
Abstract
Syntheses of a polydentate ligand based on the second-generation hyperbranched polyester containing 3-(2-aminoethyl)amino]propionate groups and its metal complex with copper(II) ions have been elaborated. In view of the IR, electronic absorption, and EPR spectroscopy data, it has been suggested that the coordination sites in the metal-polymer complex are paramagnetic sites with the CuN4Solv2 or CuN2O2Solv2 composition (Solv = H2O, DMSO).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Multidentate ligands are widely used in the preparation of functional self-assembling systems for molecular recognition, nanosized molecular devices, and in catalysis applications. Macromolecular chemistry of dendritic polymers opens the possibility for the development of highly efficient nanosized polydentate chelating agents with predefined molecular composition, size, and shape. Metal complexes of dendritic chelating agents exhibit diverse opportunities for practical applications; fundamental study of their complex structure is important as well [1–5].
Knowledge of chemical structure and properties of polynuclear coordination metal complexes is important in the study of magnetic materials, modeling and understanding of the importance of multinuclear reactive sites in biological processes [6–8]. For example, the metal ions in copper(II) complexes with poly(propylene imine) and polyamidoamine dendrimers with terminal amino and carboxyl groups can be bound to the terminal groups or to the internal amide and tertiary amino groups, depending on the structure of the polydentate macroligand and the medium pH [4, 9–15]. Such complexes serve as model compounds in the study of metal ions transport in biological systems [16, 17], they can be used in metals recovery [10], and can be applied as contrasting agents in magnetic resonance imaging [5, 17].
Hyperbranched polymers can be successfully used for the creation of modern materials based on polynuclear metal complexes exhibiting biological activity, along with dendrimers. Macromolecules of hyperbranched polymers are readily soluble in majority of organic solvents and exhibit high specific concentration of surface groups. These properties emerge the use of hyperbranched polymers as nanocontainers hosting diverse high- and low-molecular compounds [2, 3, 18]. Nontoxic biodegradable hyperbranched polyester polyols can be applied as core structure in the synthesis of novel hybrid materials via sequential functionalization with organic coordinating fragments and metal ions [3, 19–24].
We have earlier synthesized polyamines and polyester polycarboxylic acids capable of complex formation with d-metal ions and pharmaceuticals basing on hyperbranched polyester polyols [25–30]. Investigation of their physico-chemical properties has revealed that the amphiphilic reactants such as amino derivatives of hyperbranched polyesters are the most promising in this instance. Extending the studies aimed to produce polydentate macroligands and polynuclear metal complexes based on hyperbranched polyesters, we herein describe hyperbranched second-generation polyester polyol with terminal ester groups, containing ethylenediamine moieties. This structure allowed combining the amphiphilic properties and complexing activity in a single macromolecular reactant.
Synthesis of the hyperbranched polyester with terminal ethylenediamine fragments included two stages (Scheme 1). The first stage consisted in the modification of polyol 1 via the substitution of the hydroxyl groups with acrylate ones to afford the polyacrylate 2. The second stage was the addition of ethylenediamine at polyacrylate 2 (the aza-Michael reaction) yielding the ethylenediamine derivative 3.
Analysis of IR spectra of the starting hyperbranched polyester polyol 1 and the synthesized compounds revealed that the IR spectrum of polyacrylate 2 contained weak O–H stretching band at 3452 cm–1 which confirmed the partial substitution of hydroxyl groups with the acrylate moieties [25, 29]. IR spectrum of hyperbranched polyester poly{3-[(2-aminoethyl)amino]propionate} 3 contained stretching bands of the OH, NH2, and NH groups at 3352‒3293 cm–1; strengthening and broadening of those bands was due to the formation of a system of intra- and intermolecular hydrogen bonds. The bands of the NH group deformation vibrations were observed at 1658, 1648, and 1556 cm–1. Stretching bands of the C−H bonds in the CH3 and CH2 groups (2970‒2880 cm–1) were accompanied by the band at 2829 cm–1, likely assigned to the NHCH2 fragment stretching [25, 29, 31].
