
Abstract
The reaction of 9-iodo-ortho-carborane with n-butylmagnesium bromide affords 9-n-butyl-ortho-carborane. The reaction of the latter with alkali in boiling ethanol gives new nido-carborane [5-Bu-7,8-C2B9H11]– (I) containing the n-butyl substituent at the lower rim of the basket. The reaction of compound I with RuCl2(PPh3)(Ph2P(CH2)4PPh2) results in the formation of the corresponding ruthenium(IV) closo complex 3,3-(Ph2P(CH2)4PPh2)-3-H-3-Cl-9-Bu-closo-3,1,2-RuC2B9H10 (II) characterized by 2D NMR spectroscopy. On heating compound II can react with carbon tetrachloride to form the 17-electron complex 3,3-(Ph2P(CH2)4PPh2)-3-Cl-9-Bu-closo-3,1,2-RuC2B9H10 (III). The structure of complex III is solved by X-ray diffraction (XRD) (CIF file CCDC no. 2180761). The electrochemical studies show that complex III undergoes the reversible transition Ru(II) → Ru(III) similarly to the earlier studied ruthenacarboranes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
The C2B9-nido-carborane ligand is an isolobal analog of the cyclopentadienyl anion and can form complexes of a similar structure with a broad range of transition metals [1–6]. A substantial distinction of the dicarbollide ligand is its double negative charge and spatial aromaticity. The introduction of substituents to the carbon and boron atoms is a traditional method for changing the reactivity and properties of carborane clusters of transition metals [7, 8]. Since the carborane ligand is nonplanar, the introduction of substituents into the upper rim results in additional steric hindrances capable of leading to the catalytic activity loss by the carborane-based complexes [9]. In this respect, approaches that make it possible to prepare metallocarboranes substituted at the lower rim are most interesting.
The enhancement of the solubility is one of the problems than can be solved by the introduction of substituents into the structure of the complex. It is known that the presence of aliphatic fragments in the transition metal complexes substantially enhances their solubility in nonpolar solvents, which is especially important for homogeneous catalysis, including polymerization processes. The ruthenium carborane clusters were shown to be capable of efficiently catalyzing the polymerization of a number of methacrylic monomers via the atom-transfer mechanism [10]. A restricted solubility of compounds of this class in nonpolar solvents does not allow one to use the proposed systems in the polymerization of low-polarity monomers. The introduction of long alkyl substituents into the lower rim of the carborane ligand could solve the stated problem.
We have previously proposed an approach to the synthesis of the complexes containing methyl substituents in the lower rim of the carborane ligand [11, 12]. Remarkably, these substituents exerted no effect on steric hindrance of the metal atom but favored an increase in the electron-donor ability of the ligand thus resulting in a higher catalytic activity. At the same time, the presence of methyl substituents exerted no substantial effect on the solubility of the compounds.
In this work, we synthesized new ruthenacarboranes bearing the butyl substituent in the lower rim of the ligand using the earlier developed approach and studied their properties.
EXPERIMENTAL
Compounds [(Dppb)(Ph3P)RuCl2] [13], 9-iodo-ortho-C2B10H11 [14], and [(Ph3P)2PdCl2] [15] were synthesized according to described procedures. Diethyl ether, benzene, and toluene were dehydrated using standard procedures [16], and 1-bromobutane was distilled at the boiling point. All other reagents were purchased at DALCHEM (Russia) and Sigma-Aldrich, respectively, and used as received. All reactions were carried out under argon unless noted otherwise. The reaction course was monitored using thin-layer chromatography (aluminum plates with supported silica gel, Merck F254 silica) and a 0.5% solution of PdCl2 in a 1% water–methanol (1 : 10) solution of HCl as the developer. Silica gel Macherey-Nagel 60 Å (0.040–0.063 mm) was used for column chromatography. NMR spectra at 400 MHz (1Н) and 128 MHz (11В) were recorded on Varian Unity Inova 400, Bruker Avance 400, and Agilent DD2 NMR 400NB spectrometers. The residual NMR signal of the solvent relative to Me4Si was accepted to be the internal standard for 1Н NMR spectra. For comparison with the 11В NMR spectra, BF3·Et2O was used as the external standard. EPR spectra were recorded in frozen toluene at 150 K on a Bruker-EMX spectrometer operating at a frequency of 9.75 GHz. The MALDI-TOF mass spectra of the complexes were detected in the linear mode using the Bruker Microflex LT system and trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]malononitrile (DCTB) as the matrix. Solutions were supported on a plate-target of stainless steel and examined in positive and negative ion modes. An HPLC analysis of metallacarboranes was carried out on a Knauer SmartLine instrument equipped with a photodiode array detector. The Kromasil 300-5-CN (4.6 × 300 mm) column was used. The eluent was n-hexane–dichloromethane (6 : 1) with a flow rate of 1 mL/min. Electrochemical experiments were carried out using cyclic voltammetry (CV) in a three-electrode cell with platinum electrodes with an IPC Pro potentiostat. Tetrabutylammonium tetrafluoroborate served as the supporting electrolyte, and potentials were measured relatively to the pseudo-reference silver electrode. To recalculate potentials relatively to ferrocene as the internal standard, ferrocene was introduced directly into the electrochemical cell after the studies of the complex, and the potential of the Fc/Fc+ transition was measured, the value of which was accepted to be zero. The IR spectra of the complexes were recorded on an Infralum FT IR spectrometer in the solid KBr matrix.
