
Abstract
New coordination polymers with lanthanide ions [Ln(Fur)3(H2O)x]n⋅Solv (Ln = Gd (I), Sm (II); Fur = 2-furoic acid anion; x = 2 (I), 3 (II); Solv = MeCN (I)) are synthesized. The structures of the synthesized compounds are determined by X-ray diffraction (CIF files CCDC nos. 2130014 (I) and 2130015 (II)). The coordination environment of the complexing agent (LnO8) corresponds to a distorted square antiprism (I) or a distorted triangular dodecahedron (II). Complexes I and II represent polymeric chains in which the Fur– anions perform the bridging function. The crystal lattice is stabilized by intra- and intermolecular hydrogen bonds between the coordinated water molecules, acid anions, and solvate molecules. The study of the thermal behavior of compound I by simultaneous thermal analysis in an argon atmosphere shows a low stability of the complex: its decomposition starts at 69°С, and the organic moiety destructs gradually without pronounced thermal effects. According to the data of X-ray photoelectron spectroscopy, gadolinium(III) oxide is the final product of thermal decomposition.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Lanthanide carboxylates attract attention of researchers due to unique coordination possibilities of Ln(III) ions to form structures of various dimensionalities and nuclearities. In addition to the magnetic and optical characteristics inherent in the complexes with lanthanide cations [1–4], interest in searching for probes based on such complexes (e.g., luminescent temperature sensors in early diagnostics of inflammatory processes, including oncological diseases) has increased in the recent years [5–7]. This phenomenon is based on significant temperature oscillations that are observed during pathological processes in living tissues and can be detected by diverse thermal imagers [8–10]. However, along with a serious demand of new detailed studies of physical and other characteristics of the known lanthanide complexes, targeted assembling of molecules with lanthanide ions of certain composition and structure remains to be one of the main problems, since it is known that the molecular (or crystal) structure often predetermines the properties and objective characteristics of similar objects.
In this study, we use 2-furoic acid as the source of carboxylate anion, because the Tb(III), Dy(III), La(III), and Eu(III) complexes of this acid are capable of manifesting interesting magnetic and luminescence properties [11–14]. However, unlike the synthetic approaches proposed in [13, 14], where Ln nitrates or chlorides serve as the starting reagents (the exchange of anions and their incomplete replacement in the case of nitrates becomes much more complicated), in the present work we use organic salts (acetate/pivalate) or carbonate facilitating ion exchange (due to their close acidities). In addition, the d-metal complexes with pyromucic acid involving the N‑donor ligands predetermine antimicobacterial activity in vitro toward the model nonpathogenic Mycolicibacterium smegmatis strain [15–20] and possess anticancer properties against SCOV-3 (ovary adenocarcinoma) [21].
The purpose of the present work is the development of synthetic procedures for the gadolinium(III) and samarium(III) complexes with furoate anions and the study of their formation conditions, structures, and thermal properties.
EXPERIMENTAL
The complexes were synthesized in air using the following commercially available reagents: 2-furoic acid (98%, Acros), gadolinium acetate hydrate (95%, Acros), samarium carbonate tetrahydrate (reagent grade, ZAO Mosreaktiv, Russia), and acetonitrile (special purity grade, Khimmed, Russia). Gadolinium pivalate [Gd2(Рiv)6(HРiv)7] was synthesized using a known procedure [22].
Elemental analysis was carried out on a Carlo Erba EA 1108 С,H,N analyzer. IR spectra were recorded on a Perkin-Elmer Spectrum 65 FT-IR spectrometer in the attenuated total reflection (ATR) mode in a frequency range of 400–4000 cm–1.
The thermal behavior of compound I was studied by simultaneous thermal analysis (STA) in an argon atmosphere with the simultaneous detection of thermogravimetry (TG) and differential scanning calorimetry (DSC) curves. The study was carried out on an STA 449 F1 Jupiter instrument (NETZSCH) in aluminum crucibles under caps with a hole providing a vapor pressure of 1 atm during the thermal decomposition of the samples. The heating rate was 10°С/min to 500°С. The sample weight was 4.96 mg. The accuracy of temperature measurement was ±0.7°С, and that of the weight change was ±1 × 10−2 mg. A correction file and temperature and sensitivity calibrations for specified temperature program and heating rate were used when recording TG and DSC curves. After thermal analysis, the qualitatively determined chemical composition and micromorphology of the residual substance were examined on a CarlZeissNVision 40 scanning electron microscope equipped with an Oxford X-Max X-ray spectral detector at an accelerating voltage of 1 and 20 kV, respectively. The magnification was ×250.
