incorporated into the synthesized terpolymer through physical interactions (e.g., electrostatic and hydrogen bonding) via solution mixing approach. The fabricated nanosystem was loaded with Dox, and its drug loading efficiency as well as pH-dependent drug release behavior were evaluated through dialysis approach.
EXPERIMENTAL
Materials
The RAFT agent (4-cyano-4-[(phenylcarbothioyl)sulfanyl] pentanoic acid) was synthesized in our laboratory [20]. The 2-hydroxyethyl methacrylate (HEMA), acrylic acid (AA), and ε-caprolactone (ε‑CL) monomers were purchased from Merck (Darmstadt, Germany), dried over calcium hydride, vacuum-distilled, and then stored at 4°C prior to use. The initiator 2,2'-azobisisobutyronitrile (AIBN, Fluka, Switzerland) was re-crystallized from ethanol at 50°C before use. The ferrous chloride tetrahydrate (FeCl2⋅4H2O) and ferric chloride hexanhydrate (FeCl3⋅6H2O) were purchased from Merck and utilized as received. Phosphate buffered saline (PBS) was purchased from Invitrogen (Carlsbad, CA, USA) and used as received. Doxorubicin hydrochloride (Dox) was prepared from Zhejiang, China and was used as received. All other reagents were purchased from Merck or Sigma-Aldrich (St. Louis, MO, USA) and were used as received.
One-pot Synthesis of POEMA-g-PCL Copolymer
A flame-dried polymerization ampule was charged with RAFT agent (100 mg, 0.37 mmol), AIBN (3.0 mg, 18 µmol), HEMA (1.87 mL, 15.3 mmol), ε-CL (2.91 mL, 26.3 mmol), Sn(Oct)2 (96 mg, 0.237 mmol), and toluene (10 mL). The ampule was degassed with several freeze–pump–thaw cycles, sealed off under vacuum, and placed in an oil bath at 100 ± 3°C for about 4 h. Afterward, the ampule was cooled to room temperature in order to stop the polymerization reaction. The content of the ampule was diluted with toluene (20 mL), and the synthesized product was precipitated in a large amount of cold methanol. The product obtained was dried under vacuum overnight.
Synthesis of PAA-b-(POEMA-g-PCL)
The PAA-b-(POEMA-g-PCL) was synthesized through the block copolymerization of AA monomer onto POEMA-g-PCL using RAFT technique as follows. A flame-dried polymerization ampule was charged with macro-RAFT agent (POEMA-g-PCL; 1.5 g, 0.10 mmol), AIBN (3.0 mg, 18 µmol), AA monomer (2 mL, 29 mmol), and dried N,N-dimethylformamide (DMF, 10 mL). The ampule was degassed with several freeze–pump–thaw cycles, sealed off under vacuum, and placed in an oil bath at 80 ± 3°C for about 12 h. At the end of this time, the ampule was cooled to room temperature in order to quench the polymerization. The product was recovered through the precipitation in large amount of diethyl ether. Finally, the product was dried under vacuum for 24 h at room temperature (Scheme 1).