Synthesis of 9-butyl-ortho-carborane (9-Bu-1,2-C2B10H11) was carried out according to a described procedure [17]. 1-Bromobutane (0.6 mL, 30% of the total amount) was added to a suspension of magnesium chips (730 mg, 30 mmol) in diethyl ether (25 mL). The obtained mixture was refluxed until a turbid solution was formed, and a solution of the remained portion of 1-bromobutane (1.0 mL, totally 2055 mg, 15.0 mmol) in diethyl ether (25 mL) was added dropwise. The mixture was refluxed for 1 h, and a solution of 9-iodo-closo-carborane (1350 mg, 5.0 mmol) in diethyl ether (25 mL) was added dropwise. The resulting suspension was stirred at room temperature for 1 h, and [(Ph3P)2PdCl2] (140 mg, 0.2 mmol) and CuI (40 mg, 0.2 mmol) were added as one portion. The mixture was refluxed for 40 h. After cooling, a 5% solution of HCl (50 mL) was added. The organic fraction was separated, and the aqueous fraction was washed with diethyl ether (3 × 50 mL). The combined organic fractions were dried over Na2SO4, filtered, and evaporated on a rotary evaporator. The product was purified by column chromatography using diethyl ether as the eluent to obtain 9-butyl-ortho-C2B10H11 as an amber-colored oil in a yield of 765 mg (72%).
1H NMR (CDCl3; 25°C; δ, ppm): 3.48 (br.s, 1H, CHcarb), 3.41 (br.s, 1H, CHcarb), 1.1–3.0 (br.m, 9H, BHcarb), 1.26 (m, 4H, CH2), 0.85 (t, 3H, J = 7.0 Hz, CH3), 0.70 (br.m, 2H, BCH2). 11B NMR (CDCl3; 25°C; δ, ppm): 9.2 (s, 1B, B–C), –2.0 (d, 1B, J = 149 Hz), –8.9 (d, 2B, J = 149 Hz), –13.7 (d, 2B, J = 162 Hz), –14.4 (d, 2B, J = 136 Hz), –15.5 (d, 2B, J = 162 Hz).
Synthesis of (Me3NH)[5-Bu-7,8-C2B9H11]. The reaction was carried out in air. A solution of 9-butyl-closo-carborane (765 mg, 3.8 mmol) and NaOH (1400 mg, 35.0 mmol) in ethanol (30 mL) was refluxed for 24 h. After cooling, a 30% solution of HCl was added dropwise to the mixture to the neutral pH of the medium, and the suspension was filtered and evaporated on a rotary evaporator. The obtained residue was dissolved in water (10 mL), and a solution of trimethylammonium chloride (570 mg, 6.0 mmol) in water (10 mL) was added. The resulting organic layer was extracted with dichloromethane (20 mL). The organic fraction was separated, and the aqueous fraction was washed with dichloromethane (3 × 20 mL). The combined organic fractions were dried over Na2SO4, filtered, and evaporated on a rotary evaporator. 5-Butyl-nido-C2B10H11 trimethylammonium salt was obtained as a beige powder in a yield of 910 mg (96%).