The X-ray photoelectron spectra (XPS) of the residual substance of compound I after thermolysis were detected on a Kratos Axis Ultra spectrometer using monochromatic Al Kα radiation with the power not higher than 180 W. A low-energy electron gun was used for charge compensation on the sample surface. The expansion of the spectra to components was performed using the Kratos Analytical program. Each spectral line was approximated by the Gaussian profile or their sum. The measurements were carried out at least two times under a pressure of ~10–9 Torr. The spectra were recorded at both the temperature of liquid nitrogen and room temperature.
Synthesis of [Gd(Fur)3(H2O)2]n⋅MeCN (I). Method 1. Compound HFur (0.18 g, 1.5 mmol) was added to a suspension of Gd(OAc)2·3H2O (0.2 g, 0.5 mmol) in acetonitrile (20 mL), and the mixture was stirred at 80°С for 90 min. The resulting solution was stored at room temperature for 24 h. The formed colorless crystals were separated from the mother liquor by decan-ting and dried in air. The yield was 0.22 g (78%).
Method 2. Compound Hfur (0.18 g, 1.5 mmol) was added to a suspension of [Gd2(Piv)6(HPiv)7] (0.4 g, 0.25 mmol) in acetonitrile (20 mL), and the mixture was stirred at room temperature for 30 min. The resulting solution was stored at room temperature for 24 h. The formed colorless crystals were separated from the mother liquor by decanting and dried in air. The yield was 0.2 g (70%).
For C17H16NO11Gd (I) | |||
Anal. calcd., % | С, 35.98 | H, 2.84 | N, 2.47 |
Found, % | C, 36.02 | H, 2.85 | N, 2.53 |
IR (ν, cm–1): 3142 w, 2990 w, 1623 w, 1582 m, 1540 s, 1472 s, 1419 s, 1373 s, 1233 m, 1197 s, 1143 w, 1078 w, 934 w, 884 s, 804 w, 782 s, 754 s, 638 w, 614 m, 593 m, 517 w, 454 s.
Synthesis of [Sm(Fur)3(H2O)3]n (II). Distilled water (30 mL) was poured to weighed samples of Sm2(CO3)3·4H2O (0.18 g, 0.33 mmol) and HFur (0.22 g, 2 mmol), and the reaction mixture was stirred at 80°C for 2 h. The solution was filtered from a precipitate and left to evaporate at room temperature. Colorless crystals formed in 3 days were separated from the mother liquor by decanting and dried in air. The yield was 0.27 g (76%).
For C15H15O12Sm (II) | ||
Anal. calcd., % | С, 33.51 | H, 2.81 |
Found, % | C, 32.17 | H, 2.84 |
IR (ν, cm–1): 3550 m, 3417 w, 3119 w, 2942 w, 2822 w, 1582 s, 1543 vs, 1471 vs, 1412 vs, 1364 vs, 1232 m, 1195 s, 1138 m, 1077 m, 1010 s, 934 m, 884 m, 849 w, 756 s, 656 m, 607 m, 587 m, 542 s, 444 s.
X-ray diffraction (XRD) of compounds I and II was carried out at 120 K on a Bruker ApexII DUO diffractometer (MoKα radiation, λ = 0.71073 Å, graphite monochromator, CCD detector). The structures were solved using the ShelXT program [23] and refined by full-matrix least squares using the Olex2 program [24] in the anisotropic approximation for non-hydrogen atoms. The hydrogen atoms of the water molecules were localized from the difference Fourier syntheses, positions of other hydrogen atoms were calculated geometrically, and all of them were refined in the isotropic approximation by the riding model. Selected crystallographic data and structure refinement parameters for compounds I and II are given in Table 1.
The full set of XRD parameters was deposited with the Cambridge Crystallographic Data Centre (CIF files CCDC nos. 2130014 (I) and 2130015 (II); deposit@ccdc.cam.uk). The geometry of the nearest environment of the metal atoms was examined using the SHAPE 2.1 program [25].
RESULTS AND DICUSSION
The entire exchange of the anions and the formation of complex [Gd(Fur)3(H2O)2]n⋅MeCN (I) occur during the exchange reaction of aqueous gadolinium acetate with 2-furoic acid (3 mol) in acetonitrile (80°С, 90 min). The use of gadolinium pivalate [Gd(Piv)3(HPiv)6]⋅3H2O (Piv– = (CH3)3COO–) as the starting reagent and performing the reaction at room temperature also afford compound I. The use of Sm2(CO3)3 as the starting substance is accompanied by the formation of [Sm(Fur)3(H2O)3]n (II).
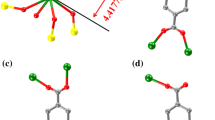
According to the XRD data (Fig. 1), compound I that crystallizes in the triclinic space group \(P\bar {1}\) is the 1D coordination polymer (Fig. 2). Its independent part contains one Gd atom, two water molecules coordinated to the Gd atom, and three carboxylate groups (Table 2), one of which acts as the chelating ligand and two remained groups are bridging. As a result, the complexing agent turns out to be bound to five carboxylate groups that build up the coordination environment to a distorted square antiprism (GdO8) (Fig. 1). Thus formed coordinately bonded chains along the crystallographic axis a are additionally stabilized by hydrogen bonds (O…O 2.714(6) and 2.768(6) Å, OHO 172.1(1)° and 172.5(2)°) between two symmetrically independent water molecules and Fur anion coordinated to the metal atom via the chelate mode (Fig. 2). Both water molecules are also involved in the formation of hydrogen bonds (O…N 2.989(6) and 3.088(8) Å, OHN 149.5(3)° and 153.3(3)°) with the solvate acetonitrile molecule.

