
Abstract
β-Aminovinyl ketone (2-nitro-3-(8-quinolylamino)prop-2-enal) was synthesized by condensation of nitromalondialdehyde with 8-aminoquinoline. The reactions of β-aminovinyl ketone with copper, nickel, and cobalt acetates in methanol gave metal complexes. The spectral and magnetic characteristics of preparatively isolated compounds were studied. The structure of cobalt(II) chelate was determined on the basis of X-ray diffraction study (CIF file CCDC no. 2109263).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
In recent years, the chemistry of amino derivatives of β-dicarbonyl compounds and their complexes with virtually all metals of the Periodic Table has been intensively developed [1–7]. The possibility of targeted control of the electronic and geometric parameters of these compounds is one of the main factors determining the broad and versatile practical applications of the complexes. They can exhibit catalytic and biological activities [4, 8–10] and can serve for activation of small molecules, models of protein active sites [11, 12], and precursors for the preparation of nanostructured compounds for electronics, energy production, and chemosensorics [3, 6, 7]. Also, they can be used for the development of optically active materials [5, 13]. The stereochemistry and physicochemical properties of metal β-ketoiminate and β-diketiminate chelates can be controlled by modifying the nature of substituents in the ketone moiety and in the amine component of the ligand. Previously, we studied the influence of such modification on the structure and properties of the products of complex formation of β-aminovinyl ketones and β-aminovinylimines [14–17]. In this communication, we describe data on the synthesis and physicochemical studies of nitromalondialdehyde β-aminovinyl ketone (2-nitro-3-(8-quinolylamino)prop-2-enal) (HL) and Cu(II), Ni(II), and Co(II) chelates of HL.
EXPERIMENTAL
The compounds were synthesized using commercially available solvents, 8-aminoquinoline (CAS 578-66-5, Sigma-Aldrich), copper acetate monohydrate (CAS 6046-93-1), and nickel (CAS 6018-89-9) and cobalt (CAS 6147-53-1) acetate tetrahydrates (Alfa Aesar). The sodium salt of nitromalondialdehyde (see Scheme 1) was synthesized by a previously described procedure [18].
Synthesis of 2-nitro-3-(8-quinolylamino)prop-2-enal (HL). Method 1. 8-Aminoquinoline hydrochloride (1.81 g, 10 mmol) dissolved in 70% ethanol (20 mL) was added with continuous stirring to a solution of nitromalondialdehyde sodium salt (1.39 g, 10 mmol) in water (30 mL). The resulting reaction mixture was left overnight. The next day, the yellow precipitate that formed was collected on a filter, washed with water, and recrystallized from toluene. The yield was 1.38 g (57%).
Method 2. A solution of sodium nitromalondialdehyde (0.695 g, 5 mmol) in ethanol (25 mL) was treated with 10% HCl to pH 6–7, and an ethanol solution (15 mL) of 8-aminoquinoline (0.721 g, 5 mmol) was added. The reaction mixture was kept at 50–55°C for 3 h with continuous stirring. After cooling to room temperature, the precipitate was collected on a filter and washed with water and ethanol. The product was recrystallized from toluene. The yield was 0.632 g (52%).
For C12H9N3O3 | |||
Anal. calcd., % | C, 59.26 | H, 3.73 | N, 17.28 |
Found, % | C, 59.19 | H, 3.80 | N, 17.19 |
IR (ν, cm–1): 3122 w, 3073 w, 1681 m,1659 s, 1632 s, 1604 s, 1513 m, 1497 s, 1464 s, 1431 m, 1394 w, 1374 m, 1338 s, 1309 s, 1232 w, 1208 w, 1173 w, 1119 m, 1019 m, 956 m, 864 m, 826 m, 805 w, 791 m, 749 m, 732 w, 622 m, 571 m. 1H NMR (DMSO-d6; δ, ppm): 7.64–7.72 (m, 2H, Cquin–H, E,Z-isomers), 7.87–7.93 (m., 1H, Cquin–H, E,Z-isomers), 8.2 (d, J = 7.7 Hz, 1H, Cquin–H, Z-isomer), 8.24 (d, J = 7.5 Hz, 1 H, Cquin–H, E-isomer), 8.47 (m, 1H, Cquin–H, E,Z-isomers), 8.79 (d, J = 15.4 Hz, 1H, CH–NH, Z-isomer), 9.00 (dd, J = 4.1, J = 1.5 Hz, 1H, Cquin–H, E,Z-isomers), 9.37 (dd, J = 14.6, J = 3.5 Hz, 1H, CH–NH, E-isomer), 9.95 (s, 1H, CH=O, Z-isomer), 10.17 (d, J = 3.5 Hz, 1H, CH=O, E-isomer), 12.81 (d, J = 15.4 Hz, 1H, NH–CH, Z-isomer) 13.37 (d, J = 14.6 Hz, 1H, NH–CH, E-isomer).
