
Abstract
A number of copolymers based on 4H-cyclopenta[2,1-b:3,4-b']dithiophene and 4H-dithieno[3,2-b:2',3'-d]silol are synthesized via direct С–Н arylation and Suzuki cross-coupling, and their properties are compared. The chemical structure of the copolymers is investigated by 1Н and 13С NMR spectroscopy, and their molecular-mass characteristics are determined by GPC. The study of absorption spectra of the copolymers in dilute solutions and thin films shows that they absorb light in a wide visible spectral range (300–800 nm). On the basis of the cyclic voltammetry data, the levels of boundary molecular orbitals—the highest occupied and the lowest unoccupied—are estimated and the width of the energy gap is shown to be in the range of 1.4–1.9 eV. Using the copolymers as donor components of the active layer, the samples of solar photocells are manufactured and their photovoltaic properties are investigated.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
An important task of modern polymer photovoltaics concerns the development of efficient donor components. Сonjugated polymers are good candidates for this purpose. One of the first thiophene-containing polymers which is still in wide use as a donor component in photovoltaics is poly(3-hexyl)thiophene (P3HT)

Photocells on its basis demonstrate an efficiency of solar light transformation of up to 3.6% [1]. However, despite many attempts to increase the efficiency of these photocells, the values of efficiency for photovoltaic cells based on P3HT are about 6.0–7.7% [2, 3]. In the case of P3HT, this fact may be attributed to both a narrow absorption spectrum of this polymer compared with the spectrum of solar radiation and a high-lying level of HOMO. This limits open-circuit voltage Uoc of the photocell, which for P3HT as a rule does not exceed 0.6 V.
Most recent publications dealing with photovoltaics are devoted to the synthesis of a new generation of narrow band-gap copolymers containing alternating donor and acceptor units [4–9]. This structure of polymer chains makes it possible to improve the efficiency of sunlight transformation by photocells based on this type of polymers via a more accurate selection of energy levels of a copolymer relative to those of the fullerene acceptor component of the photocell owing to increase in the density of short-circuit current Jsc and improvement of spectral characteristics of the polymer. Note that 4H-cyclopenta[2,1-b:3,4-b']di-thiophene (CPDT)

and its structural analog 4H-dithieno[3,2-b:2',3''-d]silol (DTS)

may be regarded as analogs of the monomer unit of regioregular polythiophene P3HT, in which each pair of thiophene rings is rigidly bound together to the annelated structures.
Because of the planar structure, the derivatives of CPDT and its analogs possess a reduced oxidation potential. This circumstance is responsible for a decrease in the width of energy gap Eg and a shift in the edge of absorption spectra to the red spectral region, where light energy emitted by sun is the highest. Just for this reason cyclopentadithiophene compounds became one of the promising donor moieties for the synthesis of conjugated polymers among the studied thiophene-containing compounds [10–13].
In this study, a number of new copolymers were synthesized on the basis of CPDT and DTS donor blocks. As acceptor units for alternating copolymers, we selected heterocyclic compounds with strong electron-acceptor properties: iso-pyrrolo[3,2-b]pyrrole-2,5-dione (IDPP)

(R = aryl or alkyl) and 4,4-difluoro-4H-cyclopenta[2,1-b:3,4-b']dithiophene (DFCPDT).

