
Abstract
The interaction of 3,3-(Ph2P(CH2)4PPh2)-3-H-3-Cl-closo-3,1,2-RuC2B9H11 (1) with isopropylamine and corresponding nitrile (NCR) in dichloromethane at 40 °C yields carborane clusters of Ru(II) with nitrile ligand [3,3-(Ph2P(CH2)4PPh2)-3-NCR-closo-3,1,2-RuC2B9H11] (R = –Ph, –CH=CH2). A similar ortho-cycloboronated clusters [3-NCR-3,3,8-{(Ph2P(CH2)4PPh-μ-(C6H4-ortho)}closo-3,1,2-RuC2B9H10] were obtained from the corresponding chlorine-containing ruthenacarboranes. The novel clusters were isolated as yellow crystal solids and characterized by NMR, mass-spectroscopy and X-ray analysis. The reaction of the obtained clusters with HCl showed the chemical reversibility of dehydrohalogenation. The performed electrochemical investigation testified the ability of novel complexes to undergo reversible oxidation to Ru(III) species.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The progress in chemistry of carboranes and its complexes with transition metals observed in recent years is governed not only by unique bonding type and possibility of formation of novel types of cluster structures [1,2,3,4,5,6,7,8,9] but also by the intensive growth of the area of its possible applications in medicine [10,11,12,13,14,15,16] organic electronics [17,18,19] catalysis [1, 20,21,22,23,24,25,26] and so on. Due to the presence of highly delocalized tri-dimensional electronic structure carboranes and its derivatives may be considered as capacitors of electronic density. It allows taking them into account as convenient building blocks in the catalysts design. Carborane-based catalysts were successfully applied in methathesis reactions [1], hydrogenation [22], radical addition [23] and Atom Transfer Radical Polymerization processes [24]. Active participation of a metal complex in catalytic cycle requires the presence of a vacant coordination site necessary for substrate addition. It is well known that such unsaturated species are unstable in a free state and are usually formed in the reaction media through the dissociation of weak-bonded two-electron ligands such as nitriles, phosphines or amines.
This paper describes the synthesis of novel ruthenacarborane clusters with nitrile ligands suitable for application as catalysts of ATRP processes.
Results and Discussion
The reaction of 3,3-(Ph2P(CH2)4PPh2)-3-H-3-Cl-closo-3,1,2-RuC2B9H11 (1) with isopropylamine and benzonitrile in CH2Cl2 solution at 40 °C afforded the compound [3,3-(Ph2P(CH2)4PPh2)-3-NCPh-closo-3,1,2-RuC2B9H11] (2) isolated in 68% yield after column separation and recrystallization. The reaction probably proceeds through the reductive elimination of HCl and further stabilization of the formed 16-electron species by benzonitrile ligand. In spite of the presence of the excess of the amine in the reaction media no amine containing complex is formed in this reaction in spite of a series of amine-containing Ru (II) complexes are known [27,28,29]. It should be mentioned that no stable product can be isolated from the reaction media in the case of conduction of the process in the absence of benzonitrile or amine. Thus, we may conclude that in this reaction amine probably acts as a base while benzonitrile as an auxiliary stabilizing ligand.

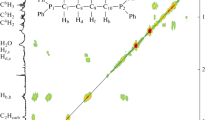
The 31P NMR spectrum of complex 2 contains only one signal at 38 ppm indicating the equivalence of two phosphorous atoms. The observed chemical shift is close to that is similar acetionitrile containing complex obtained earlier [30] corroborating the proposed structure for complex. The 1H{31P} spectrum is also similar for ones recorded for acetonitrile containing analogue of 2. It contains resonances from the protons of alkyl groups, as well as of CH-cage and phenyl rings protons from benzonitrile and diphosphine. The assignment of the proton resonances from methylene bridge was done using the 2D [1H–1H] COSY and [13C–1H] HSQC correlation spectra depicted on Fig. 1.