1Н NMR spectrum of polyacrylate 2 contained a characteristic set of signals of vinyl protons of the C(O)CH=CH2 acryloyl moiety as three multiplets at 5.78, 6.04, and 6.32 ppm, besides the signals of the protons of hyperbranched polyester polyol 1. The amount of the acryloyl fragments was calculated from the ratio of integral intensities of the signals of the CН2=CH terminal groups and the CH3 group of the hyperbranched polyester scaffold [22, 25]. According to the 1H NMR data, degree of functionalization of compound 1 with acryloyl chloride was 87.5±2%, i. e. 14 of 16 OH groups were substituted with acrylate fragments. As for the 1Н NMR spectrum of polyester poly{3-[(2-aminoethyl)amino]propionate} 3, it contained the signals of the protons of the starting hyperbranched polyester polyol and those of the protons of methylene groups of the CH2NH(CH2)2NH2 moiety (2.67‒2.89 ppm). The absence of the signals of vinyl protons (5.78‒6.32 ppm) evidenced completeness of the addition of ethylenediamine at the C=C bonds of the substrate.
Protolytic properties of polyamine 3 were evaluated from the data of potentiomentric titration processed using CPESSP software involving the Bjerrum function [25, 29, 33]. Average value of the stepwise protonation constant for compound 3 exceeded that for ethylenediamine, being pKb(av) = 6.01±0.05. Analysis of the fractional composition of the protonated forms (Fig. 1) suggested the block mechanism of protonation with addition of three protons. The major protonated forms of compound 3 were as follows: (H3L)3+, (H6L)6+, (H8L)8+, (H11L)11+, (H14L)14+, (H17L)17+, (H25L)25+, (H28L)28+.

Fractional distribution of ionized and associated forms of compounds 3. (1) H28L28+ (Ki = 8.2); (2) H25L25+ (Ki = 9.7); (3) H20L20+ (Ki = 6.3); (4) H17L17+ (Ki = 7.1); (5) H14L14+ (Ki = 10.64); (6) H11L11+ (Ki = 10.04); (7) H8L8+ (Ki = 10.77); (8) H6L6+ (Ki = 7.37); (9) H3L3+ (Ki = 8.53); (10) L.
Compound 3 (L) was used for the preparation of a water-soluble metal complex with Cu(II) ion chosen as the metal ion (Scheme 1), the suggested types of coordination nodes are shown in Scheme 2.

1.

2.
To assess the composition and structure of the metal center, we took advantage of IR, electronic absorption, and ESR spectroscopy techniques. Comparison of IR spectra of the polyester 3 and its metal complex 4 revealed the weakening of the N–H group deformation band (1552 cm–1) as well as weakening and splitting of the C=O stretching band (1729, 1708 cm–1) upon the complex formation. The complex formation likely involved the nitrogen atoms of the amino groups and the oxygen atoms of the C=O groups, the possible ligand coordination type corresponding to type B in Scheme 2. That was confirmed by the presence of the bands at 412 [ν(Cu–N)] and 532 cm–1 [ν(Cu–O)] [31, 34–36] in IR spectrum of the complex.
Parameters of electronic absorption spectra of the polydentate ligand 3 and complex 4 in a solution (in water or DMSO) over 200‒1000 nm are collected in Table 1. Electronic absorption spectra of compound 3 in water and DMSO contained several absorption bands at 275‒420 nm assignable to the n→π*-transitions of the chromophore groups of the polyester scaffold and terminal ethylenediamine fragments. The spectrum of complex 4 showed a visible-range band at 647 (water) or 660 nm (DMSO) related to the d‒d-transition of Cu2+ ion (2B1g→2A1g). Analysis of the bands position in view of the reference data [36–38] suggested the octahedral geometry of copper(II) coordination (Table 1).
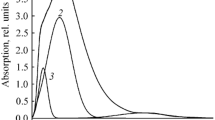
To clarify the details of the copper(II) coordination, we studied the Cu(II)–compound 3 systems at constant concentration of the ligand (10–3 mol/L) and variable M : L ratios (Fig. 2). Free Cu(II) ions in a solution in DMSO exhibited an absorption band at 850 nm (~11800 cm–1), whereas the oxygen octahedron in hexaaqua copper(II) complex has been assigned to the band at 804 nm (~12500 cm–1) [41]. The increase in the metal ions content in the Cu(II)–compound 3 system led to the shift in the d‒d-transition band reflecting the changes in the composition and structure of the coordination site (Fig. 2).