An IDPP moeity, as its analog diketopyrrolo[3,4-c]pyrrole (DPP),

where R = aryl or alkyl, which is wide use in photovoltaics [14–19], possesses a high electron-acceptor ability. This fact is of importance for the synthesis of efficient donor-acceptor copolymers. However, the synthesis of an IDPP molecule is much easier than that of DPP; therefore, it is more attractive for use in photovoltaics.
DFCPDT was chosen as another acceptor block. Owing to the presence of a difluoromethylene moiety in the CPDT structure, the character of this block changes from donor to acceptor. This choice is due to the fact that previously synthesized copolymers based on DFCPDT featured a promising photovoltaic behavior [20].
In this study, we synthesized three copolymers with the IDPP acceptor block which contained CPDT as a donor block, bithiophene (2Т) as a structural analog of the CPDT block, and quatrothiophene (4Т) block to compare the optical and electrochemical properties of these copolymers and four copolymers with the DFCPDT acceptor block and CPDT and DTS donor blocks. The copolymers based on IDPP were obtained by direct С‒Н arylation, which as described in the literature, enables the synthesis of high-molecular-mass copolymers [21–24], and the copolymers based on DFCPDT were obtained by the Suzuki cross-coupling.
EXPERIMENTAL
2.5 М n-BuLi solutions in hexane, 2-bromothiophene, N-bromosuccinimide, bromine, hydrazine hydrate, 2‑isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane, citronellol, 2-ethyl hexyl bromide, pivalic acid, N,N-dimethylcarbomoyl chloride, [1,1'‑bis(diphenylphosphino)ferrocene]palladium(II) dichloride Pd(dppf)Cl2, tetrakis(triphenylphosphine)palladium(0) Pd(PPh3)4, and palladium(II) acetate Pd(OAc)2 (Aldrich) were used as received. Solvents were diethyl ether (reagent grade), THF (reagent grade), toluene (analytical grade), hexane (reagent grade), DMF (high-purity grade), dichloromethane (reagent grade), and ethanol (high-purity grade). THF and diethyl ether were dried over calcium hydride and distilled in a flow of argon. DMF and tetramethylethylenediamine were dried over barium oxide and distilled under vacuum. Dichloromethane was boiled with phosphorus(V) oxide and distilled in a flow of argon. Other solvents were distilled on a rotary vacuum evaporator directly before use. Preparative column chromatography was performed using silica gel 60 (Merck, Germany). Thin-layer chromatography was carried out using Sorbfil plates (Sorbpolimer, Russia).
The optical properties of the synthesized copolymers were studied using spectroscopically pure THF (UV spectroscopy grade, Acros Organics). Analytical GPC was conducted with the aid of a Shimadzu instrument (Japan) equipped with RID-10A and SPD-M10AVP diode matrix detectors and Phenomenex (United States) 7.8 mm × 300 mm columns packed with a Phenogel sorbent having pore sizes of 500 and 103 Å. THF was used as an eluent. Analytical GLC was conducted on a Khromatek Analitik 5000 chromatograph (Russia) equipped with catharometer as a detector, 2 m × 3 mm columns, and SE-30 (5%) stationary phase coated on Chromaton-H-AW; helium was used as a carrier gas.
1Н NMR spectra were measured on a Bruker WP-250 SY spectrometer operating at a frequency of 250 MHz, and 13C and 29Si NMR spectra were recorded on a Bruker Avance 300 spectrometer operating at frequencies of 75 and 60 MHz, respectively. Absorption spectra were taken in the range of 200–1000 nm in dilute solutions (10–5–10–6 mol/L per monomer unit of the polymer) to avoid self-absorption. Absorption spectra were recorded on a Shimadzu UV-2501PC spectrophotometer (Japan).
Thermogravimetric studies of the samples were conducted in the dynamic regime in the range of 50–700°C using a Mettler Toledo TG50 system in a flow of nitrogen and air (200 mL/min). The accuracy of determination of the sample mass was up to 1 µg. The heating rate was 10°С/min. Phase and relaxation transitions in the copolymers were investigated on a Mettler Toledo DSC30 instrument under the dynamic regime at a heating rate of 20°С/min in a flow of nitrogen of 50 mL/min. Electrochemical measurements were conducted in an electrolyte solution containing 0.1 М tetrabutylammonium hexafluorophosphate (Bu4NPF6) in acetonitrile. In each case, the polymer film was deposited on a glassy-carbon surface used as a working electrode. An auxiliary electrode was a platinum plate arranged in a cell. The potentials were measured relative to the saturated calomel electrode (sce). The potential sweep rate was 200 mV/s. The polymer film was prepared as follows: a saturated polymer solution in о-dichlorobenzene was applied on a glassy-carbon disk electrode and dried for 30 min. Microwave synthesis was conducted in a SEM Discovery System microwave synthesizer (United States). Films were deposited using a G3 spin coater (Spin Coating Systems, United States). Metal electrodes were sprayed using a Univex 300 vacuum evaporator (Leybold, United States). The ytterbium-aluminum or calcium-aluminum bilayer system was used as a cathode material.
Synthesis of Monomers
2,2'-Bithiophene (2). This compound was synthesized as described in [20] using 195 g (1.19 mol) of 2‑bromothiophene, 14.7 g (0.61 mol) of magnesium, and 0.853 g (1.2 mmol, 2 mol %) of catalyst Pd(dppf)Cl2 with a yield of 96% as a colorless crystalline mass with Тm = 31°С (Тm = 32–22°С [20]). 1H NMR (CDCl3): δ 7.19 (m, 4Н, 2,2',4,4'-ThН), 7.01 (dd, 2H, J1 = 3.7 Hz, J2 = 1.2 Hz, 3.3'-ThH) ppm. 13C NMR (δ, CDCl3): 123.7 (4-C, 4'-C), 124.3 (3-C, 3'-C), 127.7 (5-C, 5'-C), 137.3 (2-C, 2'-C) ppm.
3,3',5,5'-Tetrabromo-2,2'-bithiophene (3). This compound was synthesized as white crystals as described in [20] using 32.7 g (0.197 mol) of compound 2 and 44 mL (0.865 mol) of bromine. The product was recrystallized from toluene and ethanol. After purification the yield of the product was 82%; Тm = 138°С (Тm= 140°С [15]). 1H NMR (CDCl3): δ 7.04 (s, 2H) ppm. 13C NMR (CDCl3): δ 112.1 (3-CBr, 3'-CBr), 114.8 (5-CBr, 5'-CBr), 129.5 (2-C, 2'-C), 133 (4-C, 4'-C) ppm.
3,3'-Dibromo-5,5'-bis(trimethylsilyl)-2,2'-bithiophene (4). This compound was synthesized as described in [20] using 21.21 g (0.044 mol) of compound 3, 36.8 mL (0.092 mol) of 2.5 М n-butyllithium solution, and 11.7 mL (0.092 mol) of trimethylchlorosilane. After three recrystallization procedures from ethanol, the product was isolated as white crystals. Yield, 65%; Тm = 85°С (Тm = 86°С [20]). 1H NMR (CDCl3): δ 7.15 (s, 2H), 0.33 (s, 18H) ppm. 13C NMR (CDCl3): δ 142.9 (2-C, 2'-C), 137 (4-C, 4'-C), 133.9 (5-C, 5'-C), 112.9 (3-CBr, 3'-CBr), −0.39 (SiMe3) ppm.
2,6-Bis(trimethylsilyl-cyclopenta[2,1-b:3,4-b']dithiophen-4-one (5). This compound was synthesized as described in [29] using 3.0 g (6.4 mmol) of compound 4, 5.6 mL (14 mmol) 2.5 М n-BuLi solution, 2.0 mL (13 mmol) of tetramethylethylenediamine, and 0.69 g (6.4 mmol) of N,N-dimethylcarbomoyl chloride. Yield, 84%; Тm = 82°С. 1H NMR (CDCl3): δ 7.07 (s, 2H), 0.31 (s, 18H) ppm. 13C NMR (CDCl3): δ 183.12 (C=O), 154.33 (4-C), 144.88 (5-C), 144.14 (2-C), 127.10 (3-С), −0.24 (SiMe3) ppm.
4H-Cyclopenta-[2,1-b:3,4-b']dithiophene (6). To the stirred suspension of 4.54 g (13.5 mmol) of compound 5 in ethylene glycol (200 mL), 3.78 g (67.4 mmol) of powdered potassium hydroxide and 5.3 mL (0.108 mol) of hydrazine hydrate were added. The reaction mixture was heated to 180°С and refluxed for 8 h under an inert atmosphere. After cooling to room temperature, 100 mL of water acidified with 25 mL 1 N hydrochloric acid and 300 mL of diethyl ether were added to the reaction mass, the organic layer was separated, and the aqueous layer was extracted with diethyl ether (2 × 100 mL). The organic layer was washed with water (2 × 200 mL) until the neutral reaction and dried over Na2SO4. The solvent was removed on the rotary evaporator, and the precipitate was dried under vacuum (0.5 mbar). The product was purified by column chromatography on silica gel using hexane as an eluent. As a result, light yellow crystals were obtained. Yield, 90%; Тm = 73–74°C (Тm = 74°С [25]). 1H NMR (CDCl3): δ 7.18 (d, 2H, J = 4.8 Hz, 4,4'Н), 7.09 (d, 2H, J = 4.8 Hz, 5,5'C), 3.54 (s, 2H) ppm.
4,4-Bis(3,7-dimethyloctyl)cyclopenta[2,1-b:3,4-b']dithiophene (7a). To 1.63 g (9.1 mmol) of compound 6 dissolved in 20 mL of DMSO, 6.06 g (27.4 mmol) of 3.7-dimethyloctyl bromide-1 and 0.17 g (1 mmol) of potassium iodide were added. The reaction mass was cooled in an ice bath, and a batch of 2.18 g (39 mmol) of ground potassium hydroxide was added. The reaction mass was stirred for 12 h under an inert atmosphere, cooled in the ice bath, and poured in a separating funnel containing 50 mL of water and 250 mL of diethyl ether. The organic phase was isolated, washed with water (2 × 100 mL) until the neutral reaction, and dried over anhydrous Na2SO4, and the solvent was removed on the rotary evaporator. Excess 3.7-dimethyloctyl bromide-1 was distilled off under vacuum (0.5 mbar). The product was purified by column chromatography on silica gel using hexane as an eluent. The product was a light yellow oil. Yield, 3.64 g (87%). 1H NMR (CDCl3): δ 7.15 (d, 2H, J = 4.9 Hz, 5,5'Н), 6.92 (d, 2H, J = 4.9 Hz, 4,4'C), 1.83 (m, 4H, CН2), 1.52 (m, 2H, CH), 1.3–1.0 (br. m, 10H, CH2), 0.83 (d, 12H, J = 6.7 Hz, 4CH3), 0.74 (d, 6H, J = 6.4 Hz, 2CH3) ppm.
4,4-Didecylcyclopenta[2,1-b:3,4-b']dithiophene (7b). This compound was synthesized in a manner similar to that described above for compound 7a using 1.5 g (8.4 mmol) of 4H-cyclopenta-[2,1-b:3,4-b′]dithiophene (6), 5.58 g (25.3 mmol) of decyl bromide, 2.01 g (35.8 mmol) of potassium hydroxide, and 0.15 g (0.9 mmol) of potassium iodide. The product was purified by column chromatography on silica gel using hexane as an eluent to obtain a yellow viscous oil. Yield, 3.47 g (90%). 1H NMR (CDCl3): δ 7.13 (d, 2H, J = 4.9 Hz, 5,5'Н), 6.65 (d, 2H, J = 4.9 Hz, 4,4'C), 1.62 (m, 4H, CH2), 1.26 (m, 29H, CH2), 0.83 (m, 9H, CH3) ppm.
2,6-Dibromo-4,4-bis(3,7-dimethyloctyl)-cyclopenta[2,1-b:3,4-b']dithiophene (8а). To a solution of 2.84 g (6.2 mmol) of compound 7a in 90 mL of DMF protected from light, a solution of 2.42 g (13.6 mmol) of N-bromosuccinimide in 5 mL of DMF was slowly added dropwise. The reaction mixture was stirred at room temperature for 4 h. When the reaction was completed, 100 mL of water was added and the reaction product was extracted with diethyl ether (2 × 150 mL). The ether layer was washed water until the neutral reaction (2 × 200 mL) and dried over anhydrous Na2SO4. The solvent was distilled off on the rotary evaporator. The product was purified by passing through a Schott filter filled with silica gel using hexane as an eluent. A bright yellow oil was isolated. Yield, 3.43 g (90%). 1H NMR (CDCl3): δ 6.95 (s, 2H, 5,5'Н), 1.82(m, 4H, 2CН2), 0.94–1.79 (m, 16H, CH2), 0.87–0.61 (m, 4H, CH), 0.95–0.82 (m, 18H, CH3) ppm.
2,6-Dibromo-4,4-didecylcyclopenta[2,1-b:3,4-b']di-thiophene (8b). This compound was synthesized in a manner similar to that of compound 8a using 1.8 g (3.92 mmol) of 4,4-didecylcyclopenta[2,1-b:3,4-b']dithiophene (7b) and 1.54 g (8.63 mmol) of N-bromosuccinimide. The product was a yellow-green oil. Yield, 2.25 g (93%). 1H NMR (CDCl3): δ 6.96 (s, 2H, 5,5'Н), 1.8 (m, 4H, CН2), 0.90–1.84 (m, 32H, CH2), 1.05–0.80 (m, 6H, CH3) ppm.
