
Abstract
An efficient metal free high yield method of synthesis of bis(indolyl)methanes (BIMs) and bis(aryl)alkanes analogs in the presence of B(C6F5)3 by condensation of primary amines with pyruvates has been successfully developed. Some analogs of BIMs display good in vitro antitumor activity, screened by MTT assay. Compound 3b demonstrates the best anti-cancer activity against lung carcinoma cell A549 cells with IC50 of 4.52 µM.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
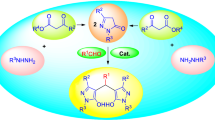
Bis(indolyl)methane derivatives (BIMs) as well as bis(aryl)alkane moieties demonstrate various types of biological and pharmacological activities, including antibacterial, anti-inflammatory [1], and anti-cancer [2]. Subsequently, a big number of practical methods of synthesis of BIMs has been reported, using Lewis acids, heteropoly acids, solid acids, ionic liquids, metal complexes, or biocatalysts [3–5]. Though some of such approaches proved to be efficient, some drawbacks still limited their application, among those were long reaction time, expensive or toxic metal ions, harsh acidity, and high temperature [3]. Most of the methods gave an accesses to the covalent C–C bond between C(sp2) of indoles or pyrroles and the acceptors, and only few catalytic protocols of activation of primary amines led to formation of covalent C–N bond (Scheme 1). Therefore, a demand of mild, efficient, metal-free catalytic method for various BIMs or bis(aryl)alkanes analogs was of considerable importance.

Catalytic systems for BIMs derivatives.
B(C6F5)3, (Frustrated Lewis Pairs, FLPs) [6] has been successfully applied to several important organic transformations as a Lewis acid catalyst without any metal ion involved, including hydrogenation, hydrosilylation of unsaturated organic functional groups, dehydrogenation, polymerization, and some more [7–9]. In general, B(C6F5)3 as a strong Lewis acid, is able to activate an imine via π-coordination to the C–N double bonds. Accordingly, B(C6F5)3 is used as a π-Lewis acid activating the imine to facilitate addition of primary amines leading to novel BIMs and bis(aryl)alkanes derivatives. Herein we report for the first time application of B(C6F5)3 as an efficient metal-free catalyst activating primary amine and leading to diverse BIMs and bis(aryl)alkanes analogs (Scheme 1,c).
RESULTS AND DISCUSSION
Initially, the process conditions were optimized for the reaction of tryptamine with methyl pyruvate (Table 1). Some solvents were tested in the presence of 5 mol % of B(C6F5)3, and it was found that 72% yield of 3a was obtained in DCM at room temperature (Table 1, entry 4). When the same reaction was carried out in DCM at 40°C, the desired product was synthesized in 81% yield within 6 h. Subsequently, a series of synthetic approaches to 3a was carried out using some common metal catalysts. In all cases the yield was poor, even at high temperature (Table 1, entries 6–10). The product yield 79% was achieved with 2 mol % of B(C6F5)3 in DCM at 40°C, while only 61% yield of 3a was obtained with 1 mol % of a catalyst (Table 1, entries 11–12).

With the optimized reaction conditions in hand, other substrates were selected to explore the scope and limitations of the method using this catalytic system. Under the worked out conditions, BIMs and bis(aryl)alkanes analogs were obtained in good to excellent yields (Table 2, 3a–3k). Presence of the methoxy group in tryptamine supported the yield of the corresponding products3c and 3d as high as 86 and 82%, respectively. For further broadening the scope of the reaction, we explored the condensation reaction of aromatic primary amines with pyruvate under optimum condition. The length of aromatic primary amine side chain had slight impact on the yield of products. For example, benzylamine and methyl pyruvate 3e had a slight higher yield than phenylethylamine3h. The process of α-methylbenzylamine with pyruvate could not proceed smoothly probably due to the steric effect. Heterocyclic primary amine, such as thiophenamine and methyl pyruvate gave the expected product3i with good yield. The above results demonstrated that the protocol of B(C6F5)3 catalyzed condensation reaction of primary amine with pyruvate provided an efficient and convenient approach to BIMs and bis(aryl)alkanes derivatives.