Electronic absorption spectra of compound 3 in the absence (1) and in the presence of metal ions in the ratio of M : L = 3 : 1 (2), 4 : 1 (3), 7 : 1 (4), 15 : 1 (5), 16 : 1 (6), 20 : 1 (7).
Gradual increase in the amount of Cu(II) to the 3 : 1 ratio (corresponding to the excess of the binding sites in the ligand) was accompanied by the appearance of a weak band at 550 nm (~18200 cm–1), pointing at the population of the nodes C with the CuN4 coordination (Scheme 2) in the equatorial positions. Further increase in the metal content [the ratio of (4 : 1) to (7 : 1)] led to the shift of the said band to 675 nm (~14800 cm–1), corresponding to the A and C node with the equatorial CuN2O2 coordination; further increase in the metal concentration (to the ratios corresponding to the deficit in the ligand nitrogen atoms) led to the appearance of the absorption band at λ ~800 nm, evidencing the formation of the CuO4 equatorial node D. At M : L ratios of (15 : 1) to (20 : 1), the CuN2O2 and CuO4 nodes coexisted in the dissolved complex, as evidenced by the shape of the absorption band (Fig. 2).
To elucidate and refine the equatorial surrounding in the coordination node of Cu(II) ion in the synthesized complex with compound 3, we studied the solid samples of complex 4 (Fig. 3) and its solutions in DMSO (Fig. 4) by means of ESR. The optical spectroscopy data coincided with the ESR results, taking into account that the concentration of the solutions studied by ESR was 10 times higher to overcome the decrease in the resonator goodness (and, thus, the ESR signal amplitude) in the presence of DMSO.

ESR spectrum of solid complex 4 (200 K, frequency 9.424 GHz). (1) experimental spectrum, (2) theoretical spectrum with parameters given in Table 2.

ESR spectrum of complex 4 in DMSO (с4 = 1× 10–2 mol/L, 200 K, frequency 9.445 GHz). (1) experimental spectrum, (2) theoretical spectrum with parameters given in Table 2.
The spectrum of exchange-bound paramagnetic copper(II) sites was observed in the case of solid samples (Fig. 3). The hyperfine structure in the ESR spectrum was not observed, since the hyperfine interaction of the electron spin of copper(II) ion with its own nucleus was likely shielded by the exchange interaction between the copper ions. The ESR spectrum was recorded over the 100‒300 K temperature range; the spectrum shape was found temperature-independent. The ESR line was asymmetric due to the presence of both exchange-bound sites and the mononuclear sites with the nitrogen equatorial surrounding in the sample, in the about 2 : 1 ratio. Hence, somewhat less than 20% of the copper ions in the sample existed as the mononuclear sites, other ions being involved in the exchange.
Dissolution of the samples in DMSO led to the disappearance of the exchange copper complexes, and the signals of isolated mononuclear paramagnetic copper sites were exclusively observed, revealing distinct hyperfine structure (Fig. 4). The ESR spectrum of the frozen solution was complex and evidenced the coexistence of two types of the complexes differing in the ESR parameters and the coordination node structure. The spectrum shape remained unchanged over the 100‒ 270 K temperature range.
The ESR spectra could be well described by the axial-symmetrical Hamiltonian. Analysis of the spectra of the frozen solutions and their simulation gave the g||, g┴ parameters and the hyperfine structure parameter А|| for those two types of the complexes (Table 2). The ESR spectrum was a superposition of two spectra of copper complexes with nitrogen surrounding in the equatorial plane (C) and with purely oxygen equatorial surrounding (D) (Scheme 2).
The hyperfine structure А|| ≈ 187×10–4 cm–1 and g|| 2.252 parameters of the complex C in DMSO pointed at purely nitrogen surrounding of the copper atom in the equatorial plane (СuN4) [39–41], whereas the |g||/А||| < 135 cm value evidenced its planar structure (the absence of the pseudo tetrahedral distortion) [42–44]. The parameters of the complex D in the solution, corresponding to the oxygen surrounding of the copper ion in the equatorial plane (СuO4), were very close to those of the hexaaqua complex Cu(H2O)6 [45]. The ratio of the amounts of copper ions in the nodes C and D in the solution was of 2.5 : 1.
