
Abstract
A series of cyclometalated platinum(II) complexes with nitrile and isocyanide ligands (RCN and RNC; R = t-Bu, Bn, Ph) have been synthesized in 60–80% yields from the dimer [{Pt(ppy)Cl}2] (Hppy is 2-phenylpyridine) and the corresponding nitriles and isocyanides. The structure of the synthesized complexes was determined by mass spectrometry, IR and NMR spectroscopy, and X-ray diffraction. The contributions of different intermolecular interactions to the crystal packing were estimated by Hirshfeld surface analysis. Photophysical properties of the synthesized complexes in solution and solid state were studied.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
In recent years, studies of C,N-cyclometalated platinum(II) complexes have become a rapidly developing field of coordination and organometallic chemistry due to interesting photophysical properties of this class of compounds [1–4]. Rational design of cyclometalating (C^N) and auxiliary ligands (L1, L2) in compounds like [Pt(C^N)(L1,L2)]Z allows fine tuning of photophysical properties, so that such complexes can be used as materials for emitting layer in light-emitting devices [5, 6], photocatalysts in organic synthesis and hydrogen generation processes [7], sensitized dyes in solar cells [8], and biosensors for diagnostics of diseases [9].
Nitriles and isocyanides are auxiliary ligands with a linear structure which ensures the absence of steric hindrances to various intermolecular noncovalent interactions [10] and hence increases structural rigidity of molecules in crystal, thus reducing nonradiative energy dissipation [4, 11–14]. Reported examples of cyclometalated platinum(II) complexes with nitrile or isocyanide ligands are highly efficient luminophores [1, 15–20] and photocatalysts [21–23]. However, only a few published studies on cyclometalated platinum(II) complexes with such ligands are available. Moreover, no simultaneous studies of C,Ncyclometalated platinum(II) complexes with isostructural nitrile and isocyanide ligands have been performed.
In this work we synthesized two series of cyclometalated platinum(II) complexes with nitrile [Pt(ppy)Cl{NCR}] and isocyanide [Pt(ppy)Cl{CNR}] (R = t-Bu, Bn, Ph) ligands (Scheme 1) and examined their photophysical properties in solid phase and in solution.
Nitrile platinum(II) complexes [Pt(ppy)Cl{NCR}] [R = t-Bu (2a), Bn (2b), Ph (2c)] were prepared by stirring a suspension of the μ-chloro dimer [{Pt(ppy)Cl}2] (1) in the corresponding nitrile (excess) at 40°C for 24 h (Scheme 1). Complexes 2a–2c were isolated as analytically pure yellow finely crystalline powders in 60–70% yield. Crystalline complexes 2a–2c are stable on exposure to air at room temperature; however, they are decomposing to initial dimer 1 and nitrile in CH2Cl2, CH(D)Cl3, or MeOH solutions. According to the 1H NMR data, the complete decomposition was achieved in 6– 24 h. As reported previously for the complex [Pt(ppy)Cl{NCMe}] [24], the formation of nitrile complexes 2a–2c and their decomposition in solution are equilibrium processes. This was indirectly confirmed by the presence of the [Pt(ppy)Cl{NCR}]+ ion peaks in the mass spectra of suspensions of stoichiometric amounts of dimer 1 and nitriles in methanol.

1.
Isocyanide complexes [Pt(ppy)Cl{CNR}] [R = t-Bu (3a), Bn (3b), Ph (3c)] were prepared by reacting dimer 1 with 2 equiv of CNR in 1,2dichloroethane. Compounds 3a–3c were isolated in 72–80% yield as analytically pure yellow finely crystalline powders. Unlike nitrile complexes 2a–2c, their isocyanide analogs 3a–3c are stable both in the crystalline state and in solution (CH2Cl2, CHCl3, MeOH).
Newly synthesized complexes 2b, 2c, and 3a–3c were characterized by high-resolution mass spectrometry and IR and 1H, 13C–{1H}, 195Pt–{1H}, 1H–1H COSY/1H–1H NOESY, and 1H–13C HSQC/1H–13C HMBC NMR spectroscopy. In addition, the structures of 2c and 3b were determined by X-ray analysis. As shown by 1H NMR spectroscopy, compound 2a in CDCl3 solution is partially converted to dimer 1 and free nitrile ligand even in 15 min after dissolution; therefore, this complex in solution was studied only by mass spectrometry and 1H NMR. The spectral characteristics of 3c fully agree with those reported previously [20].