In order to demonstrate the protocol of B(C6F5)3 catalyzed condensation reaction, a scale-up reaction at 5 mmol level was carried out between tryptamine 1a and methyl pyruvate 2a under optimized conditions, affording 3a with a slight increase in yield (Scheme 2), thus indicating that this catalytic system could be applied as an environmently friendly and effective method for the larger scale synthesis of BIMs derivatives.

The proposed mechanism of the reaction.
Based on our results and the previous work on B(C6F5)3 catalyzed reactions [10], a proposed mechanism for this reaction is presented in Scheme 2 using tryptamine 1a and methyl pyruvate 2a as template substrates. Firstly, nucleophilic attack of 1a on the ketocarbonyl of2a results in the intermediate A. Subsequently, elimination of one molecule of water leads to formation of B in the presence of B(C6F5)3. Coordination of B(C6F5)3 with the amine could activate the C–N double bonds, then another nucleophilic attack between tryptamine and intermediate B, resulted in the desired product 3a along with release of the catalyst. However, the condensation reaction could not occur between 1a and methyl 2-oxo-2-phenylacetate 4. Probably because the large conjugated system stabilized the intermediate D and it could not be activated by B(C6F5)3.
The synthesized compounds were tested for their anticancer activity using the MTT assay [11]. The standard anticancer drug camptothecin was used as a positive control. According to the accumulated data, six most active compounds (3a, 3b, 3c, 3d, 3j, 3k) were singled out according to their IC50 values against lung carcinoma A549 cells (Fig. 1). Generally, BIMs derivatives demonstrated better anti-cancer activity than bis(aryl)alkanes derivatives. However, most of those were less efficient against HCT 116 cells, the best IC50 of 3b was only 17.14 µM. The compound3b was determined to have the optimum structural requirement for anticancer activity. The experimental data indicated BIMs nucleus as an important structural feature to demonstrate good anticancer activity. Therefore, this compound will be modified further in the studies to follow for creating the more potent molecules.

Anti-cancer activity of some synthesized compounds against A549 cells.
EXPERIMENTAL
All reactions were carried out in an oven-dried glassware with magnetic stirrer in the atmosphere of air unless otherwise mentioned. All other commercially available reagents were used without purification. Solvents were purified and dried according to the standard methods prior to use. All reactions were monitored by TLC. Purification of reaction products was carried out by flash chromatography on silica gel. Chemical yields refer to pure isolated substances.
NMR spectra were measured on a 400 MHz spectrometer using TMS as an internal standard and CDCl3 as a solvent. Mass spectra were measured on a Thermo Finnigan LCQ-Advantage. High resolution mass spectra (HRMS) were measured on a Bruker microTOF-Q II instrument using ESI techniques.
Standard reaction conditions. An aromatic primary amine 1 (1.0 mmol) and pyruvate 2 (0.6 mmol) were mixed with 2 mL of DCM and 2 mol % of B(C6F5)3, and then heated at 40°C for 6–12 h. Upon completion of the process, the reaction mixture was purified by flash column chromatography (ethyl acetate–petroleum ether = 1 : 15 to 1 : 5) to give the corresponding pure product 3a–3k.