Synthesis of Cu(II) (I), Ni(II) (II), and Co(II) (III) complexes. Metal acetate (0.5 mmol) (Cu(OAc)2·H2O (0.1 g), Ni(OAc)2·4H2O (0.125 g), or Co(OAc)2·4H2O (0.125 g)) in methanol (15 mL) was added to a hot solution of ligand HL (0.243 g, 1 mmol) in methanol (15 mL). The reaction mixture was refluxed for 3 h. After cooling, the precipitate was collected on a filter, washed three times with hot methanol (10 mL), and recrystallized from toluene or a chloroform–methanol mixture (1 : 1). The resulting crystalline compounds were dried in a vacuum drying oven at 100°C.
Bis[2-nitro-3-(8-quinolylimino)prop-1-enoxy]cop-per(II) (I). Light green powder. The yield was 0.183 g (67%).
For C24H16N6O6Cu | |||
Anal. calcd., % | C, 52.61 | H, 2.94 | N, 15.34 |
Found, % | C, 52.49 | H, 2.87 | N, 15.29 |
IR (ν, cm–1): 3064 w, 1621 s, 1596 m, 1558 m, 1525 s, 1494 s, 1470 s, 1376 s, 1343 s, 1295 s, 1259 s, 1214 m, 1180 w, 1120 w, 1082 w, 1060 w, 1039 w, 974 m, 830 m, 811 m, 788 m, 753 m, 723 w, 650 m, 620 m, 581 m.
µeff = 1.89 μB (294 K).
Bis[2-nitro-3-(8-quinolylimino)prop-1-enoxy]ni-ckel(II) (II). Brown powder. The yield was 0.155 g (57%).
For C24H16N6O6Ni | |||
Anal. calcd., % | C, 53.07 | H, 2.97 | N, 15.47 |
Found, % | C, 52.99 | H, 2.93 | N, 15.44 |
IR (ν, cm–1): 3063 w, 1615 с, 1594 m, 1551 w, 1524 s, 1494 s, 1469 s, 1376 s, 1340 s, 1316 m, 1294 s, 1259 s, 1239 m, 1211 m, 1180 w, 1122 w, 1080 w, 1064 w, 1044 w, 971 m, 831 m, 811 m, 788 m, 753 m, 722 w, 650 m, 620 m, 579 m.
µeff = 3.07 μB (294 K).
Bis[2-nitro-3-(8-quinolylimino)prop-1-enoxy]co-balt(II) (III). Orange powder. The yield was 0.147 g (54%).
For C24H16N6O6Co | |||
Anal. calcd., % | C, 53.05 | H, 2.97 | N, 15.47 |
Found, % | C, 52.87 | H, 2.94 | N, 15.31 |
IR (ν, cm–1): 3065 w, 1611 s, 1594 m, 1553 w, 1524 s, 1494 s, 1469 s, 1396 m, 1377 s, 1341 s, 1293 s, 1259 s, 1211 m, 1180 w, 1121 w, 1081 w, 1063 w, 1042 w, 973 m, 830 m, 811 m, 788 m, 753 m, 723 w, 646 m, 620 m, 577 m.
µeff = 4.47 μB (294 K).
Elemental analysis for C, H, N was performed using a TCM 480 Carlo Erba instrument.