In the mass spectra of solutions of 2a–2c and 3a–3c in methanol, the base peak was that of the [M – Cl]+ ion with a characteristic isotope distribution. The IR spectra of isocyanide complexes 3a–3c showed a strong C≡N stretching band in the region 2180–2200 cm–1, which is typical of isocyanide platinum(II) complexes [18, 25–28]. In the IR spectra of nitrile complexes 2a–2c, the C≡N stretching band was located at 2218–2277 cm–1.
The 1H, 13C{1H}, and 195Pt–{1H} NMR spectra of 2a–2c and 3a–3c displayed only one set of signals, indicating the presence of only one isomer in solution. The formation of a single geometric isomer of 2a–2c and 3a–3c with trans-(Cppy,Cl) configuration can be rationalized by the thermodynamic trans-effect which makes trans orientation of the donor atoms Cppy/Cl and Nppy/CCNR (or Nppy/NNCR) preferred [26]. Signals of the ppy moiety in the 1H and 13C{1H} NMR spectra were assigned using 2D NMR correlation techniques (1H–1H COSY, 1H–13C HSQC, 1H–13C HMBC), and our assignment was consistent with published data for other complexes containing a [Pt(ppy)] fragment [29–31]. In the 1H NMR spectraFootnote 1 of 2a–2c and 3a–3c, a characteristic signal was that located at δ 9.50–9.70 ppm, which was assigned to the H11 proton of the phenylpyridine fragment; its downfield position is determined by the strong deshielding effect of the pyridine nitrogen atom [30–32], and its multiplicity originates from spin–spin coupling with the magnetically active 195Pt nucleus [30]. Likewise, the C2–C6 and C8 atoms of isocyanide complexes 3a–3c resonated as multiplets in the 13C NMR spectra. The platinum signal in the 195Pt NMR spectra appeared in the δPt range from –3945 to –3893 ppm, which is typical of cyclometalated platinum(II) complexes with isocyanide ligands [18, 25].
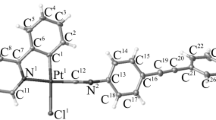
The structure of complexes 2c and 3b in the solid state was studied by X-ray diffraction (Fig. 1). The crystal structure of 3c was reported by us previously [20], and the corresponding data are used here for comparison. The crystallographically independent part of unit cells of 2c and 3b is represented by one [Pt(ppy)Cl(L)] molecule (Fig. 1) against two molecules for complex 3c. The selected bond lengths and bond angles in the complexes are given in Table 1 (the molecule of 3c with shorter bonds was selected). In all structures, the nitrile/isocyanide ligand and phenylpyridine carbon atom coordinated to Pt(II) are oriented cis, which is consistent with the X-ray diffraction data for other isocyanide and nitrile complexes of platinum halides with cyclometalated phenylpyridine ligand [16, 18]. The Pt–C bond with the isocyanide ligand of complexes 3b and 3c is shorter by about 0.1 Å than the Pt–C bond with the phenylpyridine ligand. A similar pattern is observed for complex 2c where the Pt–N bond with the nitrile ligand is shorter by ~0.05 Å than the Pt–N bond with the phenylpyridine fragment. In all cases, the nitrile or isocyanide C≡N triple bond length is similar to the lengths of analogous bonds in other platinum(II) complexes [34, 35]. All other bond lengths were in good agreement with interatomic distances in other cyclometalated nitrile and isocyanide platinum(II) complexes [15, 19, 36].

Structures of complexes (a) 2c and (b) 3b in crystal according to the X-ray diffraction data.
In order to estimate contributions of different intermolecular contacts to the crystal packing, Hirshfeld surface analysis was performed for complexes 2c and 3b, and previously reported complex 3c [20] (Table 2). It was found that intermolecular interactions involving hydrogen atoms contribute most to the crystal packing of all these complexes. However, it should be noted that these contacts predominate due to higher fraction of hydrogen atoms in all structures. Apart from hydrogen contacts, interactions between carbon atoms, on the one hand, and carbon and nitrogen atoms, on the other, that are responsible for π–π interactions, as well as contacts with the platinum atoms (which could affect photophysical properties of the complex) were revealed. All complexes displayed Pt–H and Pt–C interactions, whereas no Pt–Pt contacts typical of bis-isocyanide and bis-nitrile platinum(II) complexes [26, 34–36, 39, 40] were found.
Photophysical properties of complexes 2a–2c and 3a–3c were studied in the solid state and CH2Cl2solution (Table 3). In particular, their electronic absorption and emission spectra were recorded, and the lifetimes excited state of luminescence were measured. The electronic absorption spectra of the complexes in solution (Table 3, Fig. 2) showed several types of optical transitions. In keeping with published data [15, 16, 18, 36, 41–43], the strong high-energy absorption bands in the region λ 200–300 nm correspond to intraligand π–π* transitions. Less intense low-energy bands at λ 320– 380 nm were assigned to spin-allowed charge transfer between the ligands (1LLCT, ppy, π→π*) and from the metal to ligand [1MLCT, dπ(Pt)→π*(ppy)] [5–7, 18, 29].