Scheme 1.
Synthesis of Fe3O4 NPs
The Fe3O4 NPs were synthesized by chemical co-precipitation approach as described in our previous work [28]. In brief, solutions of ferric chloride (40 mL, 0.1 mol/L) and ferrous chloride (20 mL, 0.1 mol/L) was mixed in a 250-mL three-neck round-bottom flask, and degassed through the bubbling of highly pure argon gas for some minutes. Afterward, the reactor was placed in an oil bath at 80 ± 3°C under argon protection. At this time, NH4OH (10 mL, 25 wt %) was added dropwise under vigorous stirring. The resultant suspension was maintained at mentioned temperature for about 1 h while being stirred under argon atmosphere. The black NPs were precipitated through centrifugation for about 15 min at 10000 rpm, washed several times with water followed by ethanol until its pH becomes completely neutral. The product obtained was dried under vacuum at room temperature.
Fabrication of PAA-b-(POEMA-g-PCL)/Fe3O4 Nanocomposite
The PAA-b-(POEMA-g-PCL)/Fe3O4 nanosystem was fabricated through physical mixing as follows. The synthesized PAA-b-(POEMA-g-PCL) (200 mg) and Fe3O4 NPs (100 mg) were dispersed in DMSO (15 mL) by sonication for about 20 min, and then stirred for about 24 h at room temperature. The un-coated Fe3O4 NPs were separated by centrifugation at 10000 rpm for 5 min. The fabricated nanosystem was further purified through an external magnetic field and decantation process for several times in order to remove the un-adsorbed polymeric chains [28, 13].
Formulation of PAA-b-(POEMA-g-PCL)/Fe3O4-Dox Nanomedicine
A 10-mL round-bottom flask was charged with PAA-b-(POEMA-g-PCL)/Fe3O4 (200 mg) and deionized water (10 mL). The content of the flask was sonicated for about 15 min. The drug (Dox, 20 mg) was added to the flask, and then stirred for about 48 h in darkness at room temperature. The formulated nanomedicine collected by centrifugation at 10000 rpm for about 20 min. The loading (LE) and encapsulation (EE) efficiencies of PAA-b-(POEMA-g-PCL)/Fe3O4 were determined by analyzing the unloaded drug concentration using UV–Vis spectroscopy at 480 nm according to following equations [13]. It is worth noting that the experiment was conducted in triplicate and the results were reported as a mean ± standard deviation (SD). It should be pointed out that Dox is a water-soluble anticancer drug (50‒52 mg per mL).
In Vitro pH-dependent Drug Release Behavior
The in vitro drug release behavior of the developed DDS was evaluated at different pH values (4 and 7.4) at 37°C. For this purpose, the fabricated PAA-b-(POEMA-g-PCL)/Fe3O4-Dox nanomedicine (300 mg) was suspended in PBS (0.01 mol/L, 50 mL), transferred into a dialyze bag with cut-off of 10 KDa, and then exposed to dialyze against buffer solution (200 mL) in pH values of 4 and 7.4 at 37°C with gentle stirring (500 rpm). At the definite time intervals, 1.00 mL of buffer solution was taking out from release media and drug concentration was determined by UV–Vis spectrophotometry at 480 nm. The aliquots were brought back into the reactor after analyzing in order to prevent volume change. The rate of drug release was calculated using the following equation:
In this equation, M1 is the cumulative mass of Dox released from the sample at a given time, and M0 is the total loading amount of drug in the sample [13, 15].
Characterization
The molecular weights of the synthesized polymers were obtained using a Waters 1515 (USA) gel permeation chromatography (GPC) instrument equipped with Breeze 1515 isocratic pump and 7725 manual injector. N,N-Dimethylformamide (DMF) was used as eluent at a flow rate of 1 mL min–1. Fourier transform infrared (FTIR) spectra of the samples were obtained in a Shimadzu 8101M FTIR (Shimadzu, Kyoto, Japan) in the pellet form with dried potassium bromide (KBr) powder. Proton nuclear magnetic resonance (1H NMR) spectra were obtained at room temperature using an FT-NMR (400 mHz) Bruker spectrometer (Bruker, Ettlingen, Germany). Ultraviolet-visible (UV–Vis) spectroscopy was taken in a Shimadzu 1650 PC UV–Vis spectrophotometer (Shimadzu, Kyoto, Japan). The particle sizes were measured by a dynamic light scattering (DLS) (Zetasizer Nano ZS90; Malvern Instruments, Malvern, UK). The powder X-ray diffraction (XRD) analysis was performed on a Siemens D5000 diffractometer (Aubrey, Texas, USA), X-ray generator (CuKα radiation with λ = 1.5406 Å) at room temperature. Thermal properties of the samples were examined using thermogravimetric analyzer (TGA-PL STA 1640 equipment (Polymer Laboratories, Shropshire, UK)). The thermogravimetric analysis (TGA) experiment was conducted under nitrogen atmosphere from room temperature to 700°C with heating rate of 10 deg/min. Magnetic properties of the samples were investigated using vibrating sample magnetometer (VSM, AGFM, Iran) at room temperature.
RESULTS AND DISCUSSION
Characterization of PAA-b-(POEMA-g-PCL)
The synthesized PAA-b-(POEMA-g-PCL) terpolymer was characterized using FTIR as well as 1H NMR spectroscopies as depicted in Figs. 1 and 2. The FTIR spectrum of the POEMA-g-PCL (Fig. 1, curve 1) exhibited the finger print absorption bands related to both POEMA and PCL segments including, the stretching vibrations of aliphatic C–H at 2941 and 2862 cm−1, the stretching vibration of the carbonyl group at 1728 cm−1, the bending vibration of –CH2 at 1467 cm−1, the bending vibration of –CH3 at 1369 cm−1, asymmetric and symmetric stretching vibrations of C–O–C at 1244 and 1182 cm–1, respectively, and the hydroxyl stretching vibration as a broad band centered at 3510 cm–1.

