
Abstract
Phosphates of the new formula type A5/3MgE4/3(PO4)3 (A = K, Rb; E = Ti, Zr) with a langbeinite structure have been synthesized via the sol-gel method followed by heat treatment and studied using X-ray diffraction, IR spectroscopy, and electron microscopy methods.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Phosphates structurally analogous to natural mineral langbeinite K2Mg2(SO4)3 [1, 2] are attractive due to the possibility to combine various chemical elements in a single lattice which can lead to interesting optical, luminescent, magnetic, and photoelectrical properties [3−12]. Owing to wide isomorphism of the cations in the substances with the langbeinite structure as well as their radiation and chemical stability, they have been considered as inert stable ceramic matrices for nuclear waste immobilization [13, 14].
The langbeinite M2L2(XO4)3 crystal structure contains the {[L2(XO4)3]p−}3∞ framework built of the vertex-linked LO6 octahedrons and XO4 tetrahedrons, each octahedron being linked to six tetrahedrons, and each tetrahedron being linked to four octahedrons. The framework determines the geometry of the interstitial closed voids. The crystallochemical formula of this type of phosphates is as follows: (М1)[9](M2)[6−12][(L1)[6](L2)[6](PO4)3]3∞. Position L of the framework usually contains the same or different small cations with +2, +3, or +4 oxidation state. There are two void positions (М1) and (М2) per a formula unit which can be occupied by two or (more seldom) less of the cations, being in most cases nonacoordinate with +1 or +2 oxidation state. Most of the compounds with the langbeinite structure are cubic with the P213 space group or (less commonly) orthorhombic (for example, certain sulfates and vanadates) with the P212121 space group; some of them can undergo the P213→P212121 phase transitions [15−17].
Theoretically possible formula types (compositions) of phosphates with the langbeinite structure could be calculated from the conditions of the framework formation and the correspondence between its negative charge to the total positive charge of two populated void positions. General formula of the framework with two cations (identical or different) in positions L being in the m and n oxidation states is as follows: [Lrm+Lsn+(PO4)3]p−. The r, s, m, n, and p variables are related as r + s = 2 and 9 – (m·r + n·s) = p. The latter expression follows from the electroneutrality principle. If two void positions are occupied, the p charges of the langbeinite framework can be of 2 to 4. In view of the mentioned limitations, we have earlier explored the following langbeinite-type phosphates: A+2[M2+0.5E4+1.5(PO4)3][Cs2Mn0.5Zr1.5(PO4)3] [18] with r = 0.5, m = 2, s = 1.5, n = 4, p = 2 and A+M2+[MgE4+(PO4)3] (A = K, Rb, Cs; M = Sr, Pb, Ba; E = Ti, Zr) [19] with r = 1, m = 2, s = 1, n = 4, p = 3. If positions L of the framework are populated with M2+ and E4+ cations (as in the latter case) yet introduce the compensating cations in oxidation states +1 and +4 in the void positions (M1 and M2) an earlier unknown formula type of the phosphates with should be crystallized in the considered structural type: A+5/3E41+/3[M2+E4+(PO4)3] = A5/3M2+E44+/3(PO4)3.
This study aimed to synthesize the A5/3MgE4/3(PO4)3 (A = K, Rb; E = Ti, Zr) phosphates with expected langbeinite structure, investigate their phase formation and thermal behavior, and refine their crystal structure.
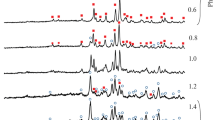
The X-ray diffraction analysis revealed the formation of single-phase A5/3MgE4/3(PO4)3 (A = K, Rb; E = Ti, Zr) phosphates with the langbeinite structure. The X-ray diffraction patters of the samples annealed at 600−700°С contained strong reflections of the langbeinite-type phases as well as of TiP2O7, K3PO4, Rb3PO4, ZrO2, and Mg3(PO4)2, which were completely transformed into the single-phase phosphates upon stepwise heating to до 850°С (Fig. 1). Further heating improved the samples crystallinity. The unite cell parameters of the synthesized compounds were increased with the increase in radius of the alkali and(or) transition metal cation (Table 1). The compounds were crystallized in the Р213 space group, the formula units number being 4.