The fragments of 2D [1H–1H] COSY (left) and [13C–1H] HSQC (right) NMR spectra for complex 2. Solvent—CD2Cl2
The signals of four protons from two central methylene units appear as a broad multiplet at δ 1.74 ppm. At the same time the geminal hydrogen atoms at terminal methylene units connected with phosphorous atoms are magnetically nonequivalent giving resonances at 2.98 and 2.46 ppm respectively. The signal from from two hydrogen atoms bonded to carbons in carborane cage appears as a broad singlet at 2.53 ppm. Signals from phenyl rings of benzonitrile and diphosphine moieties appear as a complicated multiplet in the range of 7.28–7.70 ppm.
MALDI TOF mass-spectrum of cluster 2 recorded in positive mode contains a signal at 660 Da with the characteristic envelope-type pattern corresponding to [2-PhCN]+. The absence of the strong signal of molecular ion in the mass-spectra indicates the low energy of Ru–N bond in the cluster. At the same time the presence of coordinated nitrile ligand in cluster 2 is confirmed by the C-N strethcing band vibration at 2212 cm−1 in the IR spectrum.
The crystals suitable for X-ray study were obtained by slow crystallization of 2 from concentrated benzonitrile solution on cooling. The performed X-ray analysis confirmed the structure, proposed basing on the spectral investigation of 2 (Fig. 2).

The molecular structure of cluster 2
The obtained cluster is a typical example of closo-ruthenacarboranes. The metal atom is coordinated on the open C2B3-facet of the carborane ligand and coordinated with two psophorous atom of the diphosphine moiety and the nitrogen atom of the benzonitrile ligand. In spite of the equivalence of phosphorous atoms in solution observing in the 31P NMR spectrum the Ru–P bonds in solid state are slight differ: (2.316 and 2.330 Å). Coordination of benzonitrile ligand into trans-position relative to the B(2) boron atom is typical for metallacarborane clusters [30, 31]. It should be mentioned that the observed Ru–N distance is slight shorter (2.045 Å) than in the corresponding acetonitrile derivative [3-NCMe-3,3,8-{Ph2P(CH2)4PPh2}-closo-3,1,2-RuC2B9H11] (2.088 Å) [30]. This fact may be attributed to the less steric hindrances in the benzonitrile complex due to the planarity of the phenyl ring and the donation of electron density from the phenyl ring through the conjugated π-system.
To expand the scope of the possible nitrile containing clusters we examined the reaction of complex 1 with acrylonitrile in the similar conditions. A particular interest to this reaction was governed by the presence of double bond in acrylonitrile ligand and possible η2-coordination of this ligand to ruthenium atom. Such η2-ruthenium complexes were earlier described in literature [32]. Another point of interest to these complexes is determined by its behavior in polymerization processes. The information about the stability of the nitrile complex is necessary for evaluation of the possibility of its application in polymerization of unsaturated nitriles via ATRP mechanism [24, 33].
Cluster 3 was isolated as a yellow solid in a quantitative yield from the reaction of 1 with isopropylamine and acrylonitrile in CH2Cl2 solution in the conditions similar to that for obtaining of 2:

Cluster 3 is characterized by good solubility in ethyl acetate, acetone or CH2Cl2 but these solutions are unstable on air. Solution of 3 in acrylonitrile is stable on air due to the excess of stabilizing ligand. Complex 3 is insoluble in methanol, alkanes and diethyl ether.
The characterization of complex 3 by NMR and IR spectroscopy allowed us to propose its structure and the type of acrylonitrile coordination. The 31P NMR spectrum (Fig. S2) contains one signal at − 41 ppm indicating the preservation of diphosphine ligand in the cluster and the chemical equivalence of two phosphorous atoms. The 1H NMR spectrum of 3 contains the signals of the aromatic protons of phenyl rings at 7.87–7.39 ppm, the signals of methylene units and C–H groups of carborane cage at 2.52–2.30 and 1.58 ppm respectively. The assignment of the proton signals was done with the use of the correlation 1H–1H COSY and 1H–13C HSQC spectrometry. The corresponding spectra are provided as electronic supplementary materials and are very similar to ones depicted on Fig. 1. The presence of acrylonitrile molecule in the cluster was confirmed by the signals at 6.01, 6.04 and 5.80 ppm (Fig. 3). The character of the spectra and the observed chemical shifts values allow to suppose that acrylonitrile molecule is probably coordinated to the ruthenium atom via nitrogen atom forming nitrile—type complex. The similar consideration may be done in accordance with the value of 31P chemical shift similar to that for the complex 2.