The FTIR spectra of (1) POEMA-g-PCL and (2) PAA-b-(POEMA-g-PCL).

The 1H NMR spectra of (1) POEMA-g-PCL and (2) PAA-b-(POEMA-g-PCL).
The most important absorption band in the FTIR spectrum of the PAA-b-(POEMA-g-PCL)terpolymer (Fig. 1, curve 2) can be listed as the hydroxyl stitching vibration centered at 3500 cm–1 as a strong and broad band, the stretching vibrations of aliphatic C–H at 2931 and 2866 cm−1, the stretching vibration of the carbonyl group at 1718 cm−1,the bending vibration of –CH2 at 1473 cm−1, the bending vibration of –CH3 at 1398 cm−1, and the stretching vibrations of C–O–C at 1253 and 1089 cm–1.
The POEMA-g-PCL and PAA-b-(POEMA-g-PCL) samples were further characterized by means of 1H NMR spectroscopy as illustrated in Fig. 2. The most important chemical shifts in the 1H NMR spectrum of the POEMA-g-PCL sample are the chemical shift of methyl group related to POEMA segment (f) at 0.83 ppm, methylene backbone of POEMA (g) at 1.35 ppm, and –OCH2 groups of POEMA (h and l) at 4.15 ppm. The 1H NMR spectrum also exhibited all characteristic chemical shifts of PCL segment as labeled. In addition, the chemical shift at 7.95 ppm (n) is related to the aromatic protons of RAFT agent.
The most important difference after block copolymerization of AA monomer onto POEMA-g-PCL is the appearance of chemical shift of carboxylic acid group (m) of PAA segment at 11.70 ppm.
The fabricated POEMA-g-PCL and PAA-b-(POEMA-g-PCL) samples were also characterized using GPC analysis in order to investigated molecular weight parameters as illustrated in Fig. 3. As seen, both samples exhibited mono-modal GPC chromatograms, which suggest there is no homo-polymer contamination. The average molecular weights of POEMA-g-PCL (curve 1) and PAA-b-(POEMA-g-PCL) (curve 2) were obtained to be 1.5 × 104 and 2.6 × 104 g/mol, respectively. The dispersity (Ð) values of the POEMA-g-PCL and PAA-b-(POEMA-g-PCL) samples are 1.14 and 1.19, respectively. These dispersity values are below 1.2, indicating good control during the polymerization process.

The GPC chromatograms of (1) POEMA-g-PCL and (2) PAA-b-(POEMA-g-PCL).
Characterization of PAA-b-(POEMA-g-PCL)/Fe3O4 Nanocomposite
The fabricated PAA-b-(POEMA-g-PCL)/Fe3O4 nanocomposite as a DDS was characterized using FTIR, XRD, VSM, and TGA as follows. The FTIR spectra of the Fe3O4 NPs and PAA-b-(POEMA-g-PCL)/Fe3O4 nanocomposite are shown in Fig. 4 (curves 1 and 2, respectively). The characteristic absorption bands of Fe3O4 NPs can be listed as the absorption band of Fe‒O at 566 cm−1, and the bending and stretching vibrations of the surface hydroxyl groups at 1596 and 3367 cm−1, respectively. As seen, the FTIR spectrum of the PAA-b-(POEMA-g-PCL)/Fe3O4nanocomposite shows the main absorption bands of PAA-b-(POEMA-g-PCL) and Fe3O4 NPs as labeled in the spectrum.