Methyl-2,2-bis{[2-(1H-indol-3-yl)ethyl]amino}propanoate (3a). White solid, yield 79%, mp 199–201°C. IR spectrum, ν, cm–1: 1733 (C=O), 3215–3451 (NH).1H NMR spectrum, δ, ppm: 1.47 s (3H, CH3), 3.01–3.05 m (2H, CH2), 3.06–3.12 m (1H, CH2), 3.15–3.21 m (1H, CH2), 3.28–3.33 m (2H, CH2), 3.35–3.41 m (1H, CH2), 3.63 s (3H, OMe), 3.76–3.84 m (1H, CH2), 4.29–4.32 m (1H, CH2), 6.97 dd (1H, HAr, J = 8.0, 4.0 Hz), 7.10–7.19 m (4H, HAr), 7.34 t (2H, HAr, J = 8.0 Hz), 7.58 d (1H, HAr, J = 8.0 Hz), 7.65 d (1H, HAr, J = 8.0 Hz), 8.23 d (2H, HAr, J = 16.0 Hz). 13C NMR spectrum, δC, ppm: 172.66, 140.17, 136.51, 136.28, 127.37, 127.20, 122.39, 122.21, 121.99, 119.40, 119.33, 118.80, 118.58, 113.05, 112.69, 111.43, 111.26, 102.72, 67.66, 44.08, 42.82, 24.68, 24.26, 21.41. HRMS: m/z: 405.2229 [M + H]+.
Ethyl-2,2-bis{[2-(1H-indol-3-yl)ethyl]amino}propanoate (3b). White solid, yield 81%, mp 200–203°C. IR spectrum, ν, cm–1: 1736 (C=O), 3219-3450 (NH).1H NMR spectrum, δ, ppm: 1.20 t (3H, CH3, J = 8.0 Hz), 1.48 s (3H, CH3), 3.03–3.06 m (2H, CH2), 3.08–3.11 m (1H, CH2), 3.18–3.20 m (1H, CH2), 3.18–3.45 m (3H, CH2), 3.78–3.86 m (1H, CH2), 4.06–4.15 m (2H, CH2), 4.29–4.32 m (1H, CH2), 6.98–6.99 m (2H, HAr), 7.12–7.15 m (4H, HAr), 7.35 t (2H, HAr, J = 8.0 Hz), 7.60 d (1H, HAr,J = 8.0 Hz), 7.68 d (1H, HAr, J = 8.0 Hz), 8.24 d (2H, HAr, J = 16.0 Hz).13C NMR spectrum, δC, ppm: 171.95, 136.50, 136.27, 127.40, 127.21, 122.37, 122.19, 122.09, 121.98, 119.40, 119.33, 118.83, 118.58, 113.13, 112.73, 111.42, 111.25, 102.92, 67.75, 44.12, 42.83, 24.63, 24.28, 21.40, 14.11. HRMS: m/z: 419.2433 [M + H]+.
Ethyl-2,2-bis{[2-(5-methoxy-1H-indol-3-yl)ethyl]amino} propanoate (3c). White solid, yield 86%, mp 214–216°C. IR spectrum, ν, cm–1: 1739 (C=O), 3207–3442 (NH).1H NMR spectrum, δ, ppm: 1.20 t (3H, CH3, J = 8.0 Hz), 1.43 s (3H, CH3), 2.98–3.15 m (4H, CH2), 3.27–3.41 m (4H, CH2), 3.84 s (3H, OMe), 3.87 s (3H, OMe), 4.07–4.15 m(2H, CH2), 6.83–6.87 m (2H, HAr), 6.94–6.96 m (2H, HAr), 7.02 d (1H, HAr, J = 4.0 Hz), 7.13 d (1H, HAr, J = 4.0 Hz), 7.21–7.26 m (2H, HAr), 8.14 br. s (1H, NH), 8.19 br. s (1H, NH). 13C NMR spectrum, δC, ppm: 172.10, 153.96, 153.94, 140.13, 131.65, 131.39, 127.84, 127.60, 123.16, 122.90, 112.96, 112.48, 112.21, 112.16, 112.11, 111.96, 102.93, 100.59, 100.56, 67.76, 56.01, 55.90, 44.10, 42.77, 24.64, 24.23, 21.32, 14.11. HRMS: m/z: 479.2637 [M + H]+.