Fragment of the polymer chain of compound I. Only hydrogen atoms of water molecules are shown; non-hydrogen atoms are presented as ellipsoids of thermal vibrations (p = 50%), and numeration is presented only for the heteroatoms of the independent part of the unit cell. Hereinafter hydrogen bonds are shown by dashed lines.

Fragment of the crystal packing of compound I.
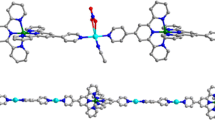
Compound II (Fig. 3) that crystallizes in the monoclinic space group P21/c, like compound I, is the 1D coordination polymer (Fig. 4). Unlike compound I, the independent part in compound II contains three water molecules coordinated to the Sm atom, one of three carboxylate groups linked to the Sm atom acts as the terminal ligand, and other molecules are bridging (Table 2). As a result, the coordination environment of samarium (SmО8) formed by three water molecules and five Fur anions has the shape of a distorted triangular dodecahedron (Fig. 3). As in the case of compound I, infinite chains along the crystallographic axis с formed due to coordination with four bridging ligands (Fig. 4) are additionally stabilized by hydrogen bonds between two symmetrically independent water molecules (O…O 2.812(6) Å, OHO 172.9(3)°) and between the discussed water molecules and furoic acid anion (O…O 2.642(6) and 3.182(8) Å, OHO 162.4(3)° and 172.4(3)°) acting as the terminal ligand. The latter is also involved in hydrogen bond formation (O…O 2.775(7) and 2.855(7) Å, OHO 155.1(3)° and 163.3(3)°) with two water molecules of the adjacent chains of the 1D coordination polymer resulting in the formation in the crystal of compound II of hydrogen-bonded layers perpendicular to the crystallographic axis a.

Fragment of the polymer chain of compound II. Only hydrogen atoms of water molecules are shown; non-hydrogen atoms are presented as ellipsoids of thermal vibrations (p = 50%), and numeration is presented only for the heteroatoms of the independent part of the unit cell.

Fragment of the crystal packing of compound II.
The thermal behavior of compound I was studied by STA in an inert atmosphere. The first indications to the mass loss start at 69°С and correspond to the elimination of two coordinated water molecules (mexp/theor = 6.4/6.3%) (Fig. 5, curve 1). The intense endothermal peak with an extremum at 96°С corresponds to this effect in the DSC curve (Fig. 5, curve 2). On the whole, the thermolysis of the organic moiety of the complex, including the decarboxylation and furan fragment destruction, occurs gradually without pronounced thermal effects and reaches the maximum rate in a range of 404–461°С. The DSC curve exhibits an exothermal effect with an extremum at 443°С (Fig. 5, curve 2). The final weight corresponds to the formation of Gd2O3 (mexp/theor = 61/63%). The chemical composition and micromorphology were qualitatively determined and examined using scanning electron microscopy (SEM) and XPS (Fig. 6). The energy dispersive spectrum contains the peaks from the oxygen and gadolinium atoms (Fig. 6b).

(1) TG and (2) DSC curves for compound I.