The FTIR spectra of (1) Fe3O4 NPs and (2) PAA-b-(POEMA-g-PCL)/Fe3O4 nanocomposite.
The magnetic properties of the synthesized Fe3O4 NPs and PAA-b-(POEMA-g-PCL)/Fe3O4 nanocomposite were analyzed by the VSM equipment at room temperature as illustrated in Fig. 5. The saturation magnetization (δs) of Fe3O4 NPs and PAA-b-(POEMA-g-PCL)/Fe3O4 nanocomposite are obtained to be 58.3 and 18.7 emu g–1, respectively, which means that the developed DDS has good potential for targeted drug delivery.

The magnetization curves of (1) the Fe3O4 NPs and (2) PAA-b-(POEMA-g-PCL)/Fe3O4 nanocomposite.
The XRD pattern of the Fe3O4 NPs and PAA-b-(POEMA-g-PCL)/Fe3O4nanocomposite are depicted in Fig. 6. The XRD pattern of Fe3O4 NPs exhibited diffraction peaks at about 2θ = 30.3°, 35.8°, 43.5°, 53.6°, 57.3°, and 62.8° corresponding to (220), (311), (400), (422), (511), and (440) prisms of Fe3O4 crystalline structure, respectively (JCPDS card 19-0629 for Fe3O4). The fabricated PAA-b-(POEMA-g-PCL)/Fe3O4nanocomposite was also exhibited the same characteristic peaks as mentioned above, however, in comparison with Fe3O4 NPs the intensities of these diffraction peaks were decreased slightly due to encapsulation of Fe3O4 NPs in the non-crystalline PAA-b-(POEMA-g-PCL) matrix.

The XRD pattern of (1) Fe3O4 NPs and (2) PAA-b-(POEMA-g-PCL)/Fe3O4 nanocomposite.
The thermal properties of the fabricated magnetite nanocomposite were investigated using TGA and DTA as shown in Fig. 7. The mass loss was started with initial evaporation of any organic solvent or adsorbed water (2.5 wt %) at 60 to 140°C followed by de-carboxylation and anhydride formation in the PAA segments (14 wt %) at 200 to 300°C. The final weight loss process is related to the degradation of polymeric chains (22.5 wt %) at 300 to 750°C. The residue at 750°C for the fabricated magnetite nanocomposite is 61 wt %, which main portion of this residue is related to the most thermal stable Fe3O4 NPs [29].

(1) TGA and (2) DTA curves of PAA-b-(POEMA-g-PCL)/Fe3O4 nanocomposite.
Self-Assembly Behavior
The synthesized PAA-b-(POEMA-g-PCL) is an amphiphilic terpolymer mainly due to hydrophobic feature of POEMA-g-PCL block and hydrophilic property of PAA block. Therefore, in all conditions the formation of micelles with POEMA-g-PCL as core is expected. In detail, at pH value of above 4.2 due to deprotonation of PAA, these segments are soluble and micelles with POEMA-g-PCL and PAA as core and shell, respectively, can be formed. However, in pH values below 4.2 the PAA segments are collapse due to fully protonation of carboxyl groups, so, the sizes of the micelles are decreased significantly. A possible chemical structure of the synthesized terpolymer under pH stimuli is shown in Scheme 2.

Scheme 2.
The above-mentioned theory was evaluated using DLS measurement as illustrated in Fig. 8. According to above discussion, at pH 7.4 the PAA chains is soluble and the formation of larger NPs than those of the acid pH (pH <4.2) is expected. In contrast, at pH 4 all polymeric chains aggregated onto Fe3O4 NPs. As seen in Fig. 8, in pH values of 7.4 and 4 the sizes of particles were obtained to be 131.5 and 82 nm, respectively, that confirmed self-assembly of the fabricated PAA-b-(POEMA-g-PCL) terpolymer under pH stimuli.