1H NMR spectra were recorded on an Avance-600 spectrometer (Bruker, Germany) at 20°C. The proton chemical shifts were referred to the residual solvent signals (DMSO-d6). The IR spectra of the compounds (as mineral oil mulls) were measured on a Varian 3100-FTIR Excalibur instrument in the 4000–400 cm–1 range.
The specific magnetic susceptibility was determined by the relative Faraday method at room temperature using Hg[Co(CNS)4] as the calibration standard.
Quantum chemical calculations were carried out using the density functional theory (DFT) with the B3LYP hybrid exchange correlation potential [19] in the 6-311++G** split valence Gaussian basis set extended by polarization and diffusion functions for all atoms. The Gaussian’09 software was used [20]. The geometry optimization was carried out without symmetry constraints; the potential energy surface (PES) minima were characterized by the absence of imaginary frequencies of the calculated normal vibrations. The data and presentation graphics were prepared and calculation results were visualized using the Chemcraft software [21].
X-ray diffraction study of cobalt complex III was carried out on a Bruker APEX2 diffractometer equipped with a CCD array detector and a source of monochromatic radiation (MoKα radiation, λ = 0.71073 Å, graphite monochromator, ω scan mode). The structure was solved by direct methods and refined in the full-matrix anisotropic approximation for all non-hydrogen atoms. The hydrogen atoms in III were located from difference Fourier maps and refined in the isotropic approximation. All calculations were carried out using the SHELXS-97 program package [22]. The main crystallographic data for cobalt complex III and refinement details are summarized in Table 1; selected interatomic distances and bond angles are given in Table 2.
The full set of X-ray diffraction parameters is deposited with the Cambridge Crystallographic Data Centre (CCDC no. 2109263; deposit@ccdc.cam. ac.uk).
RESULTS AND DISCUSSION
As a continuation of studies into the effect of the natures of metal atom and substituent in the β-aminovinyl ketone and β-aminovinylimine molecules on the structures and properties of the complexes they form [14–17], we synthesized 2-nitro-3-(8-quinolylamino)prop-2-enal (HL) and copper(II) (I), nickel(II) (II), and cobalt(II) (III) chelates of this ligand (Scheme 1).

Scheme 1 .
Ligand HL was prepared by two methods. The first method was based on the reaction between the nitromalondialdehyde sodium salt and 8-aminoquinoline hydrochloride in aqueous ethanol for 15–18 h. The second method included condensation in situ of pre-synthesized 3-hydroxy-2-nitroprop-2-enal (see Scheme 1) and 8-aminoquinoline in ethanol at 50–55°C for 3 h. Previously, it was shown that aromatic amines can react with β-dicarbonyl compounds to give β-aminovinylimines [17, 23–25]. However, despite numerous attempts, we were unable to obtain nitromalondialdehyde β-aminovinylimine (HL1) (see Scheme 1). However, two examples of compounds of this type based on malondialdehyde and 2-(4-tolyl)malondialdehyde have been reported in the literature [26, 27]. Probably, the presence of the nitro group in the aldehyde moiety of the molecule affects the addition of the second quinoline substituent.
β-Aminovinyl ketone HL was identified by elemental analysis and IR and NMR spectroscopy.
The compound HL can exist as, at least, two tautomers (A and B) and E,Z-isomers (Scheme 2).

Scheme 2 .
The IR spectrum of β-aminovinyl ketone HL did not show absorption band at 1700 cm–1, characteristic of non-conjugated C=O bond. Meanwhile, the spectrum exhibited a weak band at 1681 cm–1, which was assigned to the ν(CO) mode of Z-isomer, and three intense bands at 1658–1604 cm–1, which corresponded to the O=CH–CR=C–NH moiety and ‒HC=CH– and –CH=N– groups of the quinoline substituent. These bands were also detected in the 1650–1560 cm–1 range of the spectra of close analogues of the HL ligand, quinoline-containing benzoylacetone (HL2) and 3-hydroxy-1-phenylprop-2-ene-1-one (HL3) enamines [14]. This attests to participation of the C=O and C=N bonds of the aldehyde moiety in conjugation, as can be seen in structures A and B. The highest-frequency band of this group at 1659 cm–1 corresponds to the ν(CO) stretching mode of the E-isomer and is shifted by, on average, ~30 cm–1 to shorter wavelengths relative to the bands of enamines HL2 and HL3. The nitro group is manifested in the IR spectrum of HL as two symmetric and antisymmetric stretching bands at 1340 and 1500 cm–1. However, it is difficult to make an unambiguous choice between the keto-amine and imino-enol tautomers from IR spectroscopic data.