2,6-Bis(4,4,5,5-tetramethyl-1,3,2-dioxoborolan-2-yl)-4,4-bis(3,7-dimethyloctyl)cyclopenta-[2,1-b:3,4-b']dithiophene (9a). To a solution of 1.32 g (2.14 mmol) of compound 8a in THF (50 mL) cooled to –78°С, 1.9 mL of 2.5 M n-BuLi solution in hexane (4.71 mmol) was added dropwise. The reaction mixture was stirred for one hour at a temperature below ‒65°С, and 0.96 g (5.14 mmol) of 2‑isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (I) was added. Cooling was stopped, and stirring was continued at room temperature for another 4 h. When the reaction was completed, the reaction mixture was poured in a separating funnel containing 50 mL of NH4Cl saturated solution and 250 mL of diethyl ether. The organic phase was separated, washed with water until the neutral reaction (2 × 200 mL), and dried over anhydrous Na2SO4. The solvent was distilled off on the rotary evaporator. The product was isolated as a light yellow oil (1.37 g) and used in subsequent reactions without additional purification. Yield, 90%. The purity of the product, according to GPC, was no less than 95%. 1H NMR (CDCl3): δ 7.41 (s, 2H, 5,5'Н), 1.78 (m, 4H, CН2), 1.35 (s, 24H, CH3), 1.29–0.71 (m, 20H, 8CH2, 4CH), 0.83 (s, 18H, CH3) ppm.
2,6-Bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-4,4-didecyl-cyclopenta-[2,1-b:3,4-b']dithiophene (9b). This compound was synthesized in a manner similar to that of compound 9a using 1.2 g (1.95 mmol) of 2,6-dibromo-4,4-didecyl-cyclopenta[2,1-b:3,4-b']dithiophene (8b), 1.7 mL (4.28 mmol) of 2.5 М n-BuLi solution, and 0.8 g (4.28 mmol) of 2‑isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane. The product was a yellow-green oil. Yield, 1.31 g (95%). 1H NMR (CDCl3): δ 7.45 (s, 2H, 5,5'Н), 1.80 (m, 4H, CН2), 1.35 (s, 24H, CH3), 0.93–1.74 (m, 32H, CH2), 1.05–0.80 (m, 6H, CH3) ppm.
4,4'-Bis(3,7-dimethyloctyl)-5,5'-bis(trimethylsilyl)dithieno[3,2-b:2',3'-d]silol (10a). To a solution of 12.3 g (26.3 mmol) of compound 4 in 500 mL of THF cooled to –78°С, 33.8 mL of 1.6 М n-BuLi solution in hexane was added dropwise. After stirring of the reaction mixture at a temperature of –78°С for 15 min, 11 g (0.034 mol) of bis(3,7-dimethyloctyl)dichlorosilane was poured into it. Afterwards, the cooling bath was removed and the reaction mass was stirred for 2 h at 23°С, poured into water, and extracted with diethyl ether. The organic phase was separated, washed until the neutral reaction, and dried over anhydrous Na2SO4, and the solvent was evaporated. The product was purified by column chromatography on silica gel using hexane as an eluent. A viscous light yellow mass was isolated. Yield, 11.4 g (78%). 1H NMR (CDCl3): δ 7.15 (s, 2H, 5,5'Н), 1.36 (m, 16H, CH2), 1.1 (m, 4H, CH), 0.61 (m, 4H, CH2–Si), 0.30 (s, 18H) ppm.
4,4'-Didecyl-5,5'-bis(trimethylsilyl)-dithieno[3,2-b:2',3'-d]silol (10b). This compound was synthesized in a manner similar to that of compound 10a using 4.01 g (8.56 mmol) of compound 4, 7.1 mL (17.5 mmol) of 2.5 М n-BuLi solution, and 3.27 g (8.56 mmol) of didecyldichlorosilane. The product was a viscous dark yellow mass. Yield, 4.31 g (76%). 1H NMR (CDCl3): δ 7.15 (s, 2H, 5,5'Н), 1.8 (m, 4H, CН2), 0.90–1.81 (m, 32H, CH2), 1.05–0.80 (m, 6H, CH3), 0.32 (s, 18H, SiMe3) ppm.
4,4'-Bis(3,7-dimethyloctyl)-5,5'-dibromodithieno- [3,2-b:2',3'-d]silol (11a). To a solution of 1.69 g (3 mmol) of compound 10a in 20 mL of THF, 1.1 g (6.17 mmol) of N-bromosuccinimide was added. The reaction mixture was stirred at 23°С for 4 h. When the reaction was completed, the reaction mass was poured into water and extracted with diethyl ether. The organic phase was separated, washed with water until the neutral reaction, and dried over anhydrous Na2SO4, and the solvent was evaporated. The product was purified by column chromatography on silica gel using hexane as an eluent. A viscous green oil was isolated. Yield, 1.28 g (90%). 1H NMR (CDCl3): δ 7.13 (s, 2H, 5,5'Н), 1.34 (m, 16H, CH2), 1.10 (m, 4H, CH), 0.61 (m, 4H, CH2–Si), 0.83 (s, 18H, CH3) ppm.
4,4'-Didecyl-5,5'-dibromodithieno[3,2-b:2',3'-d]silol (11b). This compound was synthesized in a manner similar to that of compound 11a using 5.6 g (9 mmol) of compound 10b and 3.54 g (19.9 mmol) of N-bromosuccinimide. The product was a green viscous mass. Yield, 5.3 g (94%). 1H NMR (CDCl3): δ 7.15 (s, 2H, 5,5'Н), 1.8 (m, 4H, CН2), 0.90–1.81 (m, 32H, CH2), 1.05–0.8 (overlapping signals, 6H, CH3) ppm.
4,4'-Bis(3,7-dimethyloctyl)-5,5'-bis(4,4,5,5-tetra-methyl-1,3,2-dioxo-borolan-2-yl)-dithieno-[3,2-b:2',3'-d]silol (12a). This compound was synthesized in a manner similar to that of compound 9a using 1.10 g (1.73 mmol) of compound 11а, 1.5 mL (3.81 mmol) of 2.5 М n-BuLi solution, and 0.71 g (3.81 mmol) of 2‑isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane. The product was a green viscous mass. Yield, 1.14 g (91%). 1H NMR (CDCl3): δ 7.16 (s, 2H, 5,5'Н), 1.72 (m, 4H, CН2), 1.35 (s, 24H, CH3), 1.32–1.93 (m, 16H, CH2), 1.10–0.71 (m, 4H, CH), 1.37–0.80 (s, 18H, CH3) ppm.
4,4'-Didecyl-5,5'-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-dithieno-[3,2-b:2',3'-d]silol (12b). This compound was synthesized in a manner similar to that of compound 9a using 1.77 g (2.8 mmol) of compound 11b, 2.5 mL (6.15 mmol) of 2.5 М n-BuLi solution, and 1.26 g (6.15 mmol) of 2-isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane. The product was a green mass. Yield, 1.78 g (88%). 1H NMR (CDCl3): δ 7.17 (s, 2H, 5,5'Н), 1.74 (m, 4H, CН2–Si), 1.35 (s, 24H, CH3), 0.93–1.74 (m, 32H, CH2), 1.12–0.60 (m, 6H, CH3) ppm.
3,7-Dimethyloctanol-1 (13). Fifty grams (0.32 mol) of citronellol was dissolved in 500 mL of ethyl alcohol, 1.7 g (16 mmol) of 5% Pd/C was added in a flow of argon, and hydrogen was passed through the stirred solution. When the measured hydrogen volume ceased to change (13 h), the reaction mixture was filtered off through a Schott filter and the solvent was removed on the rotary evaporator. The product as a yellow oil was distilled under vacuum; Тb = 95°C (21 mbar). Yield, 48.5 g (96%). 1H NMR (CDCl3): δ 3.65 (m, 2H, CH2ОН), 1.53 (m, 2Н, СН), 1.05–1.42 (overlapping signals, 8Н, СН2), 0.86 (t, 9Н, J = 6.7 Hz, CH3) ppm.
3,7-Dimethyloctyl bromide-1 (14). To 8.84 g (56 mmol) of 3,7-dimethyloctanol-1 and 16.10 g (0.061 mol) of triphenylphosphine in anhydrous dichloromethane, 10.93 g (0.061 mol) of N-bromosuccinimide was added in portions; the temperature of the reaction mixture was maintained not above 30°C. After stirring at room temperature for 16 h, the solvent was removed on the rotary evaporator and the residue was extracted with hexane and filtered off on the Schott filter. When hexane was removed, the product was purified by fractional vacuum distillation. Yield, 12.35 g (78%). The product was a colorless transparent liquid; Тb = 91–94°C (13 mbar) (Тb = 103–105°C (23 mbar) [26]). 1H NMR (CDCl3): δ 3.52–3.33 (m, 2H, CH2Br), 1.85–1.67 (m, 2Н, СН2), 1.62 (m, 1Н, СН), 1.51 (m, 1Н, CH), 1.35–1.21 (m, 2Н, СН2), 1.19–1.05 (m, 4Н, СН2), 0.88 (d, J = 6.5 Hz, 3Н, СН3), 0.85 (d, J = 6.5 Hz, 6H, СН3) ppm. 13С NMR (CDCl3): δ 40.00, 39.11, 36.65, 32.27, 31.59, 27.90, 24.50, 22.55, 22.66, 18.91 ppm.
Bis(3,7-dimethyloctyl)dichlorosilane (15a). To 2.35 g (0.056 mol) of magnesium in 30 mL of anhydrous THF, a solution of 10.5 mL (0.05 mol) of 3,7-dimethyloctyl bromide in 30 mL of anhydrous THF was added, and the reaction mixture was boiled for 2 h. After cooling to room temperature, the resulting mixture was added to 4.3 mL (0.025 mol) of tetrachlorosilane in 50 mL of anhydrous THF at –78°С. The reaction mixture was heated to room temperature and stirred for 12 h. Inorganic admixtures were removed via precipitation into anhydrous hexane and filtration under argon. The pure product (17.1 g) was obtained as a transparent liquid; Тb = 140–143°С (3.1 mbar). Yield, 47%. The purity of the product, as evidenced by GPC chromatography, was 98%. 1H NMR (CDCl3): δ 1.54 (m, 4H, CН2), 1.32–1.93 (m, 16H, CH2), 1.10– 0.70 (m, 4H, CH2‒Si ), 1.37–0.80 (s, 18H, CH3) ppm.
Didecyldichlorosilane (15b). This compound was synthesized in a manner similar to that of compound 15a using 53.3 g (0.241 mol) of decyl bromide, 6.44 g (0.265 mol) of magnesium, and 13.8 mL (0.120 mol) of tetrachlorosilane. Тb = 198°С (5.1 mbar) (Тb = 190°С, 1.33 mbar [27]). The product was a transparent liquid. Yield, 19.7 g (43%). 1H NMR (CDCl3): δ 0.87 (s, 6H, CH3), 1.25 (s, 32H, CH2), 1.07 (t, 4H, CH2–Si) ppm.
N,N'-Bis(4-butylphenyl)oxalamide (16). A solution of 1.20 g (10 mmol) of oxalyl chloride in 20 mL of dichloromethane was added dropwise at 0°C for 20 min to a solution of 3.00 g (20 mmol) of 4-butylaniline and 1.75 g (22 mmol) of pyridine in 50 mL of anhydrous dichloromethane. The reaction mixture was stirred at room temperature for a night, and 150 mL of NaHCO3 saturated aqueous solution was added. The resulting mixture was stirred for 2 h, and a pink precipitate became colorless. The product was filtered off on the Schott filter, washed two times with water and ethyl acetate, and dried under vacuum (0.5 mbar). The product was a white powder. Yield, 3.2 g (93%). 1H NMR (CDCl3): δ 9.29 (s, 2H, NH), 7.56 (d, 4H, –NHC6H4, J = 10.1 Hz), 7.20 (d, 4H, ‒NHC6H4, J = 10.8 Hz), 2.61 (t, 4H, J = 10 Hz, ‒CH2–Ph), 1.60 (m, 4H, –CH2–CH2–), 1.36 (m, 4H, –CH2–CH3), 0.93 (t, 6H, J = 10 Hz, –CH2–CH3) ppm.
Oxalyl-bis(imidoyl)dichloride (17). To a mixture of 3.00 g (8.5 mmol) of N,N'-bis(4-butylphenyl)oxalamide (16) and 3.53 g (1.7 mmol) of fine PCl5, 200 mL of anhydrous toluene was added, and the resulting mixture was refluxed for 8 h. When the reaction was completed, the solvent was distilled off on the rotary evaporator. The residue was dissolved in 400 mL of anhydrous hexane and filtered from insoluble admixtures. Hexane was removed on the rotary evaporator, and the product was dried under vacuum (0.5 mbar). The product was a yellow crystalline mass spreading in air. Yield, 2.4 g (70%). 1H NMR (CDCl3): δ 7.24 (d, 4H, –NHC6H4, J = 10.1 Hz), 7.09 (d, 4H, –NHC6H4, J = 10.8 Hz), 2.64 (t, 4H, J = 10 Hz, –CH2–Ph), 1.61 (m, 4H, –CH2–CH2–), 1.38 (m, 4H, –CH2–CH3), 0.94 (t, 6H, J = 9.6 Hz, ‒CH2–CH3) ppm.
1,4-Bis(4-butylphenyl)-3,6-di(thiophen-2-yl)pyrrolo[3,2-b]pyrrole-2,5(1H,4H)-dione (18). A solution of 0.77 g (4.5 mmol) of thiophene ethyl acetate in 40 mL of THF was added dropwise at 0°C to the solution of lithium diisopropylamide (3.8 mL, 2 M solution in THF) in 10 mL of THF under argon. The reaction mixture was stirred at 0°C for 1 h and cooled to –78°C, and a solution of 0.8 g (2.0 mmol) of oxalyl-bis(imidoyl)dichloride in 20 mL of anhydrous THF was slowly added dropwise. When the entire volume was added, cooling was stopped and the reaction mixture was stirred at room temperature for 12 h. Upon formation of the brown mass, the reaction mixture was neutralized with water, extracted with 150 mL of dichloromethane, and dried over anhydrous sodium sulfate. When the solvent was removed under vacuum, 100 mL of hexane was added to the dry mass, filtered on the Schott filter, and washed several times with hexane. The resulting brown powder was dried under vacuum (0.5 mbar). Yield, 0.76 g (62%). 1H NMR (CDCl3): δ 7.26 (dd, J1 = 5.1 Hz; J2 = 1.0 Hz, 2H, Th), 7.20 (s, 8H, –C6H4), 6.74 (dd, J1 = 5.1 Hz, J2 = 3.8 Hz, 2H, Th), 6.52 (dd, J1 = 3.8 Hz, J2 = 1.0 Hz, 2H, Th, 2.67 (t, 4H, J = 10 Hz, –CH2–Ph), 1.64 (m, 4H, –CH2–CH2–), 1.36 (m, 4H, –CH2–CH3), 0.96 (t, 6H, J = 9.6 Hz, –CH2–CH3) ppm.
3,6-Bis(5-bromothiophen-2-yl)-1,4-bis(4-butylphenyl)pyrrolo[3,2-b]pyrrolo-2,5(1H,4H)-dione (19). To a solution of 0.20 g (0.35 mmol) of compound 18 in 50 mL of anhydrous chloroform, 0.164 g (0.9 mmol) of N-bromosuccinimide was added in portions at room temperature. After stirring for 2 h, the reaction mixture was extracted with 150 mL of dichloromethane and dried over anhydrous sodium sulfate and the solvent was distilled off on the rotary evaporator. The crude product was dissolved in 150 mL of diethyl ether and filtered off, and the solvent was distilled off on the rotary evaporator. The product was purified by column chromatography on silica gel using methylene chloride as an eluent. The resulting brick red powder was dried under vacuum. Yield, 1.23 g (80%). 1H NMR (CDCl3): δ 7.19 (m, 8H, –NHC6H4), 6.68 (d, 2H, 4-Th, J = 5.0 Hz), 6.02 (d, 2H, 3-Th, J = 5.0 Hz), 2.68 (t, 4H, J = 10 Hz, –CH2–Ph), 1.62 (m, 4H, ‒CH2–CH2–), 1.37 (m, 4H, –CH2–CH2–), 0.96 (t, 6H, J = 9.6 Hz, –CH2–CH3) ppm.