X-ray diffraction patterns of the K5/3MgTi4/3(PO4)3 (1), Rb5/3MgTi4/3(PO4)3 (2), K5/3MgZr4/3(PO4)3 (3), and Rb5/3MgZr4/3(PO4)3 (4) phosphates.
The data of electron microscopy and microprobe analysis revealed that the samples were homogeneous and contained of grains differing in shape. The grain size was of 1 to 5 μm. The microanalysis data gave, for example, the following composition: Rb1.69(4)Mg0.97(3)Zr1.38(5)P2.97(4)O12.
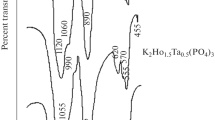
The data of IR spectroscopy which is sensitive to the local order of the atoms arrangement in the structure confirmed the orthophosphates formation. IR spectra of the obtained samples with the langbeinite structure were typical of the phosphates crystallizing in the P213 space group. The position symmetry of the PO43– ion was reduced to C1 in the langbeinite structure. The selection rules allowed the appearance of a single νs band, two δs bands, three νas bands, and δas bands in the IR spectra. Experimental spectra of the phosphates contained the allowed bands in the ranges of νas (1170−1000 cm−1) and δas (670–500 cm−1) vibrations, the band with maximum at ~920 cm−1 was assigned to νs vibration, and the bands at 450 and 420 cm−1 corresponded to δs vibrations.
Crystal structure of Rb5/3MgZr4/3(PO4)3 was refined using the powder X-ray diffraction data via the Rietveld method. The experiment conditions, unit cell parameters, and major results of the structure refinement are given in Table 2. Figure 2 displays the experimental, calculated, and difference X-ray diffraction patterns of Rb5/3MgZr4/3(PO4)3. Atomic coordinates and the isotropic heat parameters are collected in Table 3. Part of the Rb5/3MgZr4/3(PO4)3 structure is shown in Fig. 3. The structure consisted in the (Mg/Zr)O6 octahedrons and PO4 tetrahedrons linked via the vertices and forming the three-dimensional framework. Rubidium and zirconium cations were located in large closed voids.

Experimental (1), calculated (2), and difference (3) X-ray diffraction patterns of Rb5/3MgZr4/3(PO4)3. The strokes (4) point at the positions of the Braggs’ reflections.

Part of the Rb5/3MgZr4/3(PO4)3 structure.
The Mg2+ and Zr4+ ions in the framework were disordered between two crystallographic independent positions and were coordinated with six oxygen atoms. The populations of one of the positions were 55.7% (Mg) and 44.3% (Zr) and those for the other one were 44.3% (Mg) and 55.7% (Zr). The Rb+ and Zr4+ ions in the voids were also disordered between two crystallographic independent positions. One of the positions was populated by rubidium, and the populations for the other one were 66.7% (Rb) and 33.3% (Zr).
Table 4 gives the calculated bond lengths in the polyhedrons; they were typical of the langbeinite structure. The void positions populated exclusively with rubidium formed the nonacoordinate oxygen polyhedrons, the bond lengths being 2.74 to 3.39 Å. The positions populated with rubidium and zirconium formed the polyhedrons with bond lengths 3.19 to 3.48 Å. Magnesium and zirconium cations in the unequally populated positions formed oxygen octahedrons, the (Mg/Zr)−O bond lengths in the framework octahedrons being 1.92 to 2.31 Å. The PO4 tetrahedrons were distorted, forming two equivalent P−O bonds (1.66−1.69 Å) and two shorter bonds (1.46 and 1.54 Å).