Methyl-2,2-bis{[2-(5-methoxy-1H-indol-3-yl)ethyl]amino} propanoate (3d). White solid, yield 82%, mp 209–211°C. IR spectrum, ν, cm–1: 1724 (C=O), 3197–3436 (NH).1H NMR spectrum, δ, ppm: 1.43 s (3H, CH3), 2.98–3.13 m (4H, CH2), 3.27–3.40 m (4H, CH2), 3.64 s (3H, OMe), 3.84 s (3H, OMe), 3.87 s (3H, OMe), 6.82–6.87 m (2H, HAr), 6.94–6.95 m (2H, HAr), 7.02 d (1H, HAr, J = 4.0 Hz), 7.13 d (1H, HAr,J = 4.0 Hz), 7.21–7.25 m (2H, HAr), 8.16 br. s (1H, NH), 8.21 br. s (1H, NH).13C NMR spectrum, δC, ppm: 172.63, 153.96, 153.94, 131.66, 131.40, 127.82, 127.61, 123.16, 122.92, 112.89, 112.47, 112.20, 112.17, 112.11, 111.97, 102.72, 100.57, 77.40, 77.08, 76.76, 67.65, 56.01, 52.72, 44.08, 42.74, 24.70, 24.20, 21.33. HRMS: m/z: 465.2443 [M + H]+.
Methyl-2,2-bis(benzylamino)propanoate (3e). Colorless liquid, yield 87%. IR spectrum, ν, cm–1: 1731 (C=O), 3289–3315 (NH). 1H NMR spectrum, δ, ppm: 1.37 s (3H, CH3), 3.45 s (3H, OMe), 4.19–4.22 m (1H, CH2), 4.42 d (1H, CH2, J = 16.0 Hz), 4.65–4.68 m (1H, CH2), 4.80 d (1H, CH2, J = 16.0 Hz), 7.21–7.24 m (1H, HAr), 7.27–7.30 m (5H, HAr), 7.32–7.35 m (4H, HAr). 13C NMR spectrum, δC, ppm: 172.21, 139.84, 137.79, 137.31, 128.77, 128.61, 128.40, 128.21, 127.77, 127.56, 127.47, 127.32, 109.98, 104.03, 67.30, 48.44, 44.64, 21.66. HRMS: m/z: 299.1759 [M + H]+.
Ethyl-2,2-bis[(4-methoxyphenethyl)amino]propanoate (3f). Colorless liquid, yield 83%. IR spectrum, ν, cm–1: 1733 (C=O), 3285–3319 (NH).1H NMR spectrum, δ, ppm: 1.22 t (3H, CH3, J = 8.0 Hz), 1.41 s (3H, CH3), 2.80–2.99 m (4H, CH2), 3.18–3.27 m (3H, CH2), 3.66–3.69 m (1H, CH2), 3.78 s (3H, OMe), 3.79 s (3H, OMe), 6.82–6.86 m (4H, HAr), 7.10–7.16 m (4H, HAr).13C NMR spectrum, δC, ppm: 172.03, 158.29, 158.21, 131.30, 130.95, 129.88, 129.67, 114.07, 113.91, 102.69, 67.64, 61.69, 55.27, 45.69, 44.10, 34.13, 33.69, 21.35, 14.12. HRMS 401.2483 [M + H]+.
Ethyl-2,2-bis(phenethylamino)propanoate (3g). Colorless liquid, yield 89%. IR spectrum, ν, cm–1: 1734 (C=O), 3281-3316 (NH). 1H NMR spectrum, δ, ppm: 1.22 t (3H, CH3, J = 8.0 Hz), 1.41 s (3H, CH3), 2.86–2.89 m (2H, CH2), 2.93–3.07 m (2H, CH2), 3.23–3.31 m (3H, CH2), 3.69–3.77 m (1H, CH2), 7.18–7.19 m (1H, HAr), 7.21–7.25 m (5H, HAr), 7.27–7.33 m (4H, HAr).13C NMR spectrum, δC, ppm: 171.99, 139.94, 139.23, 128.94, 128.74, 128.65, 128.53, 126.53, 126.43, 102.77, 67.67, 45.54, 43.91, 35.05, 34.59, 21.31, 14.13. HRMS: m/z: 341.2266 [M + H]+.