(1) Luminescence excitation and (2) emission spectra of a solution of complex 2c in methylene chloride (c = 5× 10–4 M, 293 K).
Both nitrile (2a–2c) and isocyanide complexes (3a–3c) in solution displayed a common vibrationally structured emission profile. Taking into account available literature data, including those for cyclometalated platinum(II) complexes with isocyanide ligands [7, 15, 38, 44], this means that orbitals of the CNR and NCR ligands in the complexes do not contribute to the radiative excited state and that the emission could originate from intraligand 3LC (ppy) transitions (with participation of the metal atom) with a small contribution of 3MLCT transitions. In accordance with the mixed 3LC/3MLCT transitions, the emission maxima of all complexes are almost similar.
The solid-state emission spectra of the complexes are characterized by similar profiles with a small shift of the emission maximum to the long-wavelength region, which may be due to noncovalent interactions occurring in the solid state. The solid-state emission spectrum of isocyanide complex 3a is represented by a broad structureless band maximized at λ 592 nm [25]. The large Stokes shift and lifetime values in the microsecond domain (0.1–0.9 μs) indicate the triplet character of luminescence of the complexes, i.e., phosphorescence.
Thus, our study of C,N-cyclometalated platinum(II) complexes with isomeric nitrile [Pt(ppy)Cl{NCR}] and isocyanide ligands [Pt(ppy)Cl{CNR}] (R = t-Bu, Bn, Ph) has shown that both types of the ligands exert similar effects on the photophysical properties. However, isocyanide complexes are stable in solution, whereas their nitrile analogs decompose to the initial μ-chloro dimer and nitrile ligand in a solution. Furthermore, the synthesized isocyanide complexes are potential precursors to N-heterocyclic and acyclic aminocarbene complexes (M-NHC and M-ADC, respectively) [45–47].
EXPERIMENTAL
Commercial reagents and solvents (Aldrich) were used without further purification. 1,2-Dichloroethane and methylene chloride were distilled over P2O5, and diethyl ether was distilled over metallic sodium in the presence of benzophenone. Dinuclear complex 1 was synthesized from K2[PtCl4] according to the procedure described in [29].
The high-resolution mass spectra (electrospray ionization) were recorded on a Bruker micrOTOF instrument using methanol as solvent; m/z values are given for most abundant isotopologues. The IR spectra were measured with a Shimadzu 8400S spectrometer in the range 4000–400 cm–1 from samples prepared as KBr pellets. The 1H, 13C{1H}, and 195Pt{1H} NMR spectra were recorded at room temperature on a Bruker Avance II+ spectrometer operating at 400.13, 100.61, and 86 MHz, respectively, using CDCl3 as solvent. The electronic absorption spectra were run on a Shimadzu UV-1800 spectrophotometer. The emission and luminescence excitation spectra were recorded, and the excited state lifetimes were measured, with a Horiba Jobin Yvon Fluorolog-3 spectrofluorometer.
Complexes 2a–2c (general procedure). A suspension of 100 mg (0.13 mmol) of [{Pt(ppy)Cl}2] in 1 mL of the corresponding nitrile RCN (R = t-Bu, PhCH2, Ph) was stirred at 50°C for 24 h until it became homogeneous. The solution was diluted with 4 mL of toluene–diethyl ether (1 : 3), and the precipitate was filtered off, washed with diethyl ether (2×4 mL), and dried in air. In the reaction with t-BuCN, light green luminescing solid precipitated and was filtered off, washed with diethyl ether (2×4 mL), and dried in air.
[Pt(ppy)Cl(NCBu-t)] (2a). Yield 73 mg (60%). IR spectrum, ν, cm–1: 2231 (C≡N), 1485 (C=N). 1H NMR spectrum, δ, ppm: 1.59 s (9H, t-Bu), 7.10–7.17 m (3H, 3-H, 4-H, 10-H), 7.26–7.28 m (1H, 5-H), 7.43–7.45 m (1H, 2-H), 7.61–7.63 m (1H, 8-H), 7.82 t.d (1H, 9-H, 3JHH = 8.0, 1.5 Hz), 9.66 d.d (1H, 11-H, 3JHH = 5.9, 3JPtH = 45.3 Hz). Mass spectrum: m/z 432.1088 [M – Cl]+. Calculated for C16H17N2Pt: 432.1034.