The fragments of 1H NMR spectra of clusters 3 (top) and 6 (bottom)
The performed X-ray study has confirmed this proposition. As it may be observed on Fig. 4, cluster 3 contains acrylonitrile molecule coordinated to ruthenium center via nitrogen atom. According to the data summarized in Table 1 we may conclude that the obtained ruthenacarboranes 2 and 3 have very similar closo-structures. The interchange of benzonitrile ligand via acrylonitrile one has almost no influence on the general parameters of the cluster. It should be noted that the Ru–N bond lengths in clusters 3 and 2 are almost the same (2.044 Å) and are typical for Ru(II) acrylonitrile complexes. The similar Ru–N distance values (2.02–2.05 Å) are observed in [Ru(η5-C5H5)(acrylonitrile)(BIPHOP-F)]+ cations [34]. At the same time the corresponding Ru–N bond in Ru(III) complex with pyridinecarboxylic acid cis-[Ru(NO)(NCCH=CH2)(pyc)2]+ is significantly shorter (1.768 Å) [35].

The molecular structure of cluster 3
The interaction of cluster 4 bearing ortho-cycloboronated fragment with isopropylamine and nitrile in the above mentioned conditions allowed us to isolate corresponding complexes 5 and 6 as yellow solids in 56 and 55% yields respectively:

The structures of the obtained complexes were ascertained on the basis of NMR study. The presence of ortho-cycloboronated moiety in clusters 5 and 6 makes the phosphorous atoms in the structure of the clusters non-equivalent, and results in the presence of two signals in NMR spectra at ca 35 and 61 ppm. It should be noted that the observed phosphorous chemical shifts for cluster 5 are similar to that in complex 6 and the relative acetonitirile derivative obtained earlier [30]. It allows us to suppose that acrylonitrile ligand is coordinated on ruthenium center via nitrogen atom but not with double bond. The presence of ortho-cycloboronated fragment in the complex causes the loss of symmetry in diphosphine ligand. Due to the splitting on geminal and vicinal atoms signals of these protons appear in 1H{31P} NMR as complicated multiplets in the range from 1.5 to 3.0 ppm. Hydrogen atoms connected with carborane carbon atoms are also non-equivalent and give two broad singlets in the spectra at 1.4 and 3.1 ppm (see figures in ESM). The presence of acrylonitrile fragment in cluster 6 is unambiguously confirmed by the signals in the range of 5.73–6.12 ppm. Figure 3 represents the comparison of the spectra obtained for complexes 3 and 6. It can be concluded that the change of the ligand structure results in the increase of the values of spin–spin coupling constants Ja–c and Jb–c in complex 6. Phenyl groups of diphosphine moieties of complexes 5 and 6 give resonances in the range from 7.1 to 7.8 ppm. The presence of covalent bonding between boron framework and one phenyl ring results in complicating spectra of 5 relative to 2. Spectrum of 5 contains additional signals from phenyl ring of benzonitrile ligand.
The formation of clusters 5 and 6 containing ruthenium atom in +2 oxidation state should be considered as a reduction of initial Ru(III) complex 4. We suppose that in this process amine molecule reduces initial complex via one-electron transfer giving corresponding anion. The abstraction of chlorine atom from the cluster anions formed in this reaction generates 16-electron Ru(II) species which are further stabilized by nitrile addition [30].
It was established that novel clusters 2 and 3 may be easily converted in the initial compound 1 via reaction with hydrogen chloride in CH2Cl2 media. The addition of 10-fold excess of HCl to the cluster solution results in the fast color change from yellow to orange. The formation of cluster 1 was unambiguously confirmed by the means of HPLC and MALDI mass spectrometry. The reaction proceeds through the oxidative addition mechanism and may be considered as the reverse process towards the elimination of HCl under the action of amine described earlier. The similar reactions of HCl addition were earlier observed for Ru(0) complexes [36, 37] while the present work shows the possibility of such addition to Ru(II) complex with the formation of the corresponding Ru(IV) product. Noteworthy that this reaction may be conducted even using concentrated aqueous solution of HCl. This fact indicates high tolerance of 1 towards water.