The stoichiometry of the Cu2+–L system was investigated using the isomolar series approach (the Job’s method) by measurements of the solutions with constant total concentration of the copper ion and the ligand, yet the n : m ratio in the corresponding formula CunLm was gradually changed [25, 36, 46]. The conditional stability constant (log β') was determined and the complexing of the hyperbranched ligand with Cu(II) ions was modeled by means of the molar ratios method [25, 46] involving the mathematical processing of the experimental data using CPESSP software [25]. The modeling was performed using the stoichiometry matrix accounting for the formation of all sterically possible complex forms. Fig. 5 shows the experimental and calculated curves for the molar ratios method data and the fractional distribution of the complex forms (at 660 nm) as functions of the metal concentration. In view of the structure of the starting polyester polyol scaffold 1, the complex forms 7 : 1 (65.05%) and 10 : 1 (53.17%) with the stability constants of (log β') 31.79±0.1 and 43.10±0.8, respectively, were the most favorable for compound 3. The suggested structure of complex 4 involved the complex forming ion coordination to two ethylenediamine fragments of the same terminal branching.

Experimental and calculated curves for the molar ratios method as function of the metal ion concentration in the solution for complex 4. (■) experiment, (●) calculation.
In summary, a novel polydentate amphiphilic ligand— hyperbranched polyester poly{3-[(2-aminoethyl)amino]propionate}, a second generation polyester polyol functionalized with terminal ethylenediamine fragments—and its water-soluble complex with Cu(II) ions were synthesized. Comprehensive analysis of the data of IR, electronic absorption, and ESR spectroscopy allowed elucidation of the geometry of the complex coordination nodes. At high concentration of the complex, the dimers with the Cu(II)‒Cu(II) bonding were formed. Analysis of the hyperfine interaction parameters for copper in the frozen solutions allowed elucidation of the local structure of copper sites and suggested their model arrangements. The studied Cu(II) complexes revealed the hexacoordinate structure in the shape of elongated octahedron. Three types of the Cu(II) coordination nodes were revealed in the hyperbranched polymeric matrix: CuN4Sol2, CuN2O2Sol2, and CuO4Sol2, (Sol being H2O or DMSO) with pseudo tetrahedral distortion of the node in the equatorial plane. The dilute solutions (0.1%) additionally contained the paramagnetic sites with the copper(II) aqua complex structure.
The formation of the metal–polymer complex during the saturation of the ligand binding sites by the metal ions in the solution was accompanied by the decrease in the amount of the nitrogen atoms in the equatorial positions of copper(II) ion coordination sphere and further change in the structure of the paramagnetic sites. That was likely due to the influence of the solvent groups or their introduction in the inner coordination sphere of copper. Similar effect was observed during dissolution of the stable complexes in DMSO. The neighbor ligands in the inner coordination sphere were rearranged due to the introduction of oxygen atoms in the equatorial plane or enhancement of the rotational isomerism (the nitrogen atoms could thus be moved to the axial positions).
EXPERIMENTAL
Aliphatic hyperbranched second-generation polyester polyol based on ethoxylated pentaerythritol (core) and 2,2-bis(hydroxymethyl)propionic acid (monomer) (Sigma-Aldrich, 16 OH groups, M 1749, hydroxyl number 490–520 mg/g KOH), acryloyl chloride (97%), ethylenediamine, triethylamine (99%, Acros), Cu(NO3)2 · 3H2O, and organic solvents of analytical, ultrapure, and chemically pure grades [acetone, THF, ethanol, propan-2-ol, benzene, methylene chloride, petroleum ether, dimethyl sulfoxide (Acros)] were used in this work.