4,4-Ethylenedithio-cyclopenta[2,1-b:3,4-b']dithiophene (20). To a solution of 5.73 g (29.8 mmol) of compound 5 and 5.62 g (59.6 mmol) of 1,2-ethanediol in 200 mL of dichloromethane, 11.9 g (89.4 mmol) of AlCl3 was added at room temperature. The reaction was conducted for 1 h. Afterwards the reaction mixture was poured in the separating funnel containing 400 mL of ice distilled water and 600 mL of dichloromethane. The organic phase was separated, washed with distilled water (2 × 200 mL) until the neutral reaction, and dried over anhydrous Na2SO4. The solvent was distilled off on the rotary evaporator, and the residue was purified by column chromatography on silica gel using toluene as an eluent. The product was a light yellow powder. Yield, 7.2 g (90%). Тm =149–150°С (Тm = 148–150°С [28]). 1H NMR (CDCl3): δ 7.16 (d, 2H, J = 4.9 Hz, 3-Th), 7.08 (d, 2H, J = 5.2 Hz, 2-Th), 3.68 (s, 4H, CН2) ppm.
2,6-Dibromo-4,4-difluoro-cyclopenta[2,1-b:3,4-b']dithiophene (21). This compound was synthesized as described in [20] using 1.0 g (3.7 mmol) of 4,4-ethylenedithiocyclopenta[2,1-b:3,4-b']dithiophene, 3.31 g (0.018 mol) of N-bromosuccinimide, and 2.3 mL (0.018 mol) of 70% hydrogen fluoride solution in pyridine. Bright yellow crystals were isolated. Yield, 0.78 g (57%); Тm = 127–132 °С (Тm = 127–131°C [20]). 1H NMR (CDCl3): δ 7.08 (s, 2H, 3,5-H) ppm. 13С NMR (CDCl3): δ 143.1, 139.9, 124.1, 116.9, 114.1 ppm. 19F NMR (CDCl3): δ –121.31 ppm.
4,4-Difluoro-cyclopenta[2,1-b:3,4-b']dithiophene (22). To a solution of 0.4 g (1.1 mmol) of compound 21 in 15 mL of anhydrous THF, 0.24 g (6.5 mmol) of LiAlH4 was added in portions in a flow of nitrogen. The reaction mixture was stirred for 1 h, 50 mL of 1 N HCl solution was added, and the reaction mass was extracted with diethyl ether (2 × 100 mL). The ether layer was washed with water and dried over sodium sulfate, and the solvent was distilled off. After purification by column chromatography on silica gel using toluene as an eluent the product was isolated as a white powder. Yield, 0.21 g (89%); Тm = 85–87°С (Тm = 87°C [28]). 1H NMR (CDCl3): δ 7.15 (d, 2H, J = 4.8 Hz, 2,6-H), 7.07 (d, 2H, J = 4.8 Hz, 3,5-H) ppm.
General Procedure of Copolymer Synthesis by the Suzuki Reaction
To the equimolar mixture of comonomers in dimethoxyethane (4 mL), Pd(PPh3)4 (5 mol %) and 2 М Na2СO3 aqueous solution (1.0 mL) were added. The reaction was conducted in a microwave reactor. The temperature was gradually increased from 105 to 125°C for 10–12 h. Upon cooling, the reaction mixture was extracted with chloroform (50 mL), and the organic layer was separated and washed with water. Afterwards 100 mL of ethanol was added and the resulting mixture was centrifuged at 10 000 rpm for 15 min. The residue was dried under vacuum, and the low-molecular-mass fraction was separated via extraction in a Soxhlet apparatus successively with acetone, ethanol, and hexane. The residue was dissolved in a minimum amount of toluene and passed through a silica-gel-packed column heated to 80°C. The solution was evaporated to the minimum volume, and the polymer was precipitated via addition of the 6-fold amount of ethanol. The product was dried under vacuum (0.5 mbar). The yield of the high-molecular-mass fraction of the polymers was 54–70%.
General Procedure of Copolymer Synthesis by Direct С–Н Arylation
To the equimolar mixture of comonomers in N‑methylpyrrolidone, 10 mol % Pd(OAc)2, 30 mol % pivalic acid, and 5-fold molar excess of K2СO3 were added. The reaction was conducted in a microwave reactor at 110°C for 10–12 h. The polymers were isolated and purified according to the procedure described above for the Suzuki reaction. The yield of the polymers after fractionation was 67–87%.
Poly[{1,4-bis(4-butylphenyl)pyrrolo[3.2-b]pyrrole-2,5(1H,4H)-dione-3,6-diyl}-co-{2,2'-bithiophene-5,5'-diyl}] (P1). The polymer was synthesized according to the above–described general technique of copolymer synthesis via direct С–Н arylation using 0.88 g (1.42 mmol) of compound 18 and 1.02 g (1.42 mmol) of compound 19. The yield of high-molecular-mass fractions of the product was 75%; Mn = 44 × 103 and Mw/Mn = 2.9. 1H NMR (CDCl3): δ 7.71–7.65 (overlapping signals, 4H, Th), 7.46–7.41 (m, 4H, Ph), 7.05–6.93 (m, 4H, Ph), 2.68 (m, 4H, CH2), 1.66–1.23 (br. m, 8Н), 0.91 (s, 6Н, CH3) ppm.
Poly[{1,4-bis(4-butylphenyl)pyrrolo[3.2-b]pyrrole-2,5(1H,4H)-dione-3,6-diyl}-co-{2,2':5',2'':5'',2'''-quatrothiophene-5,5'''-diyl}] (P2). The polymer was synthesized according to the above general technique of copolymer synthesis via direct С–Н arylation using 0.3 g (1.8 mmol) of 2,2'-bithiophene 1 and 1.30 g (1.8 mmol) of 3,6-bis(5-bromothiophen-2-yl)-1,4-bis(4-butylphenyl)pyrrolo[3.2-b]pyrrole-2,5(1H,4H)-dione 19. The yield of high-molecular-mass fractions of the product was 69%; Mn = 58.5 × 103 and Mw/Mn = 3.0. 1H NMR (CDCl3): δ 7.73–7.05 (overlapping signals, 8H, Th), 7.47 (s, 4H, Ph), 6.95 (s, 4H, Ph), 2.63 (m, 4H, CH2), 1.75–0.98 (br. m, 8Н), 0.89 (s, 6Н, CH3) ppm.
Poly[{1,4-bis(4-butylphenyl)pyrrolo[3.2-b]pyrrole-2,5(1H,4H)-dione-3,6-diyl}-co-{4,4-didecylcyclopenta- [2,1-b:3,4-b']dithiophene-2,6-diyl}] (P3). The polymer was synthesized according to the above general technique of copolymer synthesis via direct С–Н arylation using 0.64 g (1.39 mmol) of 4,4-bis(decyl)cyclopenta[2,1-b:3,4-b']dithiophene and 1.00 g (1.39 mmol) of 3,6-bis(5-bromothiophen-2-yl)-1,4-bis(4-butylphenyl)pyrrolo[3.2-b]pyrrole-2,5(1H,4H)-dione. The yield of high-molecular-mass fractions of the product was 83%; Mn = 117 × 103 and Mw/Mn = 3.5. 1H NMR (CDCl3): δ 7.68–7.43 (overlapping signals, 6H, Th), (s, 4H, Ph), 7.08 (s, 4H, Ph), 2.68 (m, 4H, CH2), 1.65–0.83 (overlapping signals, 60Н) ppm.
Poly[{4,4-difluorocyclopenta[2,1-b:3,4-b']dithiophene-3,6-diyl}-со-{4,4-didecylcyclopenta-[2,1-b:3,4-b']dithiophene-3,6-diyl}] (Р4). The polymer was synthesized according to the general technique of polymer synthesis via the Suzuki cross-coupling reaction using 0.315 g (0.4 mmol) of 2,6-bis(4,4,5,5-tetramethyl-1,3,2-dioxoborolan-2-yl-4,4-didecylcyclopenta-[2,1-b:3,4-b']dithiophene 9b and 0.150 g (0.4 mmol) of 2,6-dibromo-4,4-difluoro-cyclopenta[2,1-b:3,4-b']dithiophene 21. The yield of high-molecular-mass fractions of the product was 81%; Mn = 27.4 × 103 and Mw/Mn = 2.3. 1H NMR (CDCl3): δ 7.17–7.07 (overlapping signals, 2H, Th), 6.99 (br. s, 2H, Th), 1.90–1.76 (m, 4H), 1.29–1.19 (overlapping signals, 32H), 0.83 (s, 6H) ppm.
Poly[{4,4-difluorocyclopenta[2,1-b:3,4-b']dithiophene-3,6-diyl}-со-{4,4-bis(3,7-dimethyloctyl)-cyclopenta-[2,1-b:3,4-b']dithiophene-3,6-diyl}] (Р5). The polymer was synthesized according to the general technique of copolymer synthesis via the Suzuki cross-coupling using 0.567 g (0.8 mmol) of 2,6-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-4,4-bis(3,7-dimethyloctyl)-cyclopenta-[2,1-b:3,4-b']dithiophene 9a and 0.270 g (0.8 mmol) of 2,6-dibromo-4,4-difluorocyclopenta[2,1-b:3,4-b']dithiophene 21. The yield of high-molecular-mass fractions of the product was 79%; Mn = 21.5 × 103 and Mw/Mn = 3.2. 1H NMR (δ, CDCl3): 7.15–6.96 (overlapping signals, 4H, Th), 3.68 (m, 4H), 1.35–1.09 (overlapping signals, 20H), 0.83 (br. s, 18H) ppm.
Poly[{4,4-difluorocyclopenta[2,1-b:3,4-b']dithiophene-3,6-diyl}-со-{4,4'-didecyldithieno-[3,2-b:2',3'-d]silol-3,6-diyl}] (Р6). The polymer was synthesized according to the general technique of copolymer synthesis via the Suzuki cross-coupling using 0.268 g (0.37 mmol) of 4,4'-didecyl-5,5'-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-dithieno-[3,2-b:2',3'-d]silol 12b and 0.125 g (0.37 mmol) of 2,6-dibromo-4,4-difluoro-cyclopenta[2,1-b:3,4-b']dithiophene 21. The yield of high-molecular-mass fractions of the product was 73%; Mn = 15.8 × 103 and Mw/Mn = 2.4. 1H NMR (CDCl3): δ 7.23–7.10 (overlapping signals, 4H, Th), 3.74 (m, 4H), 1.28–1.07 (overlapping signals, 32H), 0.76 (br. s, 6H) ppm.
Poly[{4,4-difluorocyclopenta[2,1-b:3,4-b']dithiophene-3,6-diyl}-со-{4,4'-bis(3,7-dimethyloctyl)-dithieno-[3,2-b:2',3'-d]silol-3,6-diyl}] (Р7). The polymer was synthesized according to the general technique of copolymer synthesis via the Suzuki cross-coupling using 0.874 g (1.2 mmol) of 4,4'-bis(3,7-dimethyloctyl)-5,5'-bis(4,4,5,5-tetramethyl-1,3,2-dioxo-borolan-2-yl)-dithieno-[3,2-b:2',3'-d]silol 12a and 0.407 g (1.2 mmol) of 2,6-dibromo-4,4-difluoro-cyclopenta[2,1-b:3,4-b']dithiophene 21. The yield of high-molecular-mass fractions of the product was 85%; Mn = 13.8 × 103 and Mw/Mn = 1.1. 1H NMR (CDCl3): δ 7.17–6.17 (overlapping signals, 4H, Th), 3.65 (m, 4H), 1.43–1.12 (overlapping signals, 20H), 0.80 (br. s, 18H) ppm.
Manufacture of Photocells and Measurement of Their Photovoltaic Characteristics
Solutions for obtaining the active layer were prepared under inert atmosphere (argon) via mixing of the copolymers with fullerene derivatives—phenyl-C61-butyric acid methyl ester (PC61BM) and phenyl-C71-butyric acid methyl ester (PC71BM)—at a mass ratio of 1 : 1 or 2 : 1 and a total concentration of 20 mg/mL in о‑dichlorobenzene. The solutions were mixed on a magnetic stirrer under heating to 75°С for 12 h and treated for 1 h in an ultrasonic bath. Using freshly prepared solutions in air, the active layer was deposited on glass substrates (23 × 23 × 1.1 mm) with the indium-tin oxide (ITO) conducting layer and poly(ethylene dioxythiophene) (PEDOT)–poly(styrenesulfonic acid) (PSS) complex via spin coating at a rotor speed of 700 rpm. Afterwards the samples were placed in a vacuum chamber, where the spraying of upper metallic electrodes—Yb or Ca (20 nm) and Al (200 nm) layers—was performed. The as-manufactured samples had the general configuration ITO/PEDOT : PSS/active layer/Mt/Al, where Мt = Yb, Ca. The photovoltaic properties—volt-ampere characteristics in the dark and under illumination—were measured in an argon box directly after the manufacture of photocells. A simulator of solar radiation with spectrum AM1.5G was used as a source of illumination in volt-ampere measurements; the intensity of incident radiation was 100 mW/cm2. Short-circuit current density Jsc, open-circuit voltage Uoc, fill factor FF, and efficiency were determined from the volt-ampere characteristics under illumination.
RESULTS AND DISCUSSION
The trimethylsilyl derivative of bithiophene 4 obtained as described in [20] (Scheme 1) is a key compound in the synthesis of initial monomers on the basis of CPDT and DTS. Among several known methods of bithiophene cycle closure [29–31] the cyclization of 3,3'-dibromo derivative of bithiophene using N,N-methylcarbamoyl chloride was chosen as the most suitable preparative method of obtaining the CPDT moiety. The lithiation of bithiophene derivative 4 was conducted in the presence of tetramethylethylenedimaine (TMEDA) needed to enhance the activity of n-butyllithium. To the obtained dilithium derivative, 1 equiv. of N,N-methylcarbamoyl chloride was added in situ to give rise to ketone 5. To introduce alkyl substituents in position 4 of the cyclopentadithiophene ring, the keto group of ketone 5 was reduced by the Wolf–Kizhner reaction and then alkylated by the corresponding alkyl bromide in the presence of potassium iodide in DMSO. Dibromo derivatives 8a and 8b were prepared under the action of 2 equiv. of N‑bromosuccinimide in DMF. Organoboron derivatives 9a and 9b were synthesized in two stages: replacement of bromine atoms with lithium in corresponding dibromides 8a and 8b and treatment of the synthesized dilithium derivatives in situ with 2 equiv. of 2-isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (I).