1H NMR (CD3COCD3; 25°C; δ, ppm): 3.14 (s, 9H, (CH3)3NH), 1.68 (br.s, 1H, CHcarb), 1.49 (br.s, 1H, CHcarb), 1.19 (m, 4H, CH2), –0.5–2.6 (br.m, 9H, BHcarb), 0.78 (t, 3H, J = 7.0 Hz, CH3), 0.48 (br.m, 2H, BCH2), –2.80 (br.m, 1H, BHB). 11B NMR (CD3COCD3; 25°C; δ, ppm): –3.5 (s, 1B, B–C), ‒9.4 (d, 1B, J = 134 Hz), –12.6 (d, 1B, J = 134 Hz), –18.2 (d, 1B, J = 159 Hz), –18.7 (d, 1B, J = 125 Hz), –21.2 (d, 1B, J = 141 Hz), –23.0 (d, 1B, J = 149 Hz), –31.6 (dd, 1B, J1 = 127 Hz, J2 = 44 Hz,), –36.8 (d, 1B, J = 141 Hz).
Synthesis of K[5-Bu-7,8-C2B9H11] (I). A Schlenk flask was loaded with (Me3NH)[5-Bu-7,8-C2B9H11] (47 mg, 0.188 mmol) and potassium hydroxide (10.6 mg, 0.188 mmol), methanol (5 mL) was poured to the mixture, and the resulting solution was magnetically stirred for 24 h. The solution was evaporated in vacuo and dried over P2O5 in vacuo. The yield of the final product was 40 mg (93%).
Synthesis of complex [3,3-(Ph2P(CH2)4PPh2)-3-Cl-3-H-closo-9-Bu-3,1,2-RuC2B9H10] (II). A round-bottom Schlenk flask was loaded with [(Dppb)(Ph3P)RuCl2] (138.7 mg) and compound I (40 mg), and argonized benzene (10 mL) was poured into the flask. The contents of the flask was degassed three times to a residual pressure of ~1.3 Pa. The reaction occurred at 40°С in a water bath for 3 h under an argon flow with continuous stirring. The resulting brown solution was evaporated, and the residue was chromatographed on a column packed with silica gel eluting yellow and dark red bands with benzene. Then the product was recrystallized from a dichloromethane–n-hexane mixture to obtain yellow crystals. The yield was 66.1 mg (54%).
1H NMR (CD2Cl2; 25°C; δ, ppm): 7.30–7.63 (m, 20H, Ph), 3.61, 2.92 (br.s, 2H CHcarb), 1.50, 2.12 (m, 4H PCH2CH2CH2CH2P), 2.52, 3.72 (m, 4H PCH2CH2CH2CH2P), 1.14, 1.01 (m, 4H СH3–(CH2)2–СH2B), 0.77 (m, 3H СH3), 0.53 (m, 2H ‒СH2B), –8.35 (td, 1H Ru–H). 31P{1H} NMR (CD2Cl2; 25°C; δ, ppm): 38.02 (1P), 36.31 (1P). 11B{1H} NMR (CD2Cl2; 25°C; δ, ppm): –19.6 (2B), –17.3 (1B), –5.7 (3B), –4.4 (1B), 6.5 (1B), and 8.2 (1B).
IR (KBr; ν, cm–1): 2561 ν(B–H). MALDI MS ((М–H)–, 752.2), calcd. 752.2.
Synthesis of complex [3,3-(Ph2P(CH2)4PPh2)-3-Cl-closo-9-Bu-3,1,2-RuC2B9H10] (III). Compound II (114.6 mg) was placed in a Schlenk flask. The contents of the flask was degassed three times to a residual pressure of ~1.3 Pa. Then toluene (15 mL) freshly distilled under argon and carbon tetrachloride (200 μL) were added, and the mixture was heated in an oily bath at 90°С for 1 h. The resulting bright red solution was evaporated and supported on a chromatographic column. The red band of the product was eluted with a benzene–n-hexane (2 : 1) mixture. The obtained solution was dried, and the residue was dissolved in dichloromethane and precipitated with hexane. The yield of crystals of compound III was 52.7 mg (46%).
IR (KBr; ν, cm–1): 2531 ν(B–H). MALDI MS (М–, 752.2), calcd. 752.2; EPR (toluene, 150 K): g1 = 2.496, g2 = 2.082, g3 = 1.959.