The synthesized polyester poly{3-[(2-aminoethyl)amino]propionate} 3 (L) showed amine number of 413±8 mg(HCl)/g, number of terminal ethylenediamine fragments m = 14, degree of functionalization 87.5±2% [32]. Amine number and the amino groups fraction were determined via titration with 0.1 mol/L solution of HCl prepared via dissolution of a standard sample with 50% isopropanol solution in water. The obtained solutions were diluted to obtain the demanded concentration of the titrant. The stock and working solutions of compounds 3 and 4 were prepared via dissolution of weighed amount of the sample in organic solvents; the solutions were consumed during at least a day. The working solutions (c = 1×10–4–1×10–2 mol/L) were prepared via sequential dilution. Spectrophotometric measurements were performed using 0.1 M. solution of LiClO4 (a weighed amount) in DMSO.
ATR-FT-IR spectra were recorded over the range from 4000 to 400 cm–1 using a FT-IR spectrometer Spectrum 400 (Perkin Elmer) with a universal ATR accessory and a ZnSe prism. NMR spectra (CDCl3) were recorded using an Avance 400 Bruker instrument operating at 400 (1Н) or 125.77 MHz (13С). pH-Metric titrations were performed on a Kyoto Electronics automatic potentiometric titrator AT-610 equipped with Kyoto Electronics combined glass microelectrode at 25±0.05°C using a standard 0.1 M aqueous HCl solution diluted with 1 : 1 water–propan-2-ol. ESR spectra of solid samples and frozen solutions were recorded using a Bruker-200 X-band spectrometer at frequency of 9.4 GHz, microwave power of 20 mW, and magnetic field amplitude of 5 G over 150‒300 K temperature range.
Hyperbranched polyacrylate (2) was prepared as described elsewhere [25, 29, 30] in a benzene‒acetone mixture at the 1 : acryloyl chloride ratio of 1 : 20, reaction duration 20 h at 50°С. Yield 70%. IR spectrum, ν, cm–1: 3442 (O‒H), 3061 (=C‒H), 2982–2886 (CH3, CH2), 1733 (C=O), 1635, 1619 (С=C), 1472‒1460, 1400–1372 [δ(CH3, CH2)], 1409 [δ(=CH2)], 1297‒1133 (C–Oester), 1062, 1001 (O–C), [δ(OH)], 986, 809 [δ(CH=CH2)]. 1H NMR spectrum (CDCl3), δ, ppm: 1.22‒1.30 m [36H, OC(O)CMe], 2.69 br. s (7H, CH2OH), 3.56‒3.60 m (24H, CH2OH, OCH2C), 4.26 m [48H, CH2OC(O)], 5.80 m (14H, CH2=CH), 6.07 m (14H, CH2=CH), 6.50 m (14H, CH2=CH).
Hyperbranched polyester poly{3[(2-aminoethylamino]propionate} (3) was prepared via addition of ethylenediamine to polyester polyacrylate 2 as described elsewhere [30], at the 2 : ethylenediamine ratio of 1 : 16, reaction duration 20 h at room temperature. Yield 75%. IR spectrum, ν, cm–1: 3352–3293 (O‒H, N‒H), 2970–2880 (CH3, CH2), 2829 (CH2NH), 1732 (C=O), 1662, 1648, 1556 [δ(NH)], 1464 br, 1406–1369 [δ(CH3, CH2)], 1369 [δ(СН2N)], 1297‒1125 (C–Oester), 1054–1008 [(O–C), δ(OH)]. 1H NMR spectrum (СDCl3), δ, ppm: 1.14–1.29 m [36H, OC(O)CMe], 2.37 t [(O)CCH2CH2N, 3JHH = 6.2], 2.67 t [CH2NH2, 3JHH = 5.8], 2.68 t [CH2NH2, 3JHH = 5.8], 2.80 t [CH2NH, 3JHH = 5.1], 2.81 t [CH2NH, 3JHH = 5.1], 2.87 t [CH2NH, 3JHH = 5.8], 2.89 t [CH2NH, 3JHH = 5.8], 3.26‒3.28 m [OCH2CH2O], 3.85‒3.89 m [CH2OC(O)], 6.44 br. s (CH2OH). Found, %: C 52.16; H 7.89; N 11.80. C145H268N28O59. Calculated, %: C 52.02; H 8.01; N 11.72.