Scheme 1.
To prepare dithienosylol monomers (Scheme 2), dibromide 4 was first converted into the dilithium derivative under conditions of the kinetic control for the selective substitution of bromine atoms in positions 3 and 3', and then the cycle in position 4 was closed using the appropriate dialkyldichlorosilane to afford DTS bis(trimethylsilyl) derivatives 10a and 10b. Trimethylsilyl groups in the latter derivatives were substituted under the action of N-bromosuccinimide in DMF to form dibromo derivatives 11a and 11b, which were purified by column chromatography on silica gel and converted into target organoboron derivatives 12a and 12b used in further polycondensation reactions without purification.

Scheme 2.
As opposed to the previously synthesized analogous copolymers [20], in which 2-ethylhexyl or octyl moieties are commonly used as side substituents in donor blocks, longer linear decyl and branched 3,7-dimethyloctyl moieties were applied. It was expected that the introduction of these moieties in CPDT and DTS molecules would increase ordering of the target copolymers without any loss of their solubility. 3,7-Dimethyloctyl bromide 15 was synthesized from commercial citronellol 13, in which the double bond was first reduced via hydrogenation in the presence of Pd/C, and the resulting 3,7-dimethyloctanol-1 14 was brominated by N-bromosuccinimide with PPh3 in anhydrous CH2Cl2 (Scheme 3).