XRD of compound III was carried out on an Oxford Diffraction Gemini S automated X-ray single-crystal diffractometer (MoKα radiation, λ = 0.71073 Å, graphite rod, ω scan mode) at Т = 293 K. The primary fragment of the structure was revealed by direct methods using the SHELX [18] and ShelXle [19] programs. Parameters of other atoms were determined by the difference electron density synthesis and refined for |F|2 by least squares. Positions of hydrogen atoms were determined geometrically and refined by the riding model. The main crystallographic parameters are given in Table 1. Selected bond lengths and bond angles are listed in Table 2.
The XRD results were deposited with the Cambridge Crystallographic Data Centre (CIF file CCDC no. 2180761; deposit@ccdc.cam.ac.uk; http://www.ccdc.cam.ac.uk).
RESULTS AND DISCUSSION
5-Butyl-nido-carborane potassium salt was synthesized in three stages via the scheme similar to that published previously for the methyl-substituted derivatives with resembling structures [12] (Scheme 1). 9-Methyl-ortho-carborane was synthesized at the first stage by the Pd-catalyzed cross-coupling of 9-iodo-ortho-carborane and n-butylmagnesium bromide. The introduction of the butyl substituent into the carborane structure is confirmed by the nonequivalence of the hydrogen atoms linked to the carbon atoms of the carborane ligand giving two signals in the 1H NMR spectrum at 3.48 and 3.41 ppm and by signals at 1.26, 0.85, and 0.70 ppm as well. The signal of the methylene fragment bound to the boron atom is observed in the highest-field range at 0.70 ppm.
Reflux of 9-methyl-ortho-carborane with NaOH in ethanol followed by the addition of trimethylammonium chloride made it possible to isolate trimethylammonium salt of the corresponding nido-carborane, which was further transformed into potassium salt K+[5-Bu-7,8-C2B9H11]– (I) by the treatment with KOH. The formation of nido-carborane results in the shift of the signals of protons of the butyl substituent in the 1H NMR spectrum in a high-field range (1.19, 0.78, and 0.48 ppm). The strongest shift is observed for protons of the methylene group attached directly to the boron atom, which is explained by an increase in the electron density due to the appearance of the negative charge delocalized over the carborane basket. The synthesis of substituted nido-carborane I is shown in Scheme 1.

Scheme 1 .
New ruthenium complexes bearing butyl substituents in the carborane ligand were synthesized using the procedure approbated earlier for the analogs with methyl substituents [12]. The reaction of compound I with the known complex [(Dppb)(Ph3P)RuCl2] in benzene at 40°С according to Scheme 2 gave the target product [3,3-(Ph2P(CH2)4PPh2)-3-H-3-Cl-9-Bu-closo-3,1,2-RuC2B9H10] (II) in a yield of 54%.

Scheme 2 .
The structure of compound II was assumed on the basis of the NMR spectroscopy and mass spectrometry data. The 31Р{1H} NMR spectrum of compound II in CH2Cl2 exhibits two signals at 38.02 and 36.31 ppm. These two signals are caused by the nonequivalence of the phosphorus atoms owing to the symmetry plane loss by the molecule because of the presence of the butyl substituent at the boron atom in position 9 of the carborane ligand. The 11B{1H} NMR spectrum contains signals from nine boron atoms in the range from –19.36 to –4.43 ppm. It should be mentioned that the 31Р{1H} and 11B{1H} NMR spectra detected for compound II are nearly identical to the corresponding spectra recorded for its methyl analog [3,3-(Ph2P(CH2)4PPh2)-3-H-3-Cl-9-Me-closo-3,1,2-RuC2B9H10] (IIa), which assumes a similar structure for compound II.