The performed interaction of clusters 5 and 6 with HCl in the similar conditions resulted in the formation of initial complex 4 instead of the expected diamagnetic complex of Ru (IV) containing a hydride atom at the ruthenium center. Such difference in the structures of the complexes obtained from ortho-phenylenecycloboronated and non-cycloboronated clusters may be caused by the steric hindrances determined by the presence of the Ru–P–C–C–B metallocycle which does not allow hydrogen atom to bind to the ruthenium center.

Electrochemistry is a convenient tool to evaluate the ability of complex to participate in catalytic processes incorporating redox stages [38, 39]. The performed cyclic voltammetry studies of novel clusters 2, 3, 5 and 6 showed its ability to undergo reversible oxidation corresponding to Ru(II)–Ru(III) transition. The registered cyclic voltammetry curves are depicted on Fig. 5. It should be noted that the obtained values of E1/2(RuII/RuIII) for complexes 2 and 3 provided in Table 2 are almost similar and are equal to the values observed for relative acetonitrile complex [30]. Clusters 5 and 6 bearing ortho-cycloboronated fragment in its structure are characterized by lower RuII/RuIII redox potentials relative to its non-cycloboronated analogs 2 and 3 in accordance with the dependences observed earlier [30]. Further oxidation of the obtained clusters 2, 5 and 6 to Ru(IV) state proceeds fully irreversible at potential values summarized in Table 2. At the same time we have not observed a strong signal corresponding to irreversible oxidation of the cluster 3 to the Ru(IV) species in the investigated range of the potentials. Instead of that a smooth increase of the current exceeding the drift of the baseline was observed.

The cyclic voltammograms of the obtained clusters in 1,2-dichloroethane at 25 °C. [Ru] = 0.003 M; [n-Bu4NPF6] = 0.2 M (supporting electrolyte). Scan rate υ = 100 mV/s
Thus we may conclude that the reaction of chlorine-containing closo-ruthenacarboranes with aliphatic amines and nitriles may be considered as the method to the preparation of corresponding Ru(II) clusters. The observed chemical and electrochemical reversibility of the RuII–RuIII transition allows considering such clusters as potential catalysts for various applications.
Experimental
All reactions were carried out under an atmosphere of dry, oxygen-free argon using Schlenk line techniques. Solvents were distilled from appropriate drying agents under argon prior to use. Chromatography columns (typically ca. 15 cm in length and ca. 2 cm in diameter) were packed with silica gel (Merck, 230–400 mesh). Top of the column was flushed with a flow of argon during elution process. The 1H, 31P{1H}NMR spectra were recorded on Agilent DD2 NMR 400NB spectrometer. NMR tubes were degassed before analysis, deoxygenated and argon flushed deuterated solvents were used for spectra registration. The purity of complexes was analyzed by HPLC (Knauer Smartline system equipped with diode-array UV Detector 2600) on a silica gel packed column (12.5 mm in length and 4.6 in diameter) with use of a CH2Cl2/n-hexane mixture (1:1) as eluent. Redox properties of complexes were studied in 1,2-dichloroethane (DCE) with 0.2 M n-Bu4NPF6 as the supporting electrolyte. These measurements were carried out under argon atmosphere in a conventional three-electrode cell with a Pt disk (0.8 mm in diameter) as the working electrode, a Pt wire as the counter electrode and Ag/0.01 M AgNO3 plus 0.2 M n-Bu4NPF6 in acetonitrile as the reference electrode [30]. Electrochemical measurements were performed with an IPC Pro-M potentiostat with the digital recording of the results. MALDI-TOF mass-spectra of the compounds were obtained on a Bruker Microflex LT instrument using DCTB as a matrix and a ground steel target plate. The IR spectra of complexes were recorded on Infralum FT IR spectrometer in solid KBr matrix.
Complex 1 was obtained by the procedure described earlier [24].
