
Abstract
Two new cis-dioxomolybdenum(VI) complexes, [MoO2(L1)MeOH] (I) and [MoO2(L2)MeOH] (II) with potentially tridentate ONO aroylhydrazone Schiff bases derived from 3-methoxysalicylaldehyde with 4-bromobenzohydrazide and 4-trifluoromethylbenzohydrazide, respectively, have been synthesized and fully characterized on the basis of elemental analysis, FT-IR, molar conductivity, and electronic spectra. Structures of the complexes have been accomplished by single crystal X-ray diffraction (СIF files CCDC nos. 1433352 (I) and 1433318 (II)). The complexes have distorted octahedral structures in which the aroylhydrazones behave as binegative ligands. It is also revealed from the crystal structures that the Mo(VI) center adopts NO5 donor environment, and the octahedral coordination is furnished by two oxido groups and oxygen atoms of methanol or deprotonated methanol molecules. The catalytic properties were investigated for epoxidation of cyclooctene using aqueous tert-butyl hydroperoxide as the oxidant.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
In recent years, Schiff base ligands and their transition metal complexes have attracted considerable attention, not only for their facile synthesis, but also for potential biological, catalytic and industrial applications [1–5]. As we know, catalytic epoxidation of olefins is an important reaction in organic synthesis. Many transition metal complexes are active catalysts for this process [6–10]. Yet, among the complexes, molybdenum complexes have a unique place in coordination chemistry and have displayed very high catalytic activities in the oxidation of olefins and sulfides [11–13]. To date, the relationship between structures and catalytic epoxidation activity of the molybdenum complexes is still less studied. In the present work, two similar aroylhydrazone Schiff bases, 4‑bromo-(N'-2-hydroxy-3-methoxybenzylidene)benzohydrazide (H2L1) and N'-(2-hydroxy-3-methoxybenzylidene)-4-trifluoromethylbenzohydrazide (H2L2) with different terminal substitute groups, were prepared and used to prepare dioxomolybdemum(VI) complexes with MoO2(Acac)2 in methanol. The catalytic properties were investigated for epoxidation of cyclooctene using aqueous tert-butyl hydroperoxide as the oxidant.
EXPERIMENTAL
Materials and methods. 4-Bromobenzohydrazide and 4-trifluoromethylbenzohydrazide were prepared as described in the literature [14]. 3-Methoxysalicylaldehyde was purchased from Alfa Aesar and used as received. MoO2(Acac)2 was prepared as described in the literature [15]. Reagent grade solvents were used as received. Microanalyses of the complexes were performed with a Vario EL III CHNOS elemental analyzer. Infrared spectra were recorded as KBr pellets with an FTS-40 spectrophotometer. Electronic spectra were recorded on a Lambda 900 spectrometer. 1H NMR spectra were recorded on a 500 MHz Bruker Advance instrument. The catalytic reactions were followed by gas chromatography on an Agilent 6890A chromatograph equipped with an FID detector and a DB5-MS capillary column (30 m × 0.32 mm × 0.25 μm). Molar conductance measurements were made by means of a Metrohm 712 conductometer in acetonitrile.
Synthesis of the aroylhydrazone Schiff bases H2L1 and H2L2 was carried out in a similar way by refluxing a methanolic solution (30 mL) of 3-methoxysalicylaldehyde (10 mmol, 1.52 g) with 4-bromobenzohydrazide (10 mmol, 2.15 g) and 4-trifluoromethylbenzohydrazide (10 mmol, 2.04 g), respectively. Reflux was continued for 1 h at oily bath during which a solid compound separated. It was filtered and washed with cold methanol. The crude product was recrystallized from methanol and dried over anhydrous CaCl2.
H2L1: yield was 2.89 g (83%). IR data (KBr pellet; ν, cm–1): 3384 ν(O–H), 3233 ν(N–H), 1653 ν(C=O), 1610 ν(C=N). UV-Vis data (methanol; λ, nm): 290, 355, 410. 1H NMR (300 MHz; DMSO; δ, ppm): 12.13 (s., 1H, NH), 9.73 (s., 1H, OH), 8.91 (s., 1H, CH=N), 8.15 (d., 2H, ArH), 7.87 (d., 2H, ArH), 7.31 (d., 1H, ArH), 7.22 (d., 1H, ArH), 7.01 (t., 1H, ArH), 3.82 (s., 3H, OCH3).