Complex of polyamine 3 with Cu(II) ions (4). A solution of 1.92 mmol of copper(II) nitrate in 20 mL of ethanol was added to a solution of 0.12 mmol of compound 3 in 10 mL of THF (ratio 1 : 16). The mixture was stirred at room temperature; the formed complex precipitated out. Reaction duration 8 h at 25°С. The precipitate was separated via decantation, washed with THF and ethanol, and dried in vacuum. Yield 81%. Dark-brown tar soluble in water, aqueous alcohol, and DMSO. IR spectrum, ν, cm–1: 3437‒3251 (O‒H, N‒H), 2974‒2891 (CH3, CH2), 1729, 1708 (C=O), 1664‒1578 [δ(NH)], 1458 br, 1378–1331 [δ(CH3, CH2)], 1225, 1149 (C–Oester), 1042–1010 (O–C), [δ(OH)], 532 (Cu‒O), 412 (Cu‒N). Found, %: C 21.50; H 4.70; N 11.44; Cu 16.20. C145H394Cu21N70O248. Calculated, %: C 20.67; H 4.68; N 11.65; Cu 15.85.
REFERENCES
Uflyand, I.E. and Dzhardimalieva, G.I., J. Coord. Chem., 2018, vol. 71, no. 9, p. 1272. https://doi.org/10.1080/00958972.2018.1465567
Gao, С. and Yan, D., Prog. Polym. Sci., 2004, vol. 29, p. 183. https://doi.org/10.1016/j.progpolymsci.2003.12.002
Yates, C.R. and Hayes, W., Eur. Polym. J., 2004, vol. 40, p. 1257. https://doi.org/10.1016/j.eurpolymj.2004.02.007
Diallo, M., Balogh, L., Shafagati, A., Johnson, I.H., Goddard, W.A., and Tomalia, D.A., Environ. Sci. Technol., 1999, vol. 33, no. 5, p. 820. https://doi.org/10.1021/es980521a
Jang, W.-D., Kamruzzaman Selim, K.M., Lee, C.-H., and Kang, I.-K., Prog. Polym. Sci., 2009, vol. 34, p. 1. https://doi.org/10.1016/j.progpolymsci.2008.08.003
Manoj, E., Kurup, M.R.P., and Punnoose, A., Spectrochim. Acta. (A), 2009, vol. 72, no. 3, p. 474. https://doi.org/10.1016/j.saa.2008.10.030
Wang, S.J., Brechbiel, M., and Wiener, E.C., Invest. Radiol., 2003, vol. 38, no. 10, p. 662. https://doi.org/10.1097/01.rli.0000084887.47427.75
Labieniec, M. and Watala, C., Cent. Eur. J. Biol., 2009, vol. 4, no. 4, p. 434. https://doi.org/10.2478/s11535-009-0056-7
Ottaviani, M.F., Montalti, F., Turro, N.I., and Tomalia, D.A., J. Phys. Chem. (B), 1997, vol. 101, no. 2, p. 158. https://doi.org/10.1021/jp962857h
Bosman, A.W., Schenning, A.P.H.J., Janssen, R.A.J., and Meijer, E.W., Chem. Ber. Recueil., 1997, vol. 130, p. 725. https://doi.org/10.1002/cber.19971300608
Krot, K.A., Danil de Namor, A.F., Aguilar-Cornejo, A., and Nolan, K.B., Inorg. Chim. Acta, 2005, vol. 358, p. 3497. https://doi.org/10.1016/j.ica.2005.05.001
Diallo, M.S., Christie, S., Swaminathan, P., Balogh, L., Shi, X., Um, W., Papelis, C., Goddard, W.A., and Jonson, J.H., Langmuir, 2004, vol. 20, no. 7, p. 2640. https://doi.org/10.1021/la036108k
Zhou, L., Russell, D.H., Zhao, M., and Crooks, R.M., Macromolecules, 2001, vol. 34, no. 11, p. 3567. https://doi.org/10.1021/ma001782j
Ottaviani, M.F., Bossmann, S., Turro, N.J., and Tomalia, D.A., J. Am. Chem. Soc., 1994, vol. 116, no. 2, p. 661. https://doi.org/10.1021/ja00081a029
Floriano, P.N., Noble, Schoonmaker, J.M., Poliakoff, E.D., McCarley, R.L., J. Am. Chem. Soc., 2001, vol. 123, no. 43, p. 10545. https://doi.org/10.1021/ja010549d