The 1H NMR spectrum of compound II is also similar to that of its methyl analog IIa. Six multiplets from 20 protons of the aromatic rings of the diphosphine ligand are observed in a range of 7.30–7.63 ppm. Two characteristic broadened singlets at 3.61 and 2.92 ppm correspond to the nonequivalent CH groups of the carborane basket. The protons of the CH2 groups of the bridging fragment and butyl substituent appear as multiplets in ranges of 3.7–1.5 and 1.1–0.5 ppm, respectively. The signal from the hydrogen atom bound to ruthenium appears at –8.35 ppm as a triplet of doublets caused by signal splitting on the ruthenium and phosphorus atoms. These signals in the 1Н NMR spectrum were assigned by 2D COSY 1H–1H NMR spectroscopy (Fig. 1). The signal of the hydrogen atoms of the methylene group bound to the carborane ligand is observed in a high field as a multiplet with the center at 0.53 ppm. The terminal methyl group gives a signal at 0.77 ppm. Multiplets with the centers at 1.14 and 1.01 ppm correspond to two remained methylene fragments. The protons at each carbon atom are equivalent due to a possibility of free rotation. Unlike them, all protons of the bridging diphosphine ligand are nonequivalent, which is clearly seen in the 1H–13C HSQC NMR spectrum shown in Fig. 2. The signals from the protons of the terminal groups linked to the phosphorus atoms are observed at 3.7–2.5 ppm, whereas the protons of the central groups give the signal in a higher field in a range of 1.9–1.5 ppm. The formation of compound III is shown in Scheme 3.

Scheme 3 .

Fragment of the 2D COSY (1Н–1H) NMR spectrum of compound II.

Fragment of the 2D HSQC (1Н–13C) NMR spectrum of compound II.
The reaction of complex II with carbon tetrachloride in toluene at 90°С gave new complex [3,3-(Ph2P(CH2)4PPh2)-3-Cl-9-Bu-closo-3,1,2-RuC2B9H10] (III) as dark red crystals in a yield of 46%. It should be mentioned that compounds II and III are formed in lower yields than the yield of the unsubstituted carborane derivatives. The MALDI mass spectrum detected in the negative ion mode contains a characteristic signal at 752.2 Da corresponding to the molecular anion of complex III. The signal shape (Fig. 3) corresponds to the theoretically calculated isotope distribution of the complex. The signal at 190.2 Da in the mass spectrum corresponds to nido-carborane [5-Bu–C2B9H11]– formed upon the partial decomposition of the complex under laser ionization conditions. The intense signal of the free anionic ligand indicates the absence of a covalent bond between the carborane ligand and phenyl rings of diphosphine, since the formation of 17-electron complexes is accompanied, in several cases, by the formation of such a bond.

MALDI mass spectrum recorded for compound III in the negative ion mode.
The 17-electron nature of compound III is confirmed by its EPR spectrum with the parameters g1 = 2.496, g2 = 2.082, and g3 = 1.959. The obtained values are close to those for its earlier synthesized analog based on the unsubstituted dicarbollide dianion [3,3-(Ph2P-(CH2)4PPh2)-3-Cl-closo-3,1,2-RuC2B9H11] (IV, g1 = 2.487, g2 = 2.070, g3 = 1.947) [20] and similar methyl derivative [3,3-(Ph2P(CH2)4PPh2)-3-Cl-9-Me-closo-3,1,2-RuC2B9H10] (IIIа, g1 = 2.477, g2 = 2.075, g3 = 1.950) [11]. The obtained results indicate that the introduction of the substituent into the lower rim of the carborane ligand exerts no effect on the unpaired electron density distribution and configuration of the ruthenium atom.
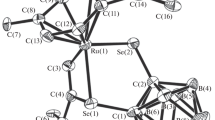
The closo structure of complex III was confirmed by XRD analysis (Fig. 4). The ruthenium atom is bound to five atoms of the open plane of the carborane ligand and one chlorine atom and two phosphorus atoms of 1,4-bis(diphenylphosphino)butane. The determined structural parameters of complex III correspond, as a whole, to those for compound IV. For example, the distance between the ruthenium atom and the center of the C2B3 plane of the carborane ligand is 1.711 Å, which is slightly longer than the analogous distance in compound IV (1.697 Å) and is characteristic of closo-ruthenacarboranes. The distance between the carbon atoms in the ligand is 1.607(7) Å, which is slightly shorter than that for compound IV (1.617(3) Å), whereas the В(9)–В(12) bond, on the contrary, is longer (1.798(8) vs. 1.777(5) Å). A similar increase in the В(9)–В(12) distance was observed upon the introduction of methyl substituents into the lower rim of the ligand [12], which suggests the regularity observed. An interesting feature of the structure of complex III in the crystalline state is a sufficiently large bond angle P(1)RuP(2) (96.38(4)°), which is substantially larger than that for compound IV (92.06(2)°). On the contrary, the P(2)RuCl angle is small (83.59(5)°). The NMR and EPR studies of compounds II and III showed that the introduction of the butyl substituent into the lower rim of the carborane ligand did not affect the configuration of the metal atom. Taking into account this fact, we can assume that the change in the bond angle is due to a less dense crystal packing of compound III. The conformation of the complex in the crystalline state is that the chlorine atom is located nearly above the С(1) carbon atom of the carborane ligand. The corresponding interplanar H–C(1)–Ru–C(1) angle is 12.28°. The phosphorus atoms are arranged above the boron atoms B(9) and B(11) of the lower rim. The P(1)–Ru–B(9)–C(3) and P(2)–Ru–B(11)–Н angles are 5.21° and 8.34°, respectively. The butyl substituent is directed from the carborane basket. No interactions with other moieties of the basket are observed in the crystalline state of the complex. At the same time, short contacts between the butyl substituent of one molecule and the phenyl rings of diphosphine of another molecule are observed in the crystalline state, which can result in a slight deformation of the metallacycle and corresponding bond angle.