Particle size dependent on pH for the fabricated PAA-b-(POEMA-g-PCL)/Fe3O4 nanocomposite. pH (1) 4.0 and (2) 7.4.
In Vitro Drug Release Behavior
The Dox loading (LE) and encapsulation (EE) efficiencies of the developed PAA-b-(POEMA-g-PCL)/Fe3O4 DDS were calculated to be 96 ± 5 and 9.65 ± 0.5%, respectively, under mentioned experimental condition [13].
As expected, in pH 4 due to fully protonation of PAA segments and their aggregation release of Dox is higher than those of the physiological condition (pH 7.4 and 37°C). In detail, in pH 7.4 the PAA segment are soluble due to fully deprotonation, thus strong physical interactions (e.g., electrostatic forces, ionic interactions, as well as hydrogen bonding) reduce the release of drug (Fig. 9). It should be pointed out that in pH 4 the protonation of Dox, which increased the dissolution of this drug is another reason for the higher drug release value at lower pH values. The release profiles of pure Dox at both pH values revealed that the pure drug has rapid and complete dissolution, mainly due to high drug solubility in the dissolution medium (especially at pH 4).

In vitro release profiles of drug from (1, 2) Dox-loaded PAA-b-(POEMA-g-PCL)/Fe3O4 nanocomposite and (3, 4) pure Dox at pH values of (1, 3) 7.4 and (2, 4) 4.0 at 37°C in PBS.
According to lower pH value in cancerous tissues, the fabricated DDS has excellent potential for cancer chemotherapy due to its pH-dependent drug release behavior [30‒32]. In conclusion, according to micellar feature of the developed DDS its intramuscular or intravenous injection may be an efficient administration approach.
CONCLUSIONS
A novel pH-responsive magnetic nanocomposite, namely PAA-b-(POEMA-g-PCL)/Fe3O4 nanocomposite, was design and developed as a DDS for the cancer chemotherapy thought the combination of ROP and RAFT polymerization techniques followed by physical incorporation of Fe3O4 NPs by solvent blending. The saturation magnetization of the fabricated DDS was obtained as 18.7 emu g–1, which represent excellent magnetic property for targeted drug delivery as well as magnetic resonance imaging (MRI). The self-assembly of the PAA-b-(POEMA-g-PCL)/Fe3O4 nanocomposite under pH stimuli was confirmed using DLS equipment. In pH values of 4 and 7.4 the sizes of the NPs were obtained to 82 and 131.5 that confirm the pH-responsivity of the developed DDS.
The Dox loading (LE) and encapsulation (EE) efficiencies of the developed PAA-b-(POEMA-g-PCL)/Fe3O4 DDS were calculated to be 96 ± 5% and 9.65 ± 0.5%, respectively. The pH-dependent drug release study exhibited that at cancerous condition (acidic environments) the developed DDS has higher drug release value than those of the physiological conditions (pH 7.4 and 37°C).
According to the results, it can be anticipated that the developed PAA-b-(POEMA-g-PCL)/Fe3O4 DDS is an acceptable nanomedicine for cancer therapy mainly due to its good magnetic property (that useful for MIR diagnosis, hyperthermia therapy, as well as isolation at the targeted area using an external magnetic field) and slow as well as pH-dependent drug release behavior.
REFERENCES
J. Hu, S. Youssefian, J. Obayemi, K. Malatesta, N. Rahbar, and W. Soboyejo, Acta Biomater. 71, 363 (2018).
D. Hanahan and R. A. Weinberg, Cell 144, 646 (2011).
A. M. Bode and Z. Dong, Nat. Rev. Cancer 9, 508 (2009),
A. Wicki, D. Witzigmann, V. Balasubramanian, and J. Huwyler, J. Controlled Release 200, 138 (2015).