Preparation of [3,3-(Ph 2 P(CH 2 ) 4 PPh 2 )-3-NCC 6 H 5 - closo -3,1,2-RuC 2 B 9 H 11 ] (2)
20 mg (0.028 mmol) of complex 1 were placed into 25-ml Schlenk tube followed by 2 ml of methylene chloride, 2 ml of benzonitrile and 71 μl (0.84 mmol) of isopropylamine. The tube was closed by a stopcock, degassed via three freeze–pump–thaw cycles and filled with argon. The reaction was conducted on water bath at 40 °C for two hours until the bright yellow color of the solution. After the reaction the reaction mixture was placed on the column filled with silica gel. A bright yellow band was eluted by 1:1 n-hexane: ethyl acetate mixture. The evaporation of the solvent under reduced pressure gave 14.8 mg (68%) of pure complex 2. 1H NMR (CD2Cl2, 25 °C), δ(ppm): 1.75 (m, 4H, PCH2CH2CH2CH2P), 2.44 (m, 1H PCH2CH2CH2CHHP), 2.47 (m, 1H PCHHCH2CH2CH2P), 2.53 (s, br, CHcarb), 2.9–3.0 (m, br 2H, PCHHCH2CH2CHHP), 7.3–7.7 (m, 25H, Ph-rings); 31P{1H} NMR (CD2Cl2, 25 °C), δ(ppm) 41.31 (s). IR (KBr): 2208 (ν(C≡N)), 2532 (ν(B–H)).
Preparation of [3,3-(Ph 2 P(CH 2 ) 4 PPh 2 )-3-NCCH=CH 2 - closo -3,1,2-RuC 2 B 9 H 11 ] (3)
19 mg (0.027 mmol) of complex 1 were placed into 25-ml Schlenk tube followed by 2 ml of methylene chloride, 2 ml of acrylonitrile and 115 μl (1.35 mmol) of isopropylamine. According the the procedure described upper for complex 2 18.86 mg (96.2%) of pure complex 3 were achieved. 1H NMR (CD2Cl2, 25 °C), δ(ppm): 1.72–1.74 (m, br, 4H, PCH2CH2CH2CH2P), 2.4–2.5 (m, br, 4H 2 × CHcarb + PCHHCH2CH2CHHP), 2.92 (m, 2H, PCHHCH2CH2CHHP), 5.79 (d, 1H NC–CH=CH2), 6.01 (d, 1H NC–CH=CH2), 6.04 (d, 1H NC–CH=CH2), 7.30–7.5 (m, 20H, Ph-rings); 31P{1H} NMR (CD2Cl2, 25 °C), δ(ppm) 41.12 (s); IR (KBr): 2213 (ν(C≡N)), 2533 (ν(B–H)).
Preparation of [3-Cl-3,3,8-{Ph 2 P(CH 2 ) 4 PPh-μ-(C 6 H 4 - o )}- closo -3,1,2-RuC 2 B 9 H 10 ] (4)
The earlier described method of preparation of 4 from 5,6,10-{Cl(Ph3P)2Ru}-[5,6,10-(μ-H)3-10-H-7,8-H-exo-nido-7,8-C2B9H8] (7) into three stages [24, 40] was modified into one-pot reaction allowed to increase total yield of 4. 136 mg (0.17 mmol) of 7 and 80 mg (0.188 mmol) of 1,4-bis(diphenyphosphino)butane were placed in a 100 ml round-bottom Schlenk flask. The flask was degassed and filled with argon. A 25 ml of toluene was distilled into the flask in argon atmosphere. The mixture was heated at 95 °C for 1 h on oil bath. After that the temperature was increased up to 105 °C and the reaction was conducted for 4 h. After that 1 ml of carbon tetrachloride was added and the reaction was conducted for additional 40 min. After cooling the solvent was evaporated in vacuum up to 3 ml and the residue was placed on a silica gel packed column. A first dark red band eluted with benzene/n-henane = 2/1 mixture yielded 63 mg (53%) of complex 4 after crystallization as a dark-red crystals. Elution of the second dark red band by benzene allowed to isolate 8 mg (6%) of 3-Cl-3,3,7,8-{Ph2P(CH2)4PPh-μ-(C6H4-o)2}-closo-3,1,2-RuC2B9H9 [24].