The most stable configurations of HL were determined by B3LYP/6-311++G** DFT calculations. The optimal conformers of tautomeric and E,Z-isomeric forms and their relative energies are depicted in Fig. 1.

Spatial structure and relative energies ∆E (kcal/mol) of isomeric and tautomeric forms of HL in the gas phase according to the B3LYP/6-311++G** data.
According to quantum chemical simulation data, tautomer A (in the E-conformation), in which all hydrogen atoms are located on the nitrogen atoms of the aldehyde moiety, is most stable (in the gas phase), which is in line with NMR data. The Z-isomer of tautomer A is slightly destabilized (by 2.98 kcal/mol) relative to the E-isomer. Thus, considering this slight energy difference, tautomer A is expected to exist simultaneously as both E- and Z-isomers. The relative energy of tautomer B in both E,Z-isomeric forms is markedly higher (10.63 and 22.44 kcal/mol) than the energies of tautomer A in E,Z-conformations, indicating low probability of formation of tautomer B.
Most detailed information about the structure of compound HL was derived from NMR studies. The 1H NMR spectrum of β-aminovinyl ketone HL in DMSO-d6 shows two sets of resonance signals with 4 : 1 intensity ratio, which makes it different from the spectra of enamines HL2 and HL3 [14]. This means that compound HL exists in solution as a mixture of either two isomers or two tautomers. However, the presence of a doublet at 8.79 ppm (J = 15.4 Hz) and a doublet of doublets at 9.37 ppm (J = 14.6 Hz, J = 3.5 Hz) for the CHNH methine proton and the presence of two low-field doublets for the resonating NH protons at 12.81 ppm (J =15.4 Hz) and 13.37 ppm (J = 14.6 Hz) unambiguously point to the existence of keto-amine tautomer A and the absence of B. Hence, the observed 1H NMR spectrum of HL is determined by the presence of E/Z-isomers in the solution, with the E-isomer predominating. It gives rise to a doublet of doublets for the CHNH methine proton at 9.37 ppm and two doublets at 10.17 and 13.37 ppm, corresponding to the resonating protons of the CH=O and NH groups, respectively. The signals of the above-listed protons of the minor Z-isomer are manifested as a doublet at 8.79 ppm, a singlet at 9.95 ppm, and a doublet at 12.81 ppm, respectively. Thus, compound HL exists as the keto-amine tautomer. Both in the solid state and in solution, the presence of both E- and Z-isomers is observed; this is in good agreement with the results of quantum chemical calculations and differentiates β-aminovinyl ketone HL from quinoline-containing analogues HL2 and HL3, which exist as single isomers.
Metal chelates I–III were obtained by reactions of β-aminovinyl ketone HL with Cu(II), Ni(II), and Co(II) acetates in methanol. According to elemental analysis data, the composition of complexes was ML2.
The IR spectra of metal chelates I–III do not exhibit the group of absorption bands in the 1680–1600 cm–1 range, present in the spectra of uncoordinated β-aminovinyl ketone HL, but exhibit new bands at 1621–1611 and 1595 cm–1. Relying on the data of [28], these bands were assigned to delocalized C=O, C=C, and C=N bonds. The results of IR investigations suggest that HL participates in the complex formation reactions in the deprotonated (L) imino-enol form. The coordination bonds are localized on the imine and quinoline nitrogen atoms and the oxygen atom to give the MN4O2 chelate unit. The spectra of all metal chelates differ little from one another, which may be indicative of their similar structure.