Mecke, A., Uppuluri, S., Sassanella, T.M., Lee, D.K., Ramamoorthy, A., Baker, J.R.Jr., Orr, B.G., and Banaszak Holl, M.M., Chem. Phys. Lipids, 2004, vol. 132, no. 1, p. 3. https://doi.org/10.1016/j.chemphyslip.2004.09.001
Kobayashi, H., Kawamato, S., Jo, S.K., Bryant, H.L.Jr., Brechbiel, M.W., and Star, R.A., Bioconjug. Chem., 2003, vol. 14, no. 2, p. 388. https://doi.org/10.1021/bc025633c
Inoue, K., Prog. Polym. Sci., 2000, vol. 25, no. 4, p. 453. https://doi.org/10.1016/S0079-6700(00)00011-3
Žagar, E. and Žigon, M., Prog. Polym. Sci., 2011, vol. 36, no. 1, p. 53. https://doi.org/10.1016/j.progpolymsci.2010.08.004
Нäußler, M., Dong, H., and Tang, B.Z., Inorganic and Organometallic Macromolecules: Desigh and Application, Springer Science+Business Media, LLC, 2008, p. 21. https://doi.org/10.1007/978-0-387-72947-3_2
Zhong, Z., Song, Y., Engbersen, J.F.J., Lok, M.C., Hennink, W.E., and Feijen, J., J. Control Release, 2005, vol. 109, no. 1–3, p. 317. https://doi.org/10.1016/j.jconrel.2005.06.022
Reul, R., Nguyen, J., and Kissel, T., Biomaterials, 2009, vol. 30, no. 29, p. 5815. https://doi.org/10.1016/j.biomaterials.2009.06.057
Arote, R., Kim, T.H., Hwang, Y.K., Jiang, H.L., Nah, J.W., Cho, M.H., and Cho, C.S., Biomaterials, 2007, vol. 28, no. 4, p. 735. https://doi.org/10.1016/j.biomaterials.2006.09.028
Reul, R., Nguyen, J., Biela, A., Marxer, E., Bakowsky, U., Klebe, G., and Kissel, T., Int. J. Pharm., 2012, vol. 436, nos. 1–2, p. 97. https://doi.org/10.1016/j.ijpharm.2012.06.065
Kutyreva, M.P., Gataulina, A.R., Kutyrev, G.A., Ulakhovich, N.A., Newman, T., Khasanova, E.M., Bondar, O.V., Yurtaeva, S.V., Ziganshina, S.A., and Khaldeeva, E.V., Inorg. Chim. Acta, 2016, vol. 450, p. 101. https://doi.org/10.1016/j.ica.2016.04.013
Khannanov, A.A., Kutyreva, М.P., Ulakhovich, N.A., Gataulina, А.R., Bondar, О.V., Zakharova, L.Y., and Kutyrev, G.A., Fluid Phase Equilibria, 2016, vol. 411, p. 93. https://doi.org/10.1016/j.fluid.2015.12.023
Kutyreva, M.P., Usmanova, G.Sh., Ulakhovich, N.A., Medvedeva, O.I., Syakaev, V.V., and Ziganshina, S.A., Polym. Sci. (B), 2013, vol. 55, nos. 3–4, p. 201. https://doi.org/10.1134/S1560090413040052
Gataulina, A.R., Khannanov, A.A., Malinovskikh, O.A., Bondar, O.V., Ulakhovich, N.A., and Kutyreva, M.P., Russ. J. Gen. Chem., 2013, vol. 83, no. 12, p. 2269. https://doi.org/10.1134/S1070363213120074
Gataulina, A.R., Khasanova, E.M., Ulakhovich, N.A., Kutyrev, G.A., and Kutyreva, M.P., Russ. J. Gen. Chem., 2018, vol. 88, no. 9, p. 1874. https://doi.org/10.1134/S1070363218090189
Kutyreva, M.P., Gataulina, A.R., Kutyrev, G.A., Nizamov, I.S., and Ulakhovich, N.A., Russ. J. Gen. Chem., 2011, vol. 81, no. 7, p. 1535. https://doi.org/10.1134/S1070363211070206
Bellamy, L.J., The Infrared Spectra of Complex Molecules Volume Two Advances in Infrared Group Frequencies, London: Methuen Inc., Chapman and Hall, 1980. https://doi.org/10.1007/978-94-011-6520-4
Toroptseva, A.M., Belogorodskaya, K.V., and Bondarenko, V.M., Laboratornyi praktikum po khimii i tekhnologii vysokomolekulyarnykh soedinenii (Laboratory Workshop on Chemistry and Technology of High-Molecular Compounds), Leningrad: Khimiya, 1972, p. 127.