[Pt(ppy)Cl(NCCH2Ph)] (2b). Yield 91 mg (70%). IR spectrum, ν, cm–1: 2218 (C≡N), 1485 (C=N). 1H NMR spectrum, δ, ppm: 4.22 s (2H, CH2), 7.01 t.d (1H, 3-H, 3JHH = 8.0, 1.2 Hz), 7.11–7.17 m (2H, 4-H, 10-H), 7.33–7.47 m (7H, Ph, 2-H, 5-H), 7.60–7.62 m (1H, 8-H), 7.81 t.d (1H, 9-H, 3JHH = 8.0, 1.4 Hz), 9.63 d.d (1H, 11-H, 3JHH = 5.3, 3JPtH = 45.8 Hz). 13C NMR spectrum, δC, ppm: 25.58 (CH2), 118.30 (C8), 121.86 (C10), 123.62 (C4), 124.13 (C5), 127.57 (Ph), 127.93 (Ph), 128.05 (Ph), 128.28 (Ph), 128.81 (Ph), 129.14, 129.60, 130.38 (C3), 131.84 (C2), 132.15 (CN), 139.34 (C9), 139.66 (C1), 143.85 (C6), 150.83 (C11), 167.69 (C7). Mass spectrum: m/z 466.0865 [M – Cl]+. Calculated for C19H15N2Pt: 466.0877.
[Pt(ppy)Cl(NCPh)] (2c). Yield 86 mg (68%). IR spectrum, ν, cm–1: 2278 (C≡N), 1486 (C=N). 1H NMR spectrum, δ, ppm: 7.10–7.16 m (3H, 3-H, 4-H, 10-H), 7.35–7.49 m (1H, 5-H, 2-H), 7.58–7.66 m (3H, Ph, 8-H), 7.77 t (1H, Ph, 3JHH = 7.6 Hz), 7.82–7.86 m (1H, 9-H), 7.90 d (2H, Ph, 3JHH = 7.6 Hz), 9.70 d.d (1H, 11-H, 3JHH = 5.8, 3JPtH = 44.8 Hz). 13C NMR spectrum, δC, ppm: 118.35 (C8), 121.91 (C10), 123.72 (C4), 124.27 (C5), 129.19, 129.58, 130.52 (C3), 131.79 (C2), 132.15 (CN), 133.14 (Ph), 134.65 (Ph), 139.42 (C9), 139.90 (C1), 143.66 (C6), 150.90 (C11), 167.70 (C7). Mass spectrum: m/z 452.0703 [M – Cl]+. Calculated for C18H13N2Pt: 452.0721.
Complexes 3a and 3b (general procedure). A suspension of 100 mg (0.13 mmol) of [{Pt(ppy)Cl}2] in 20 mL of 1,2-dichloroethane was heated to 50°C, and a solution of the corresponding isocyanide CNR (R = t-Bu, PhCH2; 0.26 mmol) in 10 mL of 1,2dichloroethane was added with stirring over a period of 1 h. The mixture was stirred at 80°C for 2 h and evaporated to a minimum volume, and 10 mL of diethyl ether was added to the residue. The precipitate was filtered off and purified by recrystallization from methylene chloride–diethyl ether (2 : 1).
Complex 3c was synthesized according to the procedure described in [20].
[Pt(ppy)Cl(CNBu-t)] (3a). Yield 88 mg (72%), decomposition point 165°C. IR spectrum: ν 2206 cm–1 (N≡C). 1H NMR spectrum, δ, ppm: 1.69 s (9H, t-Bu), 7.10 t.d (1H, 3-H, 3JHH = 7.4, 1.5 Hz), 7.16 t.d (1H, 4-H, JHH = 7.5, 1.4 Hz), 7.27 d.d.d (1H, 10-H, JHH = 7.3, 5.8, 1.4 Hz), 7.46 d.d (1H, 5-H, JHH = 7.4, 1.2 Hz), 7.53 d.d (1H, 2-H, JHH = 7.6, 1.4 Hz), 7.71–7.73 m (1H, 8-H), 7.87 t.d (1H, 9-H, JHH = 7.9, 1.6 Hz), 9.50–9.59 m (1H, 11-H). 13C NMR spectrum, δC, ppm: 30.31 (CH3), 58.50 [C(CH3)], 118.39 (C8, JCPt = 35.4 Hz), 122.13 (C10, JCPt = 26.8 Hz, Pt satellites), 124.08 s (Pt satellites), (C5, JCPt = 40.0 Hz, Pt satellites), 124.35 (C4), 131.20 (C3, JCPt = 72.9 Hz, Pt satellites), 136.81 (C2, JCPt = 106.9 Hz, Pt satellites), 139.96 (C9), 140.98 (C1), 144.11 (C6, JCPt = 34.3 Hz, Pt satellites Pt), 148.55 (C11, JCPt = 22.2 Hz, Pt satellites), 166.34 (C7); no C≡N signal was detected. 195Pt–{1H} NMR spectrum: δPt –3945 ppm. Mass spectrum: m/z 432.1038 [M – Cl]+. Calculated for C16H17N2Pt: 432.1034.