Molecular structure of complex III.
An HPLC analysis of the prepared metallacarboranes showed that the introduction of the butyl substituent into the carborane ligand structure led to a change in the polarity of the molecule. Under the chosen analysis conditions, the retention times of complexes II and III were 13.3 and 13.1 min compared to 20.5 and 25.1 min for the corresponding derivatives of unsubstituted nido-carborane, which indicated a lower polarity of the complex with the substituent.
The CV studies showed (Fig. 5) that the 18-electron complex II underwent reversible oxidation at the potential equal to 1010 mV versus ferrocene, which coincides with the value for its analog IIa bearing the methyl substituent at the B(9) atom of the carborane ligand [12]. No other transitions are observed in the studied range.

CV curves detected for complexes II (Epa = 1100 mV) and III (Epa1 = –343 mV, Epс1 = –410 mV, E1/2 = –377 mV, Epa2 = 725 mV) in a 1,2-dichloroethane (c = 0.003 mol/L) solution using Bu4NPF6 (0.2 M) as the supporting electrolyte.
The 17-electron complex III is able to the reversible reduction Ru(III)–Ru(II) at the potential equal to –377 mV. A comparison of the obtained value with that for its unsubstituted analog IV (–311 mV [21]) indicates that the introduction of the alkyl substituent into the carborane ligand decreases the oxidation potential of the metallocomplex by ~70 mV. A similar phenomenon was observed earlier for the derivatives containing methyl groups. Note that the complex is oxidized irreversibly at 725 mV versus ferrocene.
The studies performed in this work showed that the previously proposed approach can successfully be used for the synthesis of various nido-carborane derivatives and complexes based on this ligand. It should be mentioned that the introduction of the substituent into the lower rim of the carborane ligand exerts no substantial effect on the structural parameters of the complexes but decreases its redox potential, and the latter is independent of the alkyl substituent nature. At the same time, the introduction of long alkyl substituents decreases the polarity of the corresponding compounds and increases their solubility in nonpolar media.
REFERENCES
D’yachikhin, D.I., Dolgushin, F.M., Godovikov, I.A., and Chizhevsky, I.T., Mendeleev Commun., 2010, vol. 20, no. 3, p. 174. https://doi.org/10.1016/j.mencom.2010.05.018
Powley, S.L., Rosair, G.M., and Welch, A.J., Dalton Trans., 2016, vol. 45, no. 26, p. 11742. https://doi.org/10.1039/C6DT01888B
Jones, J.J., English, L.E., Robertson, A., et al., J. Organomet. Chem., 2018, vol. 865, p. 65. https://doi.org/10.1016/j.jorganchem.2018.02.007
D’yachihin, D.I., Kostyukovich, A.Yu., Godovikov, I.A., et al., Mendeleev Commun., 2019, vol. 29, no. 1, p. 69. https://doi.org/10.1016/j.mencom.2019.01.023
Kellert, M., Sarosi, I., and Rajaratnam, R., Molecules, 2020, vol. 25, no. 10, p. 2322. https://doi.org/10.3390/molecules25102322
Stogniy, M.Yu., Bogdanova, E.V., Anufriev, S.A., and Sivaev, I.B., Russ. J. Inorg. Chem., 2022, vol. 67, no. 10, p. 1537. https://doi.org/10.1134/S0036023622600858
Tutusaus, O., Vinas, C., Nunez, R., et al., J. Am. Chem. Soc., 2003, vol. 125, no. 39, p. 11830. https://doi.org/10.1021/ja036342x