Our studies of the A5/3MgE4/3(PO4)3 (A = K, Rb; E = Ti, Zr) compounds extended the knowledge of the phase formation of the known langbeinite-type phosphates. The refined structure revealed that the framework was populated with E4+ and magnesium cations, whereas the A+ and E4+ cations were located in voids. Such distribution of the cations between the framework and void positions allowed relaxation of the deformed structure, decrease in the inner strains, and, hence, increase in the thermal stability.
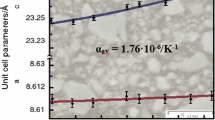
Thermal behavior of the obtained K5/3MgTi4/3(PO4)3 and K5/3MgZr4/3(PO4)3 phosphates was studied by means of high-temperature X-ray diffraction analysis. The parameter а and volume V of the cubic unit cells of the langbeinite structure grew linearly with temperature. Those materials exhibited isotropic expansion upon heating, showing positive local thermal expansion coefficients. Thermal expansion directly affected the mean kinetic energy of the vibrating particles in the body and average distance between the lattice sites and was related to asymmetry (anharmonicity) of heat vibrations of the atoms, leading to the changes in the interatomic distances with temperature. Thermal expansion coefficients calculated over 25−800°С showed that K5/3MgZr4/3(PO4)3 was an intermediate-expanding substance (αа = 5×10–6 °С–1), whereas K5/3MgTi4/3(PO4)3 was a strongly expanding substance (αа = 11×10–6 °С–1). Thermal expansion coefficient is related to the strength of the chemical bonds in the structure: stronger expansion corresponds to weaker bonding of the atoms [20, 21]. Evidently, the P−O bonds in the tetrahedrons of the isostructural and isoformula phosphates were stronger than the L−O bonds in the LO6 polyhedron (L = Zr, Ti). Zirconium and titanium formed the bonds differing in the covalent contribution. Larger size of the Zr4+ cation determined higher strength (covalent contribution) of the Zr−O bonds in comparison with the Ti−O ones. As a result, heat-induced deformation of the K5/3MgZr4/3(PO4)3 structure was less than for K5/3MgTi4/3(PO4)3. Since expansion of the substances was isotropic (αа = αb = αc), their volumetric thermal expansion coefficient (αV) was three times the linear thermal expansion coefficient.
In summary, the family of the langbeinite-type structures was complemented by a novel formula type A+5/3E41+/3[M2+E4+(PO4)3]: A5/3MgE4/3(PO4)3 (A = K, Rb; E = Ti, Zr) phosphates. We studied their phase formation, refined the crystal structure, and investigated the thermal stability as well as thermal expansion. It was shown that the structure was based in a three-dimensional framework constituted by the PO4 tetrahedrons and (Mg/E)O6 octahedrons linked via the vertexes, whereas potassium (rubidium) and zirconium atoms populated the void positions. The heating of the phosphates from 25 to 800°С led to isotropic expansion and did not induce any polymorph transitions. Linear thermal expansion coefficients of K5/3MgZr4/3(PO4)3 and K5/3MgTi4/3(PO4)3 were typical of intermediate and strongly expanding substances, respectively.
EXPERIMENTAL
The A5/3MgE4/3(PO4)3 (A = K, Rb; E − Ti, Zr) phosphates were prepared via a sol-gel method followed by heat treatment. The synthesis was performed as follows: stoichiometric amounts of aqueous solutions of the alkali metal (K or Rb) chloride, magnesium chloride, titanium or zirconium oxychloride, and ammonium dihydrophosphate (all of “chemical pure” grade) were mixed. The mixture was then dried at 90 and 150°С (12 h at each stage) and subject to stepwise heat treatment in air at 600−900°С (20−26 h at each stage). After each heating step, the mixture was homogenized by dispersion. The powder was pressed in disc to ensure more complete conversion.