W. Park, A. C. Gordon, S. Cho, X. Huang, K. R. Harris, A. C. Larson, and D. H. Kim, ACS Appl. Mater. Interfaces 9, 13819 (2017).
A. Espinosa, R. Di Corato, J. Kolosnjaj-Tabi, P. Flaud,T. Pellegrino, and C. Wilhelm, ACS Nano 10, 2436 (2016).
M. Frank, C. B. Borkner, M. Toro-Nahuelpan, H. M. Herold, D. S. Maier, J. M. Plitzko,T. Scheibel, and D. Schüler, Biomacromolecules 19, 962 (2018).
K. Soleiman, E. Arkana, H. Derakhshankhah, B. Haghshenas, R. Jahanban-Esfahlan, M. Jaymand, Carbohydr. Polym. 252, 117229 (2021).
A. A. Tregubov, I. L. Sokolov, A. V. Babenyshev, P. I. Nikitin,V. R. Cherkasov, and M. P. Nikitin, J. Magn. Magn. Mater. 449, 590 (2018).
A. Farnoudian-Habibi, S. Kangari, B. Massoumi, and M. Jaymand, RSC Adv. 5, 102895 (2015).
J. T. Jang, H. Nah, J. H. Lee, S. H. Moon, M. G. Kim, J. Cheon, Angew. Chim., Int. Ed. 48, 1234 (2009).
A. Jafarizad, A. Taghizadehgh-Alehjougi, M. Eskandani, M. Hatamzadeh, M. Abbasian, R. Mohammad-Rezaei, M. Mohammadzadeh, B. Togar, and M. Jaymand, Bio-Med. Mater. Eng. 29, 177 (2018).
Z. Mozafari, B. Massoumi, and M. Jaymand, Polym. -Plast. Technol. Eng. 58, 405 (2019).
L. H. Reddy, J. L. Arias, J. Nicolas, and P. Couvreur, Chem. Rev. 112, 5818 (2012).
N. Poorgholy, B. Massoumi, and M. Jaymand, Int. J. Biol. Macromol. 97, 654 (2017).
H. Xiong,Y. Wu, Z. Jiang, J. Zhou, M. Yang, and J. Yao, J. Colloid Interface Sci. 536, 135 (2019).
W. Lin, N. Yao, L. Qian, X. Zhang, Q. Chen, J. Wang, and L. Zhang, Acta Biomater. 58, 455 (2017).
D. Schmaljohann, Adv. Drug Delivery Rev. 58, 1655 (2006).
S. Mura, J. Nicolas, and P. Couvreur, Nat. Mater. 12, 991 (2013).
F. Mahmoodzadeh, M. Abbasian, M. Jaymand, and A. Amirshaghaghi, Polym. Int. 66, 1651 (2017).
A. Ghamkhari, B. Massoumi, and M. Jaymand, Des. Monomer Polym. 20, 190 (2017).
P. Chmielarz, M. Fantin, S. Park, A. A. Isse, A. Gennaro, A. J. Magenau, and K. Matyjaszewski, Prog. Polym. Sci. 69, 47 (2017).
G. Moad, Polym. Chem. 8, 177 (2017).
X. G. Qiao, O. Lambert, J. C. Taveau, P. Y. Dugas, B. Charleux, M. Lansalot, and E. Bourgeat-Lami, Macromolecules 50, 3796 (2017).
M. Abbasian, M. Judi, F. Mahmoodzadeh, and M. Jaymand, Polym. Adv. Technol. 29, 3097 (2018).
D. J. Keddie, Chem. Soc. Rev. 43, 496 (2014).
S. Perrier, P. Takolpuckdee, and C. A. Mars, Macromolecules 38, 2033 (2005).
B. Massoumi, S. V. Mousavi-Hamamlu, A. Ghamkhari, and M. Jaymand, Polym.-Plast. Technol. Eng. 56, 873 (2017).
B. Massoumi, Z. Mozaffari, and M. Jaymand, Int. J. Biol. Macromol. 117, 418 (2018).
R. Kulkarni, R. Boppana, G. Krishna-Mohan, S. Mutalik, and N. V. Kalyane, J. Colloid Interface Sci. 367, 509 (2012).
T. Jiang, Z. Zhang, Y. Zhang, H. Lv, J. Zhou, C. Li, L. Hou, and Q. Zhang, Biomaterials 33, 9246 (2012).
M. Jaymand, ACS Biomater. Sci. Eng. 6, 134 (2020).
Funding
The authors gratefully acknowledge the partial financial support from Payame Noor University, Tehran, Iran, and Nano Drug Delivery Research Center, Health Technology Institute, Kermanshah University of Medical Sciences, Kermanshah, Iran.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
The authors declare no competing financial interest.
Rights and permissions
About this article

Cite this article
Nazila Taghavi, Massoumi, B. & Jaymand, M. A Novel pH-Responsive Magnetic Nanosystem for Delivery of Anticancer Drugs. Polym. Sci. Ser. B 63, 408–417 (2021). https://doi.org/10.1134/S1560090421040102
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1560090421040102