Scheme 3.
Dialkyldichlorosilanes 15a and 15b were obtained from corresponding bromides via the reaction of their organomagnesium derivatives obtained in situ by the Grignard reaction with tetrachlorosilane. The reaction depicted in Schemes 1 and 2 afforded four donor blocks based on CPDT and DTS with 3,7-dimethyloctyl and decyl substituents—compounds 9a and 9b and 12a and 12b.
The synthesis of dithienopyrrolo[3.2-b]pyrrole-2,5(1H,4H)-dione (DTIDPP) acceptor block included four stages (Schemes 4). At the initial step, diamide 16 was synthesized from oxalyl chloride and п-butylaniline and was immediately transformed into imidoyl chloride 17 by boiling with phosphorus pentachloride in anhydrous toluene. The concerted cyclization of imidoyl chloride 17 with 2 equiv. of thienoacetic acid ethyl ester under mild conditions gave rise to DTIDPP 18 unsubstituted in thiophene rings. The subsequent bromination of DTIDPP 18 made it possible to prepare the dibromo derivative of DTIDPP—compound 19. Thus, because of a small number of stages and a good yield of target products, the above scheme is suitable for obtaining the DTIDPP bifunctional acceptor block.

Scheme 4.
The 4,4-difluorinated derivatives of CPDT compounds 21 and 22 (Scheme 5) were synthesized as described in [20]. Derivative 22 unsubstituted in 2,2' positions of thiophene rings was obtained to find out whether this compound may be involved in the reaction of direct С–Н arylation.

Scheme 5.
Copolymers Р1–Р3 with alternating bithiophene (2Т), quatrothiophene (4Т), and cyclopentadithiophene donor moieties and the DTIDPP acceptor block were synthesized by cross-coupling polycondensation according to the direct С–Н arylation method (Scheme 6).

Scheme 6.
Synthesis was conducted in a microwave reactor in dimethylacetamide at 110°С. A catalytic system was composed of palladium acetate, pivalic acid as a ligand, and K2CO3 as a base. The reactions were carried out to maximum monomer conversions, which were controlled by GPC. The copolymers were purified according to the standard technique described below. To the reaction mixture, 10% HCl solution was added at a temperature of 50°С for 2 h. The organic portion was extracted with chloroform, and the polymer fraction was precipitated in excess methanol. The polymeric residue obtained by centrifugation was placed in a Soxhlet apparatus, and a high-molecular-mass fraction was separated from oligomeric products via successive boiling in acetone, ethanol, and hexanе. The remaining product was dissolved in the minimum amount of toluene and passed through a silica-gel-filled column heated to 80°C. The solution was evaporated until the minimum volume, and the polymer was precipitated by adding the 6-fold amount of ethanol.
Copolymers P4–P7 with the acceptor 4,4-difluoro derivative of CPDT were synthesized by the Suzuki reaction between bifunctional pinacolborane 9a or 9b and dibromide 4,4-difluoro derivative of CPDT 21 (Scheme 7).