[Pt(ppy)Cl(CNCH2Ph)] (3b). Yield 104 mg (80%), decomposition point 160°C. IR spectrum: ν 2220 cm–1 (C≡N). 1H NMR spectrum, δ, ppm: 5.01 s (2H, CH2), 6.95 t (1H, 3-H, 3JHH = 7.1 Hz), 7.09 t (1H, 4-H, 3JHH = 7.3 Hz), 7.22–7.29 m (2H, 10-H, Ph), 7.38–7.48 m (6H, 2-H, 5-H, Ph), 7.67–7.69 m (1H, 8-H), 7.84 t (1H, 9-H, 3JHH = 7.5 Hz), 9.50 d.d (1H, 11-H, 3JHH = 5.5, 3JHPt = 32.4 Hz). 13C NMR spectrum, δC, ppm: 48.57 (CH2), 118.50 (C8, JCPt = 36.6 Hz, Pt satellites), 122.14 (C10, JCPt = 25.5 Hz, Pt satellites), 124.09 (C5), 124.42 (C4, JCPt = 15.6 Hz, Pt satellites), 127.18 (Ph), 129.05 (Ph), 129.32 (Ph), 131.08 (Ph), 131.19 (C3, JCPt = 72.6 Hz, Pt satellites), 136.13 (C2, JCPt = 106.5 Hz, Pt satellites), 140.11 (C9), 140.97 (C1), 143.94 (C6), 148.84 (C11, JCPt = 23.0 Hz, Pt satellites), 166.31 (C7); no C≡N signal was detected. 195Pt–{1H} NMR spectrum: δPt –3943 ppm. Mass spectrum: m/z 466.0875 [M – Cl]+. Calculated for C19H15N2ClPt: 466.0867. Found, %: C 45.47; H 3.01; N 5.58. C19H15N2ClPt. Calculated, %: C 44.03; H 3.12; N 5.33.
X-Ray diffraction. The X-ray diffraction data for complexes 2c (CCDC entry no. 1408594) and 3b (CCDC entry no. 1408607) were obtained with an Xcalibur Eos diffractometer at 100 K (monochromatized MoKα radiation, λ 0.71073 nm). The structures were solved by the direct method and were refined using SHELX [48] implemented in OLEX2 [49]. A correction for absorption was applied empirically by SCALE3 ABSPACK algorithm included in CrysAlisPro [50].
Complex 2c. C18H13N2ClPt, M 487.84; monoclinic crystal system, space group P21/n; unit cell parameters: a = 8.1493(3), b = 18.7526(7), c = 10.4496(4) Å; β = 105.839(4)°; V = 1536.27(11) Å3; Z = 4; dcalc = 2.109 g/cm3; μ = 9.304 mm‒1. Total of 7103 reflection intensities were measured from a 0.1×0.1×0.1-mm single crystal, including 3308 reflections with I > 2σ(I) (Rint = 0.0386); R1(|Fo| ≥ 4σF)/R1 (all reflections) 0.0326/0.0395, wR2(|Fo| ≥ 4σF)/wR2 (all reflections) 0.0678/0.0719; ρmin/ρmax 2.38/–2.68 e/Å3.
Complex 3b. C19H15N2ClPt, M 501.87; monoclinic crystal system, space group P21/n; unit cell parameters: a = 5.3540(3), b = 14.3896(10), c = 21.1589(10) Å; β = 90.169(5)°; V = 1630.11(16) Å3; Z = 4; dcalc = 2.045 g/cm3; μ = 8.771 mm‒1; Total of 7327 reflection intensities were measured from a 0.1×0.1×0.1-mm single crystal, including 3491 reflections with I > 2σ(I) (Rint = 0.0389); R1(|Fo| ≥ 4σF)/R1 (all reflections) 0.0456/0.0616, wR2(|Fo| ≥ 4σF)/wR2 (all reflections) 0.1084/0.1084; ρmin/ρmax 3.26/–1.33 e/Å3.
The Hirshfeld surface analysis of the crystal structures of complexes 2c, 3b, and 3c was performed using CrystalExplorer 3.1 [51] in terms of the normalized contact length formalism (dnorm) based on Bondi’s van der Waals radii [38].
Notes
Hereinafter, for atom numbering, see Fig. 1.