The samples were characterized by means of electron microscopy and local microanalysis, X-ray diffraction analysis, and IR spectroscopy. Chemical composition and uniformity of the phosphates were monitored using a JEOL JSM-7600F scanning electron microscope equipped with a dark-field electron gun (the Schottky cathode). The microscope was equipped with an OXFORD X-Max 80 energy-dispersive spectrometer (Premium) with semiconducting silicon-drift detector allowing local microanalysis of the specimen. The error in determining the elemental composition of the samples was no more than 2 at %.
X-ray diffraction analysis was performed using a Shimadzu XRD-6000 diffractometer (CuKα radiation, λ = 1.54178 Å, 2θ range 10°–60°). The analysis was performed upon each heat treatment step to gain information on the phase composition of the substances. High-temperature measurements were performed using the same instrument equipped with an HA-1001 Shimadzu attachment allowing discrete heating at 20–800°C in 100°С step. Temperature was measured using a platinum-rhodium thermocouple with accuracy of ±2%. Unit cell parameters were determined by the least squares method from the indicated diffraction patterns.
Structure of the Rb5/3MgZr4/3(PO4)3 sample was investigated by recording the X-ray diffraction pattern over 2θ = 10°–110° with 0.02° step (15 s per a point). The structure was refined by means of full-profile analysis of the powder diffraction data using RIETAN-97 software [22, 23]. The peaks were fitted using modified pseudo-Voight function [24]. K1.75Ti2(PO4)3 was used as the model for the structure refinement [25] .
IR spectroscopy studies were performed to confirm the functional composition of the samples. The spectra were recorded using an FSM-1201 spectrometer (400– 1400 cm−1).
REFERENCES
Sarrign, M.L.M., Clemente, A.R., and Vila, L.M., J. Solid State Chem., 1990, vol. 84, p. 308. https://doi.org/10.1016/0022-4596(90)90329-V
Zatovskii, I.V., Slobodyanik, N.S., Ushchapivskaya, T.I., Ogorodnik, I.V., and Babarik, A.A., Russ. J. Appl. Chem., 2006, vol. 79, p. 10. https://doi.org/10.1134/S1070427206010034
Pet’kov, V.I., Shchelokov, I.A., Surazhskaya, M.D., Palkina, K.K., Kanishcheva, A.S., and Knyazev, A.V., Russ. J. Inorg. Chem., 2010, vol. 55, p. 1352. https://doi.org/10.1134/S0036023610090044
Souamti, A., Martín, I.R., Zayani, L., Hernández-Rodríguez, M.A., Soler-Carracedo, K., Lozano-Gorrín, A.D., Lalla, E., and Ben Hassen Chehimi, D., J. Luminescence, 2016, vol. 177, p. 160. https://doi.org/10.1016/j.jlumin.2016.04.045
Battle, P.D., Gibb, T.C., Nixon, S., and Harrison, W.T.A., J. Solid State Chem., 1988, vol. 75, p. 21. https://doi.org/10.1016/0022-4596(88)90299-X
Ogorodnyk, I.V., Zatovsky, I.V., Slobodyanik, N.S., Baumer, V.N., and Shishkin, O.V., J. Solid State Chem., 2006, vol. 179, p. 3461. https://doi.org/10.1016/j.jssc.2006.07.015
Sadhasivam, S., Manivel, P., Jeganathan, K., Jayasankar, C.K., and Rajesh, N.P., Mater. Lett., 2017, vol. 188, p. 399. https://doi.org/10.1016/j.matlet.2016.11.091
Liu, J., Duan, X., Zhang, Y., Li, Z., Yu, F., and Jiang, H., J. Alloys Compd., 2016, vol. 660, p. 356. https://doi.org/10.1016/j.jallcom.2015.11.147