Kutyreva, M.P., Ulakhovich, N.A., Sidorov, P.O., Kutyrev, G.A., Gataulina, A.R., and Salnikov, Yu.I., World Appl Sci. J., 2013, vol. 26, no. 7, p. 973. https://doi.org/10.5829/idosi.wasj.2013.26.07.13533
Nakamoto, K., Infrared Spectra of Inorganic and Coordination Compounds, New York: John Wiley and Sons Inc., 1970. https://doi.org/10.1002/bbpc.19710750622
Kacan, M., Turkyilmaz, M., Karabulut, F., Altun, O., and Baran, Y., Spectrochim. Acta (A), 2014, vol. 118, p. 572. https://doi.org/10.1016/j.saa.2013.09.031
Basha, M.T., Alghanmi, R.M., Shehata, M.R., Abdel-and Rahman, L.H., J. Mol. Struct., 2019, vol. 1183, p. 298. https://doi.org/10.1016/j.molstruc.2019.02.001
Lever, A.B.P., Inorganic Electronic Spectroscopy in Studies in Physical and Theoretical Chemistry, Amsterdam: Elsevier, 1984. 863 p. https://doi.org/10.1002/bbpc.19850890122
Volchenskova, I.I., Teor. Eksp. Khim., 1973, vol. 9, no. 5, p. 627.
Rybak-Akimova, E.V., Nazarenko, A.Y., Chen, L., Krieger, P.W., Herrera, A.M., Tarasov, V.V., and Robinson, P.D., Inorg. Chim. Acta, 2001, vol. 324, nos. 1–2, p. 1. https://doi.org/10.1016/S0020-1693(01)00495-9
Kokorin, A.I., Vengerova, N.A., Kirsh, Yu.E., and Zamaraev, K.I., Dokl. Akad. Nauk SSSR, 1972, vol. 202, no. 3, p. 597.
Kirsh, Yu.E., Kovner, V.Ya., Kokorin, A.I., Zamaraev, K.I., Chernyak, V.Ya., and Kabanov, V.A., Eur. Polym. J., 1974, vol. 10, no. 8, p. 671. https://doi.org/10.1016/0014-3057(74)90178-5
Pilbrow, J.R., Transition Ion Electron Paramagnetic Resonance, Oxford: Clarendon Press, 1990. https://doi.org/10.1002/bbpc.19910951036
Peisach, J. and Blumberg, W.E., Arch. Biochem. Biophys., 1974, vol. 165, no. 2, p. 691. https://doi.org/10.1016/0003-9861(74)90298-7
Sakaguchi, U., Addison, A.W., J. Chem. Soc. Dalton Trans., 1979, no. 4, p. 600. https://doi.org/10.1039/DT9790000600
Lewis, W.B., Alei, M.Jr., and Morgan, L.O., J. Chem. Phys., 1966, vol. 44, no. 6, p. 2409. https://doi.org/10.1063/1.1727057
Beck, M. and Nagypál, I., Chemistry of Complex Equilibria, Budapest: Akadémiai Kiadó, 1990. https://doi.org/10.1002/prac.19913330133
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
No conflict of interest was declared by the authors.
Additional information
To the 80th Anniversary of R.A. Cherkasov
Rights and permissions
About this article

Cite this article
Gataulina, A.R., Sidorov, P.O., Yurtaeva, S.V. et al. Ionization and Complexing Properties of Hyperbranched Polyester Poly[3-(2-aminoethyl)amino)]propionate. Russ J Gen Chem 90, 425–433 (2020). https://doi.org/10.1134/S1070363220030159
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1070363220030159