Preparation of [3-C 6 H 5 CN-3,3,8-{Ph 2 P(CH 2 ) 4 PPh-μ-(C 6 H 4 - o )}- closo -3,1,2-RuC 2 B 9 H 10 ] (5)
Complex 5 was prepared by the method described for 2 from 21.4 mg (0.03 mmol) of complex 4, 2 ml of methylene chloride, 2 ml of benzonitrile and 128 μl (1.50 mmol) of isopropylamine. Product yield was 12.8 mg (54.7%).1H NMR (CD2Cl2, 25 °C), δ(ppm): 1.48 (s, br, 1H, CHcarb), 1.78 (m, 4H, o-C6H4(Ph)P(CH2)CH2(CH2)2PPh2 + o-C6H4(Ph)P(CH2)2CH2CH2PPh2), 2.30 (m, 1H, o-C6H4(Ph)P(CH2)3CHHPPh2), 2.96 (m, 3H, o-C6H4(Ph)P(CH2)3CHHPPh2 + o-C6H4(Ph)PCH2(CH2)3PPh2), 3.21 (s, br, 1H, CHcarb), 7.0–7.9 (set of m, 14 H, aromatic rings); 31P{1H} NMR (CD2Cl2, 25 °C), δ(ppm) 35.5 (d, 1P, J = 39.61), 61.5 (br. d, 1P, J = 39.61); 11B {1H} NMR (CD3CN, 25 °C), δ (ppm): − 29.9 (1B), − 20.0 (1B), − 16.0 to − 14.8(br, 3B), − 11.8(1B), − 5.34 (2B), 12.7 (1B); IR (KBr): 2214 (ν(C≡N)), 2546 (ν(B–H)).
Preparation of [3-CH 2 =CHCN-3,3,8-{Ph 2 P(CH 2 ) 4 PPh-μ-(C 6 H 4 - o )}- closo -3,1,2-RuC 2 B 9 H 10 ] (6)
Complex 6 was prepared by the method described for 2 from 19.6 mg (0.028 mmol) of complex 1, 2 ml of methylene chloride, 2 ml of acrylonitrile and 119 μl (1.40 mmol) of isopropylamine. Product yield was 11 mg (55.8%). 1H NMR (CD2Cl2, 25 °C), δ(ppm): 1.42 (s, br, 1H, CHcarb), 1.6–1.8 (m, 4H, o-C6H4(Ph)P(CH2)CH2(CH2)2PPh2 + o-C6H4(Ph)P(CH2)2CH2CH2PPh2), 2.28 (m, 1H, o-C6H4(Ph)P(CH2)3CHHPPh2), 2.91 (m, 3H, o-C6H4(Ph)P(CH2)3CHHPPh2 + o-C6H4(Ph)PCH2(CH2)3PPh2), 3.12 (s, br, 1H, CHcarb), 5.76 (4d, 1H NC–CH=CH2), 6.03 (d, 1H NC–CH=CH2), 6.1 (d, 1H NC–CH=CH2), 7.1–7.7 (set of m, 19 H, aromatic rings); 31P{1H} NMR (CD2Cl2, 25 °C), δ(ppm) 35.6 (d, 1P, J = 39.6), 61.4 (br. d, 1P, J = 39.6). 11B {1H} NMR (CD3CN, 25 °C), δ (ppm): − 29.9 (1B), − 20.2 (1B), − 16.0 (1B), − 14.5 (br, 2B), − 12.0 (1B), − 5.4 (2B), 12.7 (1B). IR (KBr): 2218 (ν(C≡N)), 2504, 2559 (ν(B–H)).
Reaction of Clusters 2, 3, 5 and 6 with HCl
11.6 mg (0.016 mmol) of complex 6 were placed in a Schlenk tube, degassed and filled with argon. After that 2 ml of degassed CH2Cl2 were added. A 2 μl of concentrated aqueous solution of hydrochloric acid were added after the full dissolution of the complex. A yellow color of the reaction mixture turned to orange in a couple of minutes. Reaction was stirred for 30 min, after that the solvent was reduced to 1 ml and the mixture was placed on a silica gel packed column. A red–orange band was eluted with CH2Cl2/n-hexane 1:1 mixture and yielded 10.3 mg (94%) of complex 4 after solvent evaporation. A similar reaction conducted with complex 5 resulted in formation of 4 in 74% yield, while the interaction HCl with 2 and 3 in the same conditions allowed isolating 1 in 55% and 65% yields respectively.