Scheme 7.
The reaction was carried out by microwave synthesis in the presence of 10 mol % Pd(PPh3)4 and 2 М Na2CO3 aqueous solution as a base. The completeness of reaction was estimated by GPC.
Attempts were made to synthesize copolymers with bifunctional acceptor block DFCPDT 22 and corresponding nonfunctional derivative CPDT 7 in a manner similar to copolymerization used to synthesize Р1–Р3; however, as was shown by GPC, both oligomeric and polymeric components were absent. A change in functionality on donor and acceptor blocks to the reverse one under the given conditions also did not cause the polycondensation of corresponding monomers:


This inert behavior of the DFCPDT derivative is possibly associated with a strong acceptor effect of a 4,4-difluoromethylene group on the reactivity of the CPDT block.
The molecular masses of the copolymers and the conversions of the monomers were measured by GPC using polystyrene standards. GPC curves are shown in Fig. 1.

GPC curves of copolymers Р1–Р3 synthesized by direct arylation and copolymers Р4–Р7 synthesized by the Suzuki reaction.
The molecular-mass characteristics of copolymers Р1–Р7 are listed in Table 1.
For copolymers Р1–Р3 synthesized by direct С–Н arylation, the values of Mn and Mw were fairly high, in agreement with the data from [32, 33], in which high Mn values of the copolymers were achieved using the direct С–Н arylation method. However, high polydispersity may provide evidence for a large number of branches of polymer chains because of a low regioselectivity under conditions of this reaction. This finding may be explained by the presence of four possible positions of addition (С–Н) in the initial nonfunctional monomer, as opposed to bifunctional monomers with two possible positions. The molecular mass of copolymers Р4–Р7 synthesized by the Suzuki reaction was much lower. This fact may be explained by a low purity (95%) of the used organodiboron derivatives; however, the polydispersity of these copolymers was also somewhat lower than that of copolymers Р1–Р3.
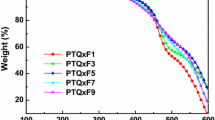
The TGA study of the thermal stability of the copolymers in nitrogen and air showed that, for copolymers Р1–Р3 (Figs. 2a, 2b), a 5% weight loss (Table 1) is observed at temperatures above 300°С both in the flow of nitrogen and in air. Note that a change of donor fragments containing dithiophene (2Т) moieties for quatrothiophene (4Т) and cyclopentadithiophene ones leads to the improvement of thermal stability from 300 to 395°С under an inert atmosphere and from 300 to 335°С in air; in addition, in a flow of nitrogen, the coke residue at 700°С grows from 52 to 63%.

TGA curves of copolymers (a, b) Р1–Р3 and (c, d) Р4–Р7. Heating at a rate of 10°С/min in (a, c) air and (b, d) nitrogen.
Copolymers Р4–Р7 are stable up to 290–330°С, at which a 5% weight loss is registered (Figs. 2c, 2d). Replacement of the С atom with the Si one on going from the copolymers based on CPDT to the copolymers based on DTS entails an insignificant reduction in thermal stability and has no effect on coke residue.
According to DSC measurements of copolymers Р1–Р3, there are no phase or relaxation transitions in the studied temperature range; for copolymers Р4–Р7, no phase transitions are detected, but in the case of copolymers Р6 and Р7, the jumps of heat capacity related to devitrification of the samples are observed at temperatures 120 and 55°С, respectively. Thus, the replacement of С with Si on going from the copolymers based on CPDT to the copolymers based on DTS promotes an increase in the mobility of molecular chains, and the replacement of n-decyl side substituents with branched 3,7-dimethyloctyl ones causes a reduction in their glass-transition temperature.
For copolymers Р1–Р7, absorption spectra were measured in the visible UV spectral region in dilute THF solutions (10–5 mol/L) and in thin films deposited on glass plates from THF solution using the rotating substrate method. The obtained data are listed in Table 2. It is seen that the copolymers efficiently absorb visible light in the range of 300–800 nm in solutions and thin films and the absorption maximum is in the range of 450–600 nm.
According to the absorption spectra of IDPP-based copolymers Р1–Р3 (Figs. 3a, 3b), it may be stated that an increase in the length of conjugation by two thiophene rings on going from Р1 to Р2 leads to the longwave shift of absorption maxima by 120 nm in solution and by 110 nm in the thin film; in the case of the annelated structure CPDT (Р3), the shift to the red spectral range is 180 nm both in solution and in the thin film relative to the unannelated structural analog Р1. For copolymers based on DFCPDT (Figs. 3c, 3d), as follows from the spectroscopy data, when the carbon atom in the donor moiety is replaced with the silicon atom, a comparison of the pair with the same 3,7-dimethyloctyl side substituents (Р4 and Р6) and the pair with the same n-decyl side substituents (Р5 and Р7) shows that the absorption maxima shift to the red spectral region by 13 and 70 nm in solutions and 41 and 59 nm in films. This fact may be explained by the stabilizing effect of the d-orbital of silicon on the π‑conjugated polymer chain. When the decyl group is replaced by the 3,7-dimethyloctyl one, the bathochromic shift is also observed for pairs Р4–Р5 and Р6–Р7. This observation may be attributed to the specific features of aggregation of macromolecules in solution and thin films.

Absorption spectra for solutions of (a) Р1–Р3 and (c) Р4–Р7 and thin films of (b) Р1–Р3 and (d) Р4–Р7.
In the series of copolymers Р1–Р2–Р3, the edges of absorption spectra for the films shift to the red spectral region; they are at 650, 720, and 850 nm, respectively. As a result, the width of the energy gap Eg decreases from 1.91 eV for Р1 to 1.72 eV for Р2 and 1.46 eV for Р3. As evidenced by cyclic voltammetry (see below), this dependence is primarily associated with increase in the levels of the highest occupied molecular orbital (HOMO). In the case of copolymers Р4–Р7 the values of Eg are close to 1.8 eV. This is related to lower lying levels of the lowest unoccupied molecular orbital (LUMO) compared with copolymers Р1–Р3.
Using oxidation and reduction potentials measured for copolymers Р1–Р3 (Fig. 4a), the energies of HOMO and LUMO were estimated through the following equations [34]:


where the charge of electron is е = 1.602 × 10–19 C.

Curves of oxidation and reduction of polymers Р1–Р7 measured by cyclic voltammetry.
For copolymers Р4–Р7 (Fig. 4b), there was no peak of reduction on the cyclic voltammograms; therefore, the energy of LUMO was derived from the difference between the energy of HOMO and the optical width of the energy gap.
For the majority of the synthesized copolymers (except P3), the levels of HOMO are fairly low lying, namely, from –5.4 to –5.7 eV (compared with –5.2 eV for P3HT). This is explained by a high oxidation potential of the CPDT structure relative to the thiophene ring (Fig. 5). Copolymers Р1–Р3 are characterized by a monotonic rise in the levels of HOMO from –5.66 to –5.22 eV because of increase in donor ability in the sequence 2Т–4Т–CPDT. Upon replacement of the carbon atom with the silicon atom, the levels of HOMO for copolymers Р6 and Р7 decrease from ‒5.61 and –5.57 eV compared with –5.42 and –5.39 eV for Р4 and Р5 copolymers, respectively. This finding may be also explained by the presence of π–d-conjugation with the silicon atom.

(Color online) Levels of HOMO (upper row) and LUMO (lower row) of copolymers Р1–Р7, P3HT, and PC61BM.
To evaluate photovoltaic properties, samples of organic solar photocells were manufactured from each of the synthesized copolymers. Measurements of the volt-ampere characteristics of photocells based on copolymers Р1–Р3 (Figs. 6a, 6b) revealed that their Uoc values are comparable with those of photocells based on P3HT; however, the values of Jsc and FF were very low because of a poor solubility of the polymers in о-dichlorobenzene and their high molecular mass. This in turn caused the nonoptimal morphology of active layer films (Fig. 7).

Photoelectric characteristics of photocells with the active layer based on the mixture of (1–3) copolymers Р1–Р3 and PC61BM at a ratio of 1 : 1 and the mixture of Р3 with PC61BM and PC71BM at a ratio of (4, 6) 1 : 1 and (5, 7) 2 : 1.