Methyl-2,2-bis(phenethylamino)propanoate (3h). Colorless liquid, yield 83%. IR spectrum, ν, cm–1: 1731 (C=O), 3280–3313 (NH). 1H NMR spectrum, δ, ppm: 1.41 s (3H, CH3), 2.86–2.89 m (2H, CH2), 2.94–3.02 m (2H, CH2), 3.22–3.29 m (3H, CH2), 3.65 s (3H, OMe), 3.72–3.75 m (1H, CH2), 7.19–7.24 m (6H, HAr), 7.27–7.32 m (4H, HAr). 13C NMR spectrum, δC, ppm: 172.53, 139.98, 139.19, 128.93, 128.74, 128.66, 128.53, 126.54, 126.45, 102.53, 77.40, 77.09, 76.77, 67.56, 52.71, 45.52, 43.89, 35.14, 34.60, 21.36. HRMS: m/z: 341.2071 [M + H]+.
Methyl-2,2-bis[(thiophen-3-ylmethyl)amino]propanoate (3i). Light green liquid, yield 67%. IR spectrum, ν, cm–1: 1701 (C=O), 3192–3208 (NH).1H NMR spectrum, δ, ppm: 1.43 s (3H, CH3), 3.51 s (3H, OMe), 4.19–4.20 m (2H, CH2), 4.44 d (1H, CH2, J = 16.0 Hz), 4.72 d (1H, CH2,J = 16.0 Hz), 7.02–7.05 m (2H, HAr), 7.12–7.16 m (2H, HAr), 7.22–7.24 m (1H, HAr), 7.28–7.30 m (1H, HAr). 13C NMR spectrum, δC, ppm: 172.18, 138.78, 137.96, 128.16, 127.13, 126.24, 125.77, 122.92, 122.05, 104.05, 77.42, 77.10, 76.78, 52.57, 43.77, 39.61, 21.51. HRMS: m/z: 311.0879 [M + H]+.
Ethyl-2,2-bis[(3,4-dimethoxyphenethyl)amino]propanoate (3j). Colorless liquid, yield 91%. IR spectrum, ν, cm–1: 1730 (C=O), 3276–3314 (NH).1H NMR spectrum, δ, ppm: 1.20 t (3H, CH3, J = 8.0 Hz), 1.40 s (3H, CH3), 2.78–2.81 m (2H, CH2), 2.89–2.95 m (2H, CH2), 3.17–3.27 m (3H, CH2), 3.65–3.72 m (1H, CH2), 3.82 s (3H, OMe), 3.83 s (3H, OMe), 3.84 s (3H, OMe), 3.85 s (3H, OMe), 6.68–6.89 m (1H, HAr), 6.73–6.74 m (3H, HAr), 6.76–6.79 m (2H, HAr). 13C NMR spectrum, δC, ppm: 171.98, 149.02, 148.89, 147.71, 147.59, 139.93, 131.76, 131.45, 120.78, 120.64, 112.14, 111.92, 111.39, 111.25, 102.76, 77.44, 77.12, 76.81, 67.64, 61.71, 55.92, 55.89, 55.87, 45.59, 44.01, 34.61, 34.17, 21.37, 14.12. HRMS: m/z: 461.2611 [M + H]+.