Crystallographic Structure Determinations
The crystallographic parameters and the X-ray-data-collection and structure-refinement statistics are given in Table 3. The initial structural fragment of 2 and 3 structures were established by direct methods. The positions of the missing non-hydrogen atoms were found in difference-electron-density maps and refined with anisotropic displacement parameters [41, 42]. The some part of H atoms were placed in calculated positions and refined in the ‘‘riding-odel” (Uiso(H) = 1.2 Ueq(carbon) Å2 for aromatic hydrogen and 1.5 Ueq(carbon) Å2 for alkyl hydrogen), and the another part was located from Fourier synthesis and refined isotropically. Hydrogen atoms in the solvate molecule of 3 were not determined. The solvate molecule of 2 was removed with PLATON/SQUEEZE [43].
References
Q. Xie, T. Wang, S. Wu, W. Guo, and H. Zhang (2018). J. Organomet. Chem. 865, 95.
A. P. Y. Chan, G. M. Rosair, and A. J. Welch (2018). Inorg. Chem. 57, 8002.
T. D. McGrath, F. G. Stone, and K. Sukcharoenphon (2005). J. Clust. Sci. 16, 201.
M. Bakarjiev, A. Ruzicka, Z. Ruzickova, O. L. Tok, J. Holub, D. Hnyk, J. Fanfrlik, and B. Stibr (2019). Inorg. Chem. 58, 2865.
A. S. Batsanov, R. C. B. Copley, M. G. Davidson, M. A. Fox, T. G. Hibbert, J. A. K. Howard, and K. Wade (2006). J. Clust. Sci. 17, 119.
D. K. Roy, S. Ghosh, and J. F. Halet (2014). J. Clust. Sci. 25, 225.
B. Pathak, S. Pandian, N. Hosmane, and E. D. Jemmis (2006). JACS 128, 10915.
N. S. Hosmane, H. Zhang, J. A. Maguire, Y. Wang, C. J. Thomas, and T. G. Gray (1996). Angew. Chem. Int. Ed. 35, 1000.
M. F. Hawthorne and A. Pushechnikov (2012). Pure Appl. Chem. 84, 2279.
D. Rozycka, Z. J. Lesnikowski, and A. B. Olejniczak (2019). J. Organomet. Chem. 881, 19.
A. A. Druzina, V. I. Bregadze, A. F. Mironov, and A. A. Semioshkin (2016). Russ. Chem. Rev. 85, 1129.
I. Fuentes, T. Garcia-Mendiola, S. Sato, M. Pita, H. Nakamura, E. Lorenzo, F. Teixidor, F. Marques, and C. Vinas (2018). Chem. Eur. J. 24, 17239.
M. Couto, I. Mastandrea, M. Cabrera, P. Cabral, F. Teixidor, H. Cerecetto, and C. Vinas (2017). Chem. Eur. J. 23, 9233.
M. F. Hawthorne and A. Maderna (1999). Chem. Rev. 99, 3421.
Y. Zhu and N. S. Hosmane (2013). Fut. Med. Chem. 5, 705.
R. Satapathy, B. P. Dash, J. A. Maguire, and N. S. Hosmane (2010). Collect. Czech. Chem. Commun. 75, 995.
O. N. Kazheva, D. M. Chudak, G. V. Shilov, E. A. Komissarova, I. D. Kosenko, A. V. Kravchenko, I. A. Shilova, E. V. Shklyaeva, G. G. Abashev, I. B. Sivaev, V. A. Starodub, L. I. Buravov, V. I. Bregadze, and O. A. Dyachenko (2018). J. Organomet. Chem. 867, 375.
I. B. Sivaev (2018). Russ. Chem. Bull. (Int. Ed.) 67, 1117.
B. P. Dash, R. Satapathy, J. A. Maguireb, and N. S. Hosmane (2011). New J. Chem. 35, 1955.
S. T. Guo, P. F. Cui, Y. Gao, and G. X. Jin (2018). Dalton Trans. 47, 13641.
N. Ma, C. Wei, S. Li, and G. Zhang (2018). Dalton Trans. 47, 14421.
L. S. Alekseev, S. E. Lyubimov, F. M. Dolgushin, V. V. Novikov, V. A. Davankov, and I. T. Chizhevsky (2011). Organometallics 30, 1942.