REFERENCES
Chen, Y., Lu, W., and Che, C.M., Organometallics, 2013, vol. 32, no. 1, p. 350. https://doi.org/10.1021/om300965b
Li, K., Tong, G.S.M., Wan, Q.Y., Cheng, G., Tong, W.Y., Ang, W.H., Kwong, W.L., and Che, C.M., Chem. Sci., 2016, vol. 7, no. 3, p. 1653. https://doi.org/10.1039/c5sc03766b
Chi, Y. and Chou, P.T., Chem. Soc. Rev., 2010, vol. 39, no. 2, p. 638. https://doi.org/10.1039/b916237b
Yam, V.W.W. and Law, A.S.Y., Coord. Chem. Rev., 2020, vol. 414, article no. 213298. https://doi.org/10.1016/j.ccr.2020.213298
Yang, S.Y., Meng, F.Y., Wu, X.G., Yin, Z., Liu, X.Z., You, C.F., Wang, Y.F., Su, S.J., and Zhu, W.G., J. Mater. Chem. C, 2018, vol. 6, no. 21, p. 5769. https://doi.org/10.1039/c8tc01142g
Chan, A.K.W., Ng, M., Wong, Y.C., Chan, M.Y., Wong, W.T., and Yam, V.W.W., J. Am. Chem. Soc., 2017, vol. 139, no. 31, p. 10750. https://doi.org/10.1021/jacs.7b04952
Schneider, J., Du, P.W., Jarosz, P., Lazarides, T., Wang, X.Y., Brennessel, W.W., and Eisenberg, R., Inorg. Chem., 2009, vol. 48, no. 10, p. 4306. https://doi.org/10.1021/ic801947v
Pashaei, B., Shahroosvand, H., Graetzel, M., and Nazeeruddin, M.K., Chem. Rev., 2016, vol. 116, no. 16, p. 9485. https://doi.org/10.1021/acs.chemrev.5b00621
Wong, K.M.C., Tang, W.S., Lu, X.X., Zhu, N.Y., and Yam, V.W.W., Inorg. Chem., 2005, vol. 44, no. 5, p. 1492. https://doi.org/10.1021/ic049079p
Kinzhalov, M.A., Kashina, M.V., Mikherdov, A.S., Mozheeva, E.A., Novikov, A.S., Smirnov, A.S., Ivanov, D.M., Kryukova, M.A., Ivanov, A.Yu., Smirnov, S.N., Kukushkin, V.Yu., and Luzyanin, K.V., Angew. Chem., Int. Ed., 2018, vol. 57, no. 39, p. 12785. https://doi.org/10.1002/anie.201807642
Solomatina, A.I., Aleksandrova, I.O., Karttunen, A.J., Tunik, S.P., and Koshevoy, I.O., Dalton Trans., 2017, vol. 46, no. 12, p. 3895. https://doi.org/10.1039/c7dt00349h
Wang, W.Z., Zhang, Y., and Jin, W.J., Coord. Chem. Rev., 2020, vol. 404, article no. 213107. https://doi.org/10.1016/j.ccr.2019.213107
Koshevoy, I.O., Krause, M., and Klein, A., Coord. Chem. Rev., 2020, vol. 405, article no. 213094. https://doi.org/10.1016/j.ccr.2019.213094
Eremina, A.A., Kinzhalov, M.A., Katlenok, E.A., Smirnov, A.S., Andrusenko, E.V., Pidko, E.A., Suslonov, V.V., and Luzyanin, K.V., Inorg. Chem., 2020, vol. 59, no. 4, p. 2209. https://doi.org/10.1021/acs.inorgchem.9b02833
Diez, A., Fornies, J., Fuertes, S., Lalinde, E., Larraz, C., Lopez, J.A., Martin, A., Moreno, M.T., and Sicilia, V., Organometallics, 2009, vol. 28, no. 6, p. 1705. https://doi.org/10.1021/om800845c
Fornies, J., Sicilia, V., Borja, P., Casas, J.M., Diez, A., Lalinde, E., Larraz, C., Martin, A., and Moreno, M.T., Chem. Asian J., 2012, vol. 7, no. 12, p. 2813. https://doi.org/10.1002/asia.201200585
Shahsavari, H.R., Aghakhanpour, R.B., Hossein-Abadi, M., Haghighi, M.G., Notash, B., and Fereidoonnezhad, M., New J. Chem., 2017, vol. 41, no. 24, p. 15347. https://doi.org/10.1039/c7nj03110f
Martinez-Junquera, M., Lara, R., Lalinde, E., and Moreno, M.T., J. Mater. Chem. C, 2020, vol. 8, no. 21, p. 7221. https://doi.org/10.1039/d0tc01163k
Sivchik, V.V., Grachova, E.V., Melnikov, A.S., Smirnov, S.N., Ivanov, A.Y., Hirva, P., Tunik, S.P., and Koshevoy, I.O., Inorg. Chem., 2016, vol. 55, no. 7, p. 3351. https://doi.org/10.1021/acs.inorgchem.5b02713