Methyl 2,2-bis[(3,4-dimethoxyphenethyl)amino]propanoate (3k). Colorless liquid, yield 89%, IR spectrum, ν, cm–1: 1727 (C=O), 3266–3309 (NH).1H NMR spectrum, δ, ppm: 1.42 s (3H, CH3), 2.79–2.82 m (2H, CH2), 2.86–2.94 m (2H, CH2), 3.18–3.27 m (3H, CH2), 3.65 s (3H, OMe), 3.66–3.72 m (1H, CH2), 3.83 s (3H, OMe), 3.84 s (3H, OMe), 3.85 s (3H, OMe), 3.86 s (3H, OMe), 6.69–6.70 m (1H, HAr), 6.72–6.75 m (3H, HAr), 6.77–6.81 m (2H, HAr).13C NMR spectrum, δC, ppm: 172.53, 149.02, 148.91, 147.72, 147.60, 139.97, 131.70, 131.42, 120.78, 120.64, 112.12, 111.93, 111.39, 111.25, 102.55, 67.55, 55.93, 55.91, 55.88, 52.71, 45.58, 43.98, 34.72, 34.18, 21.40. HRMS: m/z: 447.2439 [M + H]+.
CONCLUSIONS
An efficient metal free method of synthesis of BIMs and bis(aryl)alkanes analogs in the presence of B(C6F5)3 by the condensation of primary amines with pyruvates has been successfully developed. The products have been tested for their anti-cancer activity by the MTT assay. Some compounds display significant antitumor activity, indicating the scope for screening the relatively unexplored BIMs derivatives as potential anticancer compounds.
REFERENCES
Sarva, S., Harinath, J.S., Sthanikam, S.P., Ethiraj, S., Vaithiyalingam, M., and Cirandur, S.R., Chin. Chem. Lett., 2016, vol. 27, p. 16. https://doi.org/10.1016/j.cclet.2015.08.012
Grosso, C., Cardoso, A.L., Lemos, A., Varela, J., Rodrigues, M.J., Custodio, L., Barreira, L., and Melo, T., Eur. J. Med. Chem., 2015, vol. 93, p. 9. https://doi.org/10.1016/j.ejmech.2015.01.050
Veisi, H., Maleki, B., Eshbala, F.H., Masti, R., Ashrafib, S.S., and Baghayeri, M., RSC Adv., 2014, vol. 4, p. 30683. https://doi.org/10.1039/C4RA03194F
Das, P.J., and Das, J., Tetrahedron Lett., 2012, vol. 53, p. 4718. https://doi.org/10.4236/gsc.2013.34A002
Xiang, Z.W., Liu, Z.Q., Chen, X., Wu, Q., and Lin, X.F., Amino Acids, 2013, vol. 45, p. 937. https://doi.org/10.1007/s00726-013-1547-4
Stephan, D.W., Science, 2016, vol. 354, p. 1248. https://doi.org/10.1126/science.aaf7229
Mahdi, T., and Stephan, D.W., J. Am. Chem. Soc., 2014, vol. 136, p. 15809. https://doi.org/10.1021/ja508829x
Li, W, Werner, T., Org. Lett., 2017, vol. 19, p. 2568. https://doi.org/10.1021/acs.orglett.7b00720
Kim, D.W., Joung, S., Kim, J.G., and Chang, S., Angew. Chem. Int. Ed., 2015, vol. 54, p. 14805. https://doi.org/10.1002/anie.201507863
Ling, F., Xiao, L., Fang, L., Feng, C., Xie, Z., Lv, Y., and Zhong, W.H., Org. Biomol. Chem., 2018, vol. 16, p. 9274. https://doi.org/10.1039/C8OB02805B
Kumar, B.A. and Jagarlapudi, V.S.K., Russ. J. Gen. Chem., 2020, vol. 90, p.929. https://doi.org/10.1134/S1070363219120296
ACKNOWLEDGMENTS
We thank Jiangsu Vocational College of Medicine (20186104) and Jiangsu Provincial Higher Education Natural Science Foundation (19KJB350010) for financial support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
No conflict of interest was declared by the authors.
Rights and permissions
About this article

Cite this article
Qi, L., Xiao, L. Convenient Synthesis and Anticancer Activity of Bis(aryl)alkanes and Bis(indolyl)methane Alkaloid Analogs. Russ J Gen Chem 90, 1974–1980 (2020). https://doi.org/10.1134/S1070363220100217
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1070363220100217