The magnetochemical measurements of metal chelates I–III were performed in the 294–77 K temperature range. All compounds were paramagnetic. The μeff values (1.89 (Cu), 3.07 (Ni), and 4.47 μB (Co)) did not change as the temperature decreased down to 77.7 K. Nickel complex II was paramagnetic not only in the solid state, but also in solution, as indicated by extension of the scale of 1H NMR chemical shifts and the observed paramagnetic broadening of the proton signals of the ligand system, typical of high-spin Ni(II) chelates [29]. The high-spin nature of complex II unambiguously attests to a non-planar geometry of the complex and is related to the formation of a tetrahedral or octahedral coordination unit. The magnetic moment of metal chelate II, like those for previously studied nickel complexes of β-aminovinyl ketones HL2 and HL3 [14], was consistent with the octahedral structure and additional coordination to the nitrogen atoms of quinolyl substituents. Meanwhile, when a naphthyl substituent is present instead of the quinolyl group at the azomethine nitrogen atom, Ni(II) β-aminovinyl ketonates are square planar and diamagnetic [14]. Thus, the presence of the additional donor site, the quinoline nitrogen atom, enables the formation of octahedral chelate unit of the complexes. The data of magnetochemical measurements for copper (I) and cobalt (III) chelates were less informative. They attested to the mononuclear structure of complexes and to the d9 state of the copper ion, S =1/2, and the high-spin d7 state of cobalt, S = 3/2, and the absence of noticeable intermolecular exchange interactions in the crystalline state at temperatures above 77.7 K.
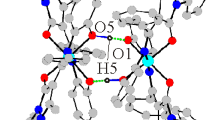
The final conclusion about the structure of Co(II) β-ketoiminate chelate III was drawn from X-ray diffraction data. The molecular structure of III is depicted in Fig. 2; selected geometric characteristics are presented in Table 2.

Molecular structure of cobalt(II) complex III with atoms represented by displacement ellipsoids (p = 50%).
Compound III crystallizes in a centrosymmetric space group, and the molecule occupies a general position. Two deprotonated tridentate β-aminovinyl ketones HL and the cobalt(II) ion form the CoN4O2 pseudo-octahedral chelate unit with cis-arrangement of the O atoms (the O(1')Co(1)O(1) angle is 90.31(12)°). The coordinated ligands are orthogonal to each other, with the dihedral angle between them being 89.65°. Two six-membered metallacycles are virtually planar. The displacements of the cobalt atom from the O(1)–C(1)–C(2)–C(3)–N(1) and O(1')–C(1')–C(2')–C(3')–N(1') planes are 0.054 and 0.062 Å, which is equivalent to folding angles along the N(1)–O(1) and O(1')–N(1') lines of 2.006° and 2.324°, respectively. Two five-membered metallacycles are flattened to a to a lesser extent, with the cobalt atom deviating from the N(1)–C(12)–C(4)–N(3) and N(1')–C(12')–C(4')–N(3') planes by 0.135 and 0.07 Å and folding angles along the N(1)–N(3) and N(1')–N(3') lines of 4.82° and 2.54°, respectively. The N(4)O(2)O(3) and N(4')O(2')O(3') nitro groups are rotated relative to the planes of metallacycles by 8.08° and 3.7°, respectively. The planarity of the ligands results in abnormally shortened C(3)H…HC(11) intramolecular contacts with the H…H distance of 1.87 Å. According to our studies, shortened contacts of this type in coordinated compounds may correspond to attractive interactions [30].
In the molecule of III, the Co(1)–O(1), Co(1)–O(1') distances are shorter than Co(1)–N(1), Co(1)–N(1') (Table 2), and are longer than the corresponding bond lengths in the square planar or tetrahedral cobalt(II) β‑ketoiminates [3, 31]. The nitrogen atoms of the quinoline rings are coordinated to the cobalt atom at distances of 2.104 and 2.105 Å, respectively. The bond lengths are, on average, ~0.18 Å longer than the corresponding Co–N distances observed in bis[2,4-di-tert-butyl-6-[8-quinolyliminomethyl]phenoxy]Co(II) [32]. The bond lengths and bond length distribution in the six-membered metallacycles (O(1)–C(1)–C(2)–C(3)–N(1), O(1')–C(1')–C(2')–C(3')–N(1')) of complex III (Table 2) indicate that the deprotonated HL ligand exists as the delocalized enol-imine form.