Katkova, S.A., Kinzhalov, M.A., Novikov, A.S., and Luzyanin, K.V., New J. Chem., 2021, vol. 45, no. 6, p. 2948. https://doi.org/10.1039/D0NJ05457G
Anderson, C., Crespo, M., Morris, J., and Tanski, J.M., J. Organomet. Chem., 2006, vol. 691, no. 26, p. 5635. https://doi.org/10.1016/j.jorganchem.2006.09.012
Wang, H., Bisoyi, H.K., Wang, L., Urbas, A.M., Bunning, T.J., and Li, Q., Angew. Chem., Int. Ed., 2018, vol. 57, no. 6, p. 1627. https://doi.org/10.1002/anie.201712781
Boterashvili, M., Lahav, M., Shankar, S., Facchetti, A., and van der Boom, M.E., J. Am. Chem. Soc., 2014, vol. 136, no. 34, p. 11926. https://doi.org/10.1021/ja5066587
Zaitceva, O., Beneteau, V., Ryabukhin, D.S., Eliseev, I.I., Kinzhalov, M.A., Louis, B., Vasilyev, A.V., and Pale, P., Tetrahedron, 2020, vol. 76, no. 14, article no. 131029. https://doi.org/10.1016/j.tet.2020.131029
Katkova, S.A., Leshchev, A.A., Mikherdov, A.S., and Kinzhalov, M.A., Russ. J. Gen. Chem., 2020, vol. 90, no. 4, p. 648. https://doi.org/10.1134/S1070363220040143
Kinzhalov, M.A., Kashina, M.V., Mikherdov, A.S., Katkova, S.A., and Suslonov, V.V., Russ. J. Gen. Chem., 2018, vol. 88, no. 6, p. 1180. https://doi.org/10.1134/S107036321806021x
Kinzhalov, M.A., Zolotarev, A.A., and Boyarskiy, V.P., J. Struct. Chem., 2016, vol. 57, no. 4, p. 822. https://doi.org/10.1134/S0022476616040302
Kinzhalov, M.A., Timofeeva, S.A., Luzyanin, K.V., Boyarskiy, V.P., Yakimanskiy, A.A., Haukka, M., and Kukushkin, V.Y., Organometallics, 2016, vol. 35, no. 2, p. 218. https://doi.org/10.1021/acs.organomet.5b00936
Kinzhalov, M.A., Katkova, S.A., Doronina, E.P., Novikov, A.S., Eliseev, I.I., Ilichev, V.A., Kukinov, A.A., Starova, G.L., and Bokach, N.A., Z. Kristallogr. Cryst. Mater., 2018, vol. 233, p. 795. https://doi.org/10.1515/zkri-2018-2075
Pawlak, T., Niedzielska, D., Vícha, J., Marek, R., and Pazderski, L., J. Organomet. Chem., 2014, vol. 759, p. 58. https://doi.org/10.1016/j.jorganchem.2014.02.016
Niedzielska, D., Pawlak, T., Wojtczak, A., Pazderski, L., and Szlyk, E., Polyhedron, 2015, vol. 92, p. 41. https://doi.org/10.1016/j.poly.2015.02.028
Pazderski, L., Pawlak, T., Sitkowski, J., Kozerski, L., and Szlyk, E., Magn. Reson. Chem., 2009, vol. 47, no. 11, p. 932. https://doi.org/10.1002/mrc.2491
Kashina, M.V., Kinzhalov, M.A., Smirnov, A.S., Ivanov, D.M., Novikov, A.S., and Kukushkin, V.Yu., Chem. Asian J., 2019, vol. 14, no. 21, p. 3915. https://doi.org/10.1002/asia.201901127
Mikherdov, A.S., Tiuftiakov, N.Yu., Polukeev, V.A., and Boyarskii, V.P., Russ. J. Gen. Chem., 2018, vol. 88, no. 4, p. 713. https://doi.org/10.1134/S1070363218040151
Dobrynin, M.V. , Sokolova, E.V., Kinzhalov, M.A., Smirnov, A.S., Starova, G.L., Kukushkin, V.Y., and Islamova, R.M., ACS Appl. Polym. Mater., 2021, vol. 3, no. 2, p. 857. https://doi.org/10.1021/acsapm.0c01190
Diez, A., Fornies, J., Larraz, C., Lalinde, E., Lopez, J.A., Martin, A., Moreno, M.T., and Sicilia, V., Inorg. Chem., 2010, vol. 49, no. 7, p. 3239. https://doi.org/10.1021/ic902094c
McKinnon, J.J., Jayatilaka, D., and Spackman, M.A., Chem. Commun., 2007, no. 37, p. 3814. https://doi.org/10.1039/b704980c