Pet’kov, V.I., Asabina, E.A., Markin, A.V., Alekseev, A.A., and Smirnova, N.N., J. Therm. Anal. Cal., 2016, vol. 124, no. 3, p. 1535. https://doi.org/10.1007/s10973-016-5319-8
Souamti, A., Kahlaoui M, Mohammed, B., LozanoGorrín, A.D., and Chehimi, D.B.H., Ceram. Int., 2017, vol. 43, no. 14, p. 10939. https://doi.org/10.1016/j.ceramint.2017.05.132
Asabina, E.A., Pet’kov, V.I., Gobechiya, E.R., Kabalov, Yu.K., Pokholok, K.V., and Kurazhkovskaya, V.S., Russ. J. Inorg. Chem., 2008, vol. 53, p. 40. https://doi.org/10.1134/S0036023608010075
Tsyrenova, G.D. and Pavlova, N.N., Inorg. Mater., 2011, vol. 47, no. 7, p. 786. https://doi.org/10.1134/S0020168511070235
Pet’kov, V.I., Asabina, E.A., Lukuttsov, A.A., Korchemkin, I.V., Alekseev, A.A., and Demarin, V.I., Radiochemistry, 2015, vol. 57, no. 6, p. 632. https://doi.org/10.1134/S1066362215060119
Pratheep Kumar, S. and Gopal, B., J. Alloys Compd., 2016, vol. 657, p. 422. https://doi.org/10.1016/j.jallcom.2015.10.088
Glogarová, M., Phys. Status Solidi (A), 1974, vol. 22, no. 1, p. 69. https://doi.org/10.1002/pssa.2210220159
Hikita, T., Kitabatake, M., and Ikeda, T., J. Phys. Soc. Japan, 1980, vol. 49, no. 4, p. 1421. https://doi.org/10.1143/JPSJ.49.1421
Trussov, I.A., Male, L.L., Sanjuan, M.L., Orera, A., and Slater, P.R., J. Solid State Chem., 2019, vol. 272, p. 157. https://doi.org/10.1016/j.jssc.2019.02.014
Zaripov, A.R., Orlova, V.A., Pet’kov, V.I., Slyunchev, O.M., Galuzin, D.D., and Rovnyi, S.I., Russ. J. Inorg. Chem., 2009, vol. 54, p. 45 https://doi.org/10.1134/S0036023609010112
Pet’kov, V.I., Alekseev, A.A., Asabina, E.A., Borovikova, E.Y., and Koval’ski, A.M., Russ. J. Inorg. Chem., 2017, vol. 62, no. 7, p. 870 https://doi.org/10.1134/S0036023617070178
Filatov, S.K., Vysokotemperaturnaya kristallokhimiya. Teoriya, metody i rezul’taty issledovanii (High Temperature Crystal Chemistry. Theory, Methods, and Research Results), Leningrad: Nedra, 1990.
Krivovichev, S.V. and Filatov, S.K., Kristallokhimiya mineralov i neorganicheskikh soedinenii s kompleksami anionotsentrirovannykh tetraedrov (Crystal Chemistry of Minerals and Inorganic Compounds with Complexes of Anion-Centered Tetrahedra), St. Petersburg: SPbGU, 2001.
Rietveld, H.M., Acta Crystallogr., 1967, vol. 22, no. 1, p. 151. https://doi.org/10.1107/S0365110X67000234
Kim, Y.I. and Izumi, F., J. Ceram. Soc. Japan, 1994, vol. 102, p. 401. https://doi.org/10.2109/jcersj.102.401
Izumi, F., The Rietveld Method, New York: Oxford University Press, 1993, ch. 13, p. 236. https://doi.org/10.1002/crat.2170300412
Leclaire, A., Benmoussa, A., Borel, M.M., Grandin, A., and Raveau, B., J. Solid State Chem., 1989, vol. 78, p. 227. https://doi.org/10.1016/0022-4596(89)90101-1
Funding
This study was financially supported by the Russian Foundation for Basic Research (project no. 18-29-12063).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
No conflict of interest was declared by the authors.
Rights and permissions
About this article

Cite this article
Pet’kov, V.I., Alekseev, A.A., Asabina, E.A. et al. New Formula Type of Phosphates with Langbeinite Mineral Structure. Russ J Gen Chem 90, 680–685 (2020). https://doi.org/10.1134/S1070363220040192
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1070363220040192