(a) SEM image of the micromorphology (×250) and (b) the energy dispersive spectrum of the final thermolysis product of compound I (high-intensity peak from carbon on the spectrum corresponds to the support material).
Thus, two 1D coordination polymers were synthesized in which each lanthanide cation coordinates three Fur– anions, which perform both the chelate and bridging functions, and water molecules to form the coordination number of the lanthanide atom equal to 8. The supramolecular level of the complexes is formed due to numerous intra- and intermolecular hydrogen bonds joining the coordination chains into the single polymer motif. The study of the thermal properties of compound I showed a relatively low stability of the compound, since the dehydration of the coordinated water molecules started already at 69°С. According to the XPS data, gadolinium(III) oxide is the final product.
REFERENCES
Peters, J.A., Nieuwenhuizen, M.S., and Raber, D.J., J. Magn. Reson., 1985, vol. 65, p. 417.
Piguet, C. and Geraldes, C.F., Handbook on the Physics and Chemistry of Rare Earths, Amsterdam: Elsevier, 2003.
Chebotar’, I.V., Novikov, I.A., Subbot, A.M., and Mayanskii, N.A., Sovremennye Tekhnologii v Meditsine, 2017, vol. 9, p. 23.
Wang, Y., Li, X., Wang, T., et al., Inorg. Chem., 2010, vol. 49, p. 969.
Babailov, S., Sens. Actuators, B, 2017, vol. 251, p. 108.
Koehler, J. and Meiler, J., Prog. Nucl. Magn. Reson. Spectrosc., 2011, vol. 59, p. 360.
Peters, J.A., Sinnema, A., Kieboom, P.G., and Bekkum, H., J. Am. Chem. Soc., 1985, vol. 107, p. 12.
Mashkovskii, M.D. Lekarstvennye sredstva (Pharmaceuticals), Moscow: Novaya volna, 2005.
Makota, H. and Gomi, T., Radiol. Contrast Ag. Radiopharm., 2015, vol. 37, p. 583.
Krylov, V.V., Drozdovskii, B.Ya., and Tsyb, A.F., Uspekhi Sovremennogo Estestvoznaniya, 2003, no. 10, p. 73.
Bartolomé. E., Bartolomé, J., Arauzo, A., et al., J. Mater. Chem. С, 2018, vol. 6, p. 5286.
Gusev, A., Kiskin, M., Lutsenko, I., et al., J. Lumin., 2021, vol. 238, p. 118305.
Li, X., Jin, L., Lu, S., et al., J. Mol. Struct., 2002, vol. 604, p. 65.
Yin, M. and Sun, J., J. Alloy. Comp., 2004, vol. 381, p. 50.
Lutsenko, I.A., Baravikov, D.E., Kiskin, M.A., et al., Russ. J. Coord. Chem., 2020, vol. 46, p. 411. https://doi.org/10.31857/S0132344X20060055
Lutsenko, I.A., Yambulatov, D.S., Kiskin, M.A., et al., Russ. J. Coord. Chem., 2020, vol. 46, p. 787. https://doi.org/10.1134/S1070328420120040
Lutsenko, I.A., Yambulatov, D.S., Kiskin, M.A., et al., Chem. Select., 2020, vol. 5, p. 11837.
Lutsenko, I.A., Kiskin, M.A., Koshenskova, K.A., et al., Russ. Chem. Bull., 2021, vol. 70, p. 463.
Uvarova, M.A., Lutsenko, I.A., Kiskin, M.A., et al., Polyhedron, 2021, vol. 203, p. 115241.
Lutsenko, I.A., Baravikov, D.E., Koshenskova, K.A., et al., RSC Adv., 2022, vol. 12, p. 5173.
Lutsenko, I.A., Nikiforova, M.E., Kosheskova, K.A., et al., Russ. J. Coord. Chem., 2021, vol. 47, p. 879. https://doi.org/10.31857/S0132344X22020049
Fomina, I.G., Kiskin, M.A., Martynov, A.G., et al., Russ. J. Inorg. Chem., 2004, vol. 49, p. 1369.
Sheldrick, G.M., Acta Crystallogr., Sect. C: Struct. Chem., 2015, vol. 71, p. 3.
Dolomanov, O.V., Bourhis, L.J., Gildea, R.J., et al., J. Appl. Crystallogr., 2009, vol. 42, p. 339.
Jordi, C., Eliseo, R., and Santiago, A., Organometallics, 2005, vol. 24, p. 1556.
ACKNOWLEDGMENTS
XRD studies were carried out using the equipment of the Center of Molecular Structure Investigation at the Nesmeyanov Institute of Organoelement Compounds (Russian Academy of Sciences). Elemental analysis, IR spectroscopy, and STA were carried out and the SEM images of the micromorphology and the energy dispersive spectra were obtained using the equipment of the Center for Collective Use of Physical Methods of Investigation at the Kurnakov Institute of General and Inorganic Chemistry (Russian Academy of Sciences).
Funding
This work was supported by the Ministry of Science and Higher Education of the Russian Federation in terms of the state assignment of the Kurnakov Institute of General and Inorganic Chemistry (Russian Academy of Sciences).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflicts of interest.
Additional information
The issue is dedicated to the 70th birthday of Academician V.I. Ovcharenko
Translated by E. Yablonskaya
Rights and permissions
About this article

Cite this article
Uvarova, M.A., Lutsenko, I.A., Nikiforova, M.E. et al. Gd(III) and Sm(III) 1D Coordination Polymers with 2-Furoic Acid: Synthesis, Structures, and Thermal Behavior. Russ J Coord Chem 48, 457–463 (2022). https://doi.org/10.1134/S1070328422080073
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1070328422080073