AFM images of the active layer of the photocell at Р3 : PC61BM = (a) 1 : 1 and (b) 2 : 1.
As follows from Table 3, despite almost equal values of Uoc, the values of Jsc, like efficiency, increase on going from photocells based on Р1 to photocells based on Р3. It is evident that these values correlate with the shift of absorption spectra to the longwave region.
For the photocells based on the most efficient copolymer Р3, we examined whether the efficiency may be further increased owing to a change in the composition of the active layer of photocells. For example, as Р3 : PC61BM was changed from 1 : 1 to 2 : 1, both Uoc and Jsc increased appreciably; as a result, the efficiency rose from 1.03 to 1.72%. The replacement of the acceptor fullerene derivative PC61BM with the PC71BM one made it possible to increase Jsc by almost 1.2 times and to double the efficiency relative to the corresponding parameters for photocells with the same PC61BM ratio. For the photocells with the active layer Р3 : PC71BM = 2 : 1 the value of Jsc increased to –6.63 mA/cm2 compared with –4.89 mA/cm2 for the photocells with the active layer Р3 : PC61BM. By varying the nature of the fullerene acceptor and its ratio to copolymer Р3, we managed to increase the efficiency of photocells by more than two times. This effect of the nature of fullerene derivative PC71BM may be attributed to a better absorption of light in the visible region and presumably to a better morphology of the active layer.
On the basis of copolymers Р4–Р7 in mixture with PC61BM 1 : 1, models of solar photocells were manufactured and their volt-ampere characteristics were measured (Table 4). The typical values of Uoc = 0.4–0.5 V were close to the corresponding values for photocells based on polymer P3HT; however, the low values of Jsc and FF provide evidence for the poor morphology of the film. This is the reason behind heterogeneities related to the low solubility (5–7 mg/mL) of these copolymers. To optimize the morphology of the active layer immediately after deposition, the substrates were annealed in air at 100°С for 10 min. Afterwards contacts were applied to them together with reference substrates without annealing and the volt-ampere characteristics were measured.
An analysis of the obtained data makes it possible to make the following conclusions. In the case of copolymers Р4 and Р6 (Figs. 8a, 8c), Uoc decreases on the cells with annealing. This fact in combination with steeper volt-ampere characteristics of the annealed samples indicates that the parallel resistance of the photocells decreases. The corresponding values of Jsc increase. This may be due to a more optimal ordering of the active layer. For cells based on Р4 and Р6, the efficiency increases upon annealing; however, in the case of Р5 and Р7 (Figs. 8b, 8d), this parameter remains almost unchanged. This fact may be attributed to different levels of order in the copolymers with different alkyl substituents upon annealing. For example, copolymers Р4 and Р6 with linear decyl substituents at CPDT moieties crystallize during the process of annealing. At the same time, copolymers Р5 and Р7 contain branched 2,3-dimethyloctyl substituents whose crystallizability is hindered considerably. Reduction in the fill factors and Uoc values of the solar photocells based on copolymers Р4 and Р6 after annealing may be related to a rise in the mobility of holes in the polymer due to the growth of its crystallinity. As was shown in [35], the value of Jsc monotonically increases with mobility, the value of Uoc monotonically decreases with a rise in the mobility of holes, and the fill factor first increases, attains a maximum value at a certain mobility level, and then decreases with its growth.

(Color online) Photoelectric characteristics of photocells based on copolymers (a) Р4, (b) Р5, (c) Р6, and (d) Р7 in the mixture with PC61BM = 1 : 1 (1) without annealing and (2) after annealing.
On the basis of the above data it may be stated that, owing to their optical properties and position of energy levels, the synthesized copolymers have further prospects in terms of optimization of their photovoltaic properties.
REFERENCES
P. Peumans and S. R. Forrest, Appl. Phys. Lett. 79, 126 (2001).
G. Zhao, Y. He, and Y. Li, Adv. Mater. 22, 4355 (2010).
D. Baran, R. S. Ashraf, D. A. Hanifi, M. Abdelsamie, N. Gasparini, J. A. Rohr, S. Holliday, A. Wadsworth, S. Lockett, M. Neophytou, C. J. M. Emmott, J. Nelson, C. J. Brabec, A. Amassian, A. Salleo, T. Kirchartz, J. R. Durrant, and I. McCulloch, Nat. Mater. 16, 363 (2017).
Y.-J. Cheng, Sh.-H. Yang, and Ch.-S. Hsu, Chem. Rev. 109, 5868 (2009).
T. Xu and L. Yu, Mater. Today 17, 11 (2014).
X. Guo, M. Baumgarten, and K. Mullen, Prog. Polym. Sci. 38, 1832 (2013).
Ch. Duan, F. Huang, and Y. Cao, J. Mater. Chem. 22, 10416 (2012).
A. V. Akkuratov and P. A. Troshin, Polym. Sci., Ser. B 56, 414 (2014).
S.-W. Chang, T. Muto, T. Kondo, M.-J. Liao, and M. Horie, Polym. J. 49, 113 (2017).
T.-Y. Chu, J. Lu, S. Beaupre, Y. Zhang, J.-R. Pouliot, S. Wakim, J. Zhou, M. Leclerc, Zh. Li, J. Ding, and Y. Tao, J. Am. Chem. Soc. 133, 4250 (2011).
C. Cui, X. Fan, M. Zhang, J. Zhang, J. Min, and Y. Li, Chem. Commun. 47, 11345 (2011).
S. C. Rasmussen and S. J. Evenson, Prog. Polym. Sci. 38, 1773 (2013).
Y. Zhang, J. Zou, H.-L. Yip, Y. Sun, J. A. Davies, K.‑Sh. Chen, O. Acton, and A. K.-J. Jen, J. Mater. Chem. 21, 3895 (2011).
K. H. Hendriks, G. H. L. Heintges, V. S. Gevaerts, M. M. Wienk, and R. A. J. Janssen, Angew. Chem., Int. Ed. Engl. 52, 8341 (2013).
S. Qu and H. Tian, Chem. Commun. 48, 3039 (2012).
X. Guo, S. R. Puniredd, B. He, T. Marszalek, M. Baumgarten, W. Pisula, and K. Müllen, Chem. Mater. 26, 3595 (2014).
W. Li, K. H. Hendriks, M. M. Wienk, and R. A. J. Janssen, Acc. Chem. Res. 49, 78 (2016).
J. Du, A. Fortney, K. E. Washington, M. C. Biewer, T. Kowalewski, and M. C. Stefan, J. Mater. Chem. A 5, 15591 (2017).
R. Dominguez, N. F. Montcada, P. de la Cruz, E. Palomares, and F. Langa, ChemPlusChem 82, 1096 (2017).
F. V. Drozdov, E. N. Myshkovskaya, D. K. Susarova, P. A. Troshin, O. D. Fominykh, M. Yu. Balakina, A. V. Bakirov, M. A. Shcherbina, J. Choi, D. Tondelier, M. I. Buzin, S. N. Chvalun, A. Yassar, and S. A. Ponomarenko, Macromol. Chem. Phys. 214, 2129 (2013).
A. Facchetti, L. Vaccaro, and A. Marrocchi, Angew. Chem., Int. Ed. Engl. 51, 3520 (2012).
S.-W. Chang, H. Waters, J. Kettle, Z.-R. Kuo, Ch.-H. Li, Ch.-Y. Yu, and M. Horie, Macromol. Rapid Commun. 33, 1927 (2012).
B. Schmatz, J. F. Ponder, and J. R. Reynolds, J. Polym. Sci., Part A: Polym. Chem. 56, 147 (2018).
J. F. Ponder, B. Schmatz, J. L. Hernandeza, and J. R. Reynolds, J. Mater. Chem. 6, 1064 (2018).
K. Endo, Synlett 25, 2184 (2014).
N. Cohen, Helv. Chim. Acta 64, 1158 (1981).
S. Seki, J. Am. Chem. Soc. 126, 3521 (2004).
Y. Ie, Org. Lett. 9, 2115 (2007).
J. Z. Brezezinski and J. R. Reynolds, Synthesis 8, 1053 (2002).
S. Gronowitz and B. Erickson, Ark. Kemi 21, 335 (1963).
P. Jordens, G. Rawson, and H. Wynberg, J. Chem. Soc. C 1970, 273 (1970).
A. Facchetti, L. Vaccaro, and A. Marrocchi, Angew. Chem., Int. Ed. Engl. 51, 3520 (2012).
S.-W. Chang, H. Waters, J. Kettle, Z.-R. Kuo, Ch.-H. Li, Ch.-Y. Yu, and M. Horie, Macromol. Rapid Commun 33, 1927 (2012).
D. M. de Leeuw, M. M. J. Simenon, A. R. Brown, and R. E. F. Einerhand, Synth. Met. 87, 53 (1997).
C. Deibel, A. Wagenpfahl, and V. Dyakonov, Phys. Status Solidi 2, 175 (2008).
H. Zhou, L. Yang, S. Stoneking, and W. You, ACS Appl. Mater. Interfaces 2, 1377 (2010).
ACKNOWLEDGMENTS
This work was supported by grant of the President of the Russian Federation for leading scientific schools NSh-5698.2018.3.
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated by T. Soboleva
Rights and permissions
About this article

Cite this article
Drozdov, F.V., Surin, N.M., Peregudova, S.M. et al. Synthesis and Properties of Alternating Copolymers Based on 4H-Cyclopenta[2,1-b:3,4-b']dithiophene and 4H-Dithieno[3,2-b:2',3'-d]silol. Polym. Sci. Ser. B 61, 56–76 (2019). https://doi.org/10.1134/S1560090419010032
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1560090419010032