Analysis of the intermolecular contacts demonstrated that the molecules in the crystal form shortened intermolecular contacts between the electron-withdrawing and electron-donating parts of the ligand π-systems with the shortest C…C distance of 3.39 Å. This contact and the parallel arrangement of the interacting ligands unambiguously attest to the presence of stacking interactions.
Thus, the reactions of nitromalondialdehyde β‑aminovinyl ketone containing a coordinatively active quinoline substituent in the amine moiety with Cu(II), Ni(II), and Co(II) acetates resulted in the synthesis of mononuclear complexes of these metals. The structures of the ligand and metal chelates were confirmed by IR and NMR spectroscopy, magnetochemistry, and X-ray diffraction data. The resulting metal-chelates have octahedral chelate unit, irrespective of the nature of the metal. The results of this study and earlier studies of quinoline-containing ketoiminate complexes indicate that substituents in the ketone moiety have little influence on the formation of the coordination polyhedron for this type of compounds. In this respect, these compounds differ from the metal chelates of β-aminovinyl ketones containing aryl substituents with an ortho-thioester group in the amine component of the molecule [15].
REFERENCES
Bourget-Merle, L., Lappert, M.F., and Severn, J.R., Chem. Rev., 2002, vol. 102, no. 9, p. 3031. https://doi.org/10.1021/cr010424r
Camp, C. and Arnold, J., Dalton Trans., 2016, vol. 45, no. 37, p. 14462. https://doi.org/10.1039/C6DT02013E
Puring, K., Zywitzki, D., Taffa, D.H., et al., Inorg. Chem., 2018, vol. 57, no. 9, p. 5133. https://doi.org/10.1021/acs.inorgchem.8b00204
Lyubov, D.M., Tolpygin, A.O., and Trifonov, A.A., Coord. Chem. Rev., 2019, vol. 392, p. 83. https://doi.org/10.1016/j.ccr.2019.04.013
Witkowska, E., Orwat, B., Oh, M.J., et al., Inorg. Chem., 2019, vol. 58, no. 22, p. 1567. https://doi.org/10.1021/acs.inorgchem.9b02785
Huster, N., Zanders, D., Karle, S., et al., Dalton Trans., 2020, vol. 49, no. 31, p. 10755.https://doi.org/10.1039/d0dt01463J
Zywitzki, D., Taffa, D.H., Lamkowski, L., et al., Inorg. Chem., 2020, vol. 59, no. 14, p. 10059. https://doi.org/10.1021/acs.inorgchem.0c01204
Allison, M., Wilson, D., Pask, C.M., et al., Chembiochem, 2020, vol. 21, no. 14, p. 1988. https://doi.org/10.1002/cbic.202000028
Lord, R.M., Hebden, A.J., Pask, C.M., et al., J. Med. Chem., 2015, vol. 58, no. 12, p. 4940. https://doi.org/10.1021/acs.jmedchem.5b00455
Bermeshev, M.V. and Chapala, P.P., Prog. Polymer Sci., 2018, vol. 84, p. 1. https://doi.org/10.1016/j.progpolymsci.2018.06.003
Yao, S. and Driess, M., Acc. Chem. Res., 2012, vol. 45, no. 2, p. 276. https://doi.org/10.1021/ar200156r
Di Francesco, G.N., Gaillard, A., Ghiviriga, I., et al., Inorg. Chem., 2014, vol. 53, no. 9, p. 4647. https://doi.org/10.1021/ic500333p
Lai, P.-N., Brysacz, C.H., Alam, M.K., et al., J. Am. Chem. Soc., 2018, vol. 140, no. 32, p. 10198. https://doi.org/10.1021/jacs.8b04841
Korshunov, O.Y., Uraev, A.I., Shcherbakov, I.N., et al., Russ. J. Inorg. Chem., 2000, vol. 45, no. 9, p. 1363.