Anufriev, S.A., Sivaev, I.B., and Bregadze, V.I., Russ. Chem. Bull., 2015, vol. 64, no. 3, p. 712. https://doi.org/10.1007/s11172-015-0924-4
Grishin, I.D., Turmina, E.S., Chizhevsky, I.T., and Grishin, D.F., Polym. Sci., 2012, vol. 54, nos. 7–8, p. 383. https://doi.org/10.1134/S1560090412080027
Grishin, I.D., Knyazeva, N.A., and Penkal’, A.M., Russ. Chem. Bull., 2020, vol. 69, no. 8, p. 1520. https://doi.org/10.1007/s11172-020-2931-3
Zimina, A.M., Derendyaeva, M.A., Knyazeva, N.A., et al., Dokl. Chem., 2021, vol. 498, no. 2, p. 97. https://doi.org/10.31857/S2686953521030110
Grishin, I.D., Zimina, A.M., Anufriev, S.A., et al., Catalysts, 2021, vol. 11, no. 11, p. 1409. https://doi.org/10.3390/catal11111409
Jung, C.W., Garrou, P.E., Hoffman, P.R., and Caulton, K.G., Inorg. Chem., 1984, vol. 23, no. 6, p. 726. https://doi.org/10.1021/ic00174a018
Andrews, J.S., Zayas, J., and Jones, M., Inorg. Chem., 1985, vol. 24, no. 22, p. 3715. https://doi.org/10.1021/ic00216a053
Handbuch der Präparativen Anorganischen Chemie, Brauer, G., Ed., Stuttgart: Ferdinand Enke, 1981.
Armarego, W.L.F. and Chai, C.L.L., Purification of Laboratory Chemicals. Burlington: Butterworth-Heinemann, 2009.
Zakharkin, L.I., Kovredov, A.I., Ol’shevskaya, V.A., and Shaugumbekova, Zh.S., J. Organomet. Chem., 1982, vol. 226, no. 3, p. 217. https://doi.org/10.1016/S0022-328X(00)83405-1
Sheldrick, G.M., Acta Crystallogr., Sect. A: Found. Adv., 2015, vol. 71, no. 1, p. 3. https://doi.org/10.1107/S2053273314026370
Hübschle, C.B., Sheldrick, G.M., and Dittrich, B., J. Appl. Crystallogr., 2011, vol. 44, no. 6, p. 1281. https://doi.org/10.1107/S0021889811043202
Grishin, I.D., D’yachihin, D.I., Piskunov, A.V., et al., Inorg. Chem., 2011, vol. 50, no. 16, p. 7574. https://doi.org/10.1021/ic200487w
Grishin, I.D., Agafonova, K.S., Grishin, D.F., et al., Russ. Chem. Bull., 2014, vol. 63, no. 4, p. 945. https://doi.org/10.1007/s11172-014-0532-8
ACKNOWLEDGMENTS
The authors are grateful to Assoc. Prof. Yu.B. Malysheva and Prof. A.V. Piskunov for recording NMR and EPR spectra of metallacarboranes, respectively. The NMR spectra of the carborane derivatives were recorded using the scientific equipment of the Center for Collective Use of the Center of Molecular Structure Investigation at the Nesmeyanov Institute of Organoelement Compounds (Russian Academy of Sciences) supported by the Ministry of Science and Higher Education of the Russian Federation.
Funding
This work was supported by the grant of the President of the Russian Federation for young scientists (doctors of sciences), project no. MD-1474.2022.1.3.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
The authors declare that they have no conflicts of interest.
Additional information
Translated by E. Yablonskaya
ADDITIONAL INFORMATION
This article is prepared for the memorial issue in tribute to the Corresponding Member of the Russian Academy of Sciences K.Yu. Zhizhin on his 50th birthday.
Rights and permissions
About this article

Cite this article
Zimina, A.M., Kolpakova, T.V., Anufriev, S.A. et al. New 5-n-C4H9-C2B9-Carborane Ligand and Its Ruthenium Complexes. Russ J Coord Chem 49, 329–337 (2023). https://doi.org/10.1134/S1070328423700604
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1070328423700604