For C15H13N2O3Br | |||
Anal. calcd., % | C, 51.60 | H, 3.75 | N, 8.02 |
Found, % | C, 51.77 | H, 3.83 | N, 7.96 |
H2L2: yield was 3.03 g (90%). IR data (KBr pellet; ν, cm–1): 3405 ν(O–H), 3226 ν(N–H), 1649 ν(C=O), 1613 ν(C=N). UV-Vis data (methanol; λ, nm): 295, 360, 418. 1H NMR (300 MHz; DMSO; δ, ppm): 12.21 (s., 1H, NH), 9.82 (s., 1H, OH), 8.90 (s., 1H, CH=N), 7.91 (d., 2H, ArH), 7.76 (d., 2H, ArH), 7.31 (d., 1H, ArH), 7.17 (d., 1H, ArH), 6.96 (t., 1H, ArH), 3.80 (s., 3H, OCH3).
For C16H13N2O3F3 | |||
Anal. calcd., % | C, 56.81 | H, 3.87 | N, 8.28 |
Found, % | C, 56.63 | H, 3.96 | N, 8.37 |
Synthesis of [MoO2(L1)MeOH] (I) and [MoO2(L2)MeOH] (II). The complexes I and II were prepared in a similar way by refluxing a methanolic solution (30 mL) of MoO2(Acac)2 (1.0 mmol, 0.33 g) with methanolic solutions of H2L1 (1.0 mmol, 0.35 g) and H2L2 (1.0 mmol, 0.34 g), respectively. Initially on mixing the two components immediately an orange yellow color was observed. The mixture was refluxed for 1 h. The respective complex separated as orange single crystals during slow evaporation of the reaction mixture. It was filtered, washed with cold methanol and dried over anhydrous CaCl2.
I: yield was 0.31 g (61%). IR data (KBr pellet; ν, cm–1): 3446 ν(O–H), 1605 ν(C=N), 1358 ν(C–Ophenolate), 1157 ν(N–N), 950 νsym(cis-MoO2), 861 νasym(cis-MoO2). UV-Vis data (acetonitrile; λ, nm): 295, 430. 1H NMR (300 MHz; DMSO; δ, ppm): 9.00 (s., 1H, CH=N), 8.20 (d., 2H, ArH), 7.90 (d., 2H, ArH), 7.35 (d., 1H, ArH), 7.27 (d., 1H, ArH), 7.05 (t., 1H, ArH), 4.10 (q., 3H, CH3OH), 3.82 (s., 3H, OCH3). Molar conductance (10–3 M, acetonitrile): 27 Ω–1 cm2 mol–1.
For C16H15N2O6BrMo | |||
Anal. calcd., % | C, 37.89 | H, 2.98 | N, 5.52 |
Found, % | C, 37.70 | H, 3.12 | N, 5.66 |
II: yield was 0.37 g (75%). IR data (KBr pellet; ν, cm–1): 3453 ν(O–H), 1607 ν(C=N), 1383 ν(C–Ophenolate), 1157 ν(N–N), 950 νsym(cis-MoO2), 850 νasym(cis-MoO2). 1H NMR (300 MHz; DMSO; δ, ppm): 8.92 (s., 1H, CH=N), 7.90 (d., 2H, ArH), 7.72 (d., 2H, ArH), 7.31 (d., 1H, ArH), 7.26 (d., 1H, ArH), 7.02 (t., 1H, ArH), 4.08 (q., 3H, CH3OH), 3.80 (s., 3H, OCH3). UV-Vis data (acetonitrile; λ, nm): 300, 430. Molar conductance (10–3 M, acetonitrile): 32 Ω–1 cm2 mol–1.
For C17H15N2O6F3Mo | |||
Anal. calcd., % | C, 41.15 | H, 3.05 | N, 5.65 |
Found, % | C, 41.31 | H, 3.14 | N, 5.53 |
X-ray structure determination. Data were collected on a Bruker SMART 1000 CCD area