Bondi, A., J. Phys. Chem., 1964, vol. 68, p. 441. https://doi.org/10.1021/j100881a503
Ivanov, D.M., Kirina, Y.V., Novikov, A.S., Starova, G.L., and Kukushkin, V.Y., J. Mol. Struct., 2016, vol. 1104, p. 19. https://doi.org/10.1016/j.molstruc.2015.09.027
Mikherdov, A.S., Orekhova, Yu.A., and Boyarskii, V.P., Russ. J. Gen. Chem., 2018, vol. 88, no. 10, p. 2119. https://doi.org/10.1134/S1070363218100158
Jamshidi, M., Babaghasabha, M., Shahsavari, H.R., and Nabavizadeh, S.M., Dalton Trans., 2017, vol. 46, no. 45, p. 15919. https://doi.org/10.1039/c7dt03599c
Forniés, J., Fuertes, S., Larraz, C., Martín, A., Sicilia, V., and Tsipis, A.C., Organometallics, 2012, vol. 31, no. 7, p. 2729. https://doi.org/10.1021/om201036z
Solomatina, A.I., Chelushkin, P.S., Abakumova, T.O., Zhemkov, V.A., Kim, M., Bezprozvanny, I., Gurzhiy, V.V., Melnikov, A.S., Anufrikov, Y.A., Koshevoy, I.O., Su, S.H., Chou, P.T., and Tunik, S.P., Inorg. Chem., 2019, vol. 58, no. 1, p. 204. https://doi.org/10.1021/acs.inorgchem.8b02204
Kui, S.C.F., Hung, F.-F., Lai, S.-L., Yuen, M.-Y., Kwok, C.-C., Low, K.-H., Chui, S.S.-Y., and Che, C.-M., Chem. Eur. J., 2012, vol. 18, no. 1, p. 96. https://doi.org/10.1002/chem.201101880
Mikherdov, A.S., Kinzhalov, M.A., Novikov, A.S., Boyarskiy, V.P., Boyarskaya, I.A., Dar’in, D.V., Starova, G.L., and Kukushkin, V.Y., J. Am. Chem. Soc., 2016, vol. 138, no. 42, p. 14129. https://doi.org/10.1021/jacs.6b09133
Kinzhalov, M.A. and Boyarskii, V.P., Russ. J. Gen. Chem., 2015, vol. 85, no. 10, p. 2313. https://doi.org/10.1134/s1070363215100175
Kinzhalov, M.A. and Luzyanin, K.V., Coord. Chem. Rev., 2019, vol. 399, article no. 213014. https://doi.org/10.1016/j.ccr.2019.213014
Sheldrick, G.M., Acta Crystallogr., Sect. A, 2008, vol. 64, no. 1, p. 112. https://doi.org/10.1107/S0108767307043930
Dolomanov, O., Bourhis, L., Gildea, R., Howard, J., and Puschmann, H., J. Appl. Crystallogr., 2009, vol. 42, p. 339. https://doi.org/10.1107/S0021889808042726
CrysAlisPro A.T., Version 1.171.36.20 (release 27-06-2012).
Wolff, S.K., Grimwood, D.J., McKinnon, J.J., Turner, M.J., Jayatilaka, D., and Spackman, M.A., Crystal Explorer, Version 3.1, University of Western Australia, 2012.
Funding
This study was performed under financial support by the Council for Science at the President of the Russian Federation (MK-1476.2019.3) using the facilities of the Magnetic Resonance Research Center, Center for X-ray Diffraction Studies, Chemical Analysis and Materials Research Center, Chemistry Educational Center, and Center for Optical and Laser Materials Research at the St. Petersburg State University.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
No conflict of interest was declared by the authors.
Additional information
Translated from Zhurnal Obshchei Khimii, 2021, Vol. 91, No. 3, pp. 430–438 https://doi.org/10.31857/S0044460X21030094.
Rights and permissions
About this article

Cite this article
Katkova, S.A., Eliseev, I.I., Mikherdov, A.S. et al. Cyclometalated Platinum(II) Complexes with Nitrile and Isocyanide Ligands: Synthesis, Structure, and Photophysical Properties. Russ J Gen Chem 91, 393–400 (2021). https://doi.org/10.1134/S1070363221030099
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1070363221030099