Uraev, A.I., Kurbatov, V.P., Nivorozhkin, A.L., et al., Russ. Chem. Bull. Int. Ed., 2002, vol. 51, no. 10, p. 1924. https://doi.org/10.1023/A:1021321022710
Uraev, A.I., Kurbatov, V.P., Tylchenko, L.S., et al., Dokl. Chem., 2002, vol. 383, no. 1, p. 57.
Uraev, A.I., Ikorskii, V.N., Bubnov, M.P., et al., Russ. J. Coord.Chem., 2006, vol. 32, no. 4, p. 287. https://doi.org/10.1134/S1070328406040105
Fanta, P.E., Organ. Synth., 1952, vol. 32, p. 95. https://doi.org/10.15227/orgsyn.032.0095
Becke, A.D., J. Chem. Phys., 1993, vol. 98, no. 7, p. 5648. https://doi.org/10.1063/1.464913
Frisch, M.J., Trucks, G.W., Schlegel, H.B., et al., Gaussian 09, Revision A.02, 2009.
Zhurko, G.A. and Zhurko, D.A., Chemcraft ver. 1.6 (build 338). http://www.chemcraftprog.com.
Sheldrick, G., Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, vol. 64, no. 1, p. 112. https://doi.org/10.1107/S0108767307043930
Uraev, A.I., Nivorozhkin, A.L., Kurbatov, V.P., et al., Russ. J. Coord. Chem., 2000, vol. 26, no. 12, p. 891. https://doi.org/10.1023/A:1026639327693
Yokota, S., Tachi, Y., Nishiwaki, N., et al., Inorg. Chem., 2001, vol. 40, no. 21, p. 5316. https://doi.org/10.1021/ic0155535
Spencer, D.J.E., Reynolds, A.M., Holland, P.L., et al., Inorg. Chem., 2002, vol. 41, no. 24, p. 6307. https://doi.org/10.1021/ic020369k
Zatka, V., Holzbecher, J., and Ryan, D.E., Anal. Chim. Acta, 1971, vol. 55, no. 1, p. 2738. https://doi.org/10.1016/S0003-2670(01)82767-7
Fritsch, J.M., Thoreson, K.A., and McNeill, K., Dalton Trans., 2006, no. 40, p. 4814.https://doi.org/10.1039/B609616F
Dorovskikh, S.I., Alexeyev, A.V., Kuratieva, N.V., et al., J. Organomet. Chem., 2013, vol. 741, p. 122. https://doi.org/10.1016/j.jorganchem.2013.05.001
Garnovskii, A.D., Nivorozhkin, A.L., and Minkin, V.I., Coord. Chem. Rev., 1993, vol. 126, nos. 1−2, p. 1. https://doi.org/10.1016/0010-8545(93)85032-y
Kotova, O., Lyssenko, K., Rogachev, A., et al., J. Photochem. Photobiol., A, 2011, vol. 218, p. 117. https://doi.org/10.1016/j.jphotochem.2010.12.011
Robson, K.C.D., Phillips, C.D., Patrick, B.O., et al., Dalton Trans., 2010, vol. 39, no. 10, p. 2573. https://doi.org/10.1039/b921153e
Gong, D., Wang, B., Jia, X., et al., Dalton Trans., 2014, vol. 43, no. 10, p. 4169. https://doi.org/10.1039/C3DT52708E
Funding
This study was supported by the Ministry of Science and Higher Education of the Russian Federation (state assignment of the Southern Federal University in the field of science, project 0852-2020-0031).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflicts of interest.
Additional information
Translated by Z. Svitanko
Rights and permissions
About this article

Cite this article
Kovaleva, T.V., Uraev, A.I., Lyssenko, K.A. et al. Synthesis, Structure, and Properties of Copper(II), Nickel(II), and Cobalt(II) Ketoiminate Chelates. Molecular and Crystal Structures of Bis[2-nitro-3-(8-quinolylimino)prop-1-enoxy]cobalt(II). Russ J Coord Chem 48, 210–217 (2022). https://doi.org/10.1134/S1070328422040029
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1070328422040029