A. Richel, S. Delfosse, C. Cremasco, L. Delaude, A. Demonceau, and A. F. Noels (2003). Tetrahedron Lett. 44, 6011.
I. D. Grishin, D. I. D’yachihin, A. V. Piskunov, F. M. Dolgushin, A. F. Smol’yakov, M. M. Il’in, V. A. Davankov, I. T. Chizhevsky, and D. F. Grishin (2011). Inorg. Chem. 50, 7574.
Y. Zhu and N. S. Hosmane (2013). J. Organomet. Chem. 747, 25.
Y. Zhu, Z. Bai, W. C. Phuan, F. A. Ghaida, N. S. Hosmane, and J. Ding (2018). J. Organomet. Chem. 865, 58.
E. A. Nyawade, H. B. Friedrich, and B. Omondi (2014). Inorg. Chim. Acta 415, 44.
E. A. Nyawade, H. B. Friedrich, and B. Omondi (2016). Inorg. Chim. Acta 441, 9.
F. L. Joslin, M. P. Johnson, J. T. Mague, and D. M. Roundhill (1991). Organometallics 10, 2781.
A. M. Penkal, D. I. D’yachihin, N. V. Somov, E. S. Shchegravina, and I. D. Grishin (2018). J. Organomet. Chem. 872, 63.
A. J. Welch (2017). Crystals 7, 234.
J. P. Qu, H. Matsuzaka, Y. Ishii, and M. Hidai (1996). Chem. Lett. 25, 767.
I. D. Grishin, E. S. Turmina, D. I. D’yachihin, I. T. Chizhevsky, and D. F. Grishin (2014). Polym. Sci. Ser. B 56, 1.
P. G. A. Kumar, P. S. Pregosin, M. Vallet, G. Bernardinelli, R. F. Jazzar, F. Viton, and E. P. Kündig (2004). Organometallics 23, 5410.
H. Nagao, T. Misawa-Suzuki, N. Tomioka, H. Ohno, and M. Rikukawa (2018). Chem. Asian J. 13, 3014.
D. Morales-Morales, R. Redon, and R. E. Cramer (2001). Inorg. Chim. Acta 321, 181.
J. K. Kim, D. H. Berry, and H. Yoo (2011). J Organomet. Chem. 696, 1895.
A. Richel, A. Demonceau, and A. F. Noels (2006). Tetrahedron Lett. 47, 2077.
W. A. Braunecker, W. C. Brown, B. C. Morelli, W. Tang, R. Poli, and K. Matyjaszewski (2007). Macromolecules 40, 8576.
D. N. Cheredilin, F. M. Dolgushin, I. D. Grishin, E. V. Kolyakina, A. S. Nikiforov, S. P. Solodovnikov, M. M. Il’in, V. A. Davankov, I. T. Chizhevsky, and D. F. Grishin (2006). Russ. Chem. Bull. Int. Ed. 55, 1163.
L. J. Farrugia (1999). J. Appl. Crystallogr. 32, 837.
G. M. Sheldrick (2008). Acta Crystallogr. Sect. A Found. Crystallogr. 64, 112.
P. V. D. Sluis and A. L. Spek (1990). Acta Crystallogr. Sect. A Found. Crystallogr. 46, 194.
Acknowledgements
The work was supported by Russian Science Foundation (Proj. No. 18-73-10092). Ivan D. Grishin thanks the Ministry of Science and higher education of Russian Federation (Task No. 4.5630.2017/6.7).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
10876_2019_1605_MOESM1_ESM.docx
Supplementary material 1: NMR spectra of complexes 2, 3, 5, 6 are provided as supplementary materials in a separate file. Atomic coordinates, bond lengths and angles, and thermal parameters for 2 and 3 have been deposited with the Cambridge Crystallographic Data Centre, CCDC Nos. 1822531 (2), 1903390 (3). (DOCX 911 kb)
Rights and permissions
About this article

Cite this article
Penkal’, A.M., Somov, N.V., Shchegravina, E.S. et al. Ruthenium Diphosphine Closo-C2B9-Carborane Clusters with Nitrile Ligands: Synthesis and Structure Determination. J Clust Sci 30, 1317–1325 (2019). https://doi.org/10.1007/s10876-019-01605-9
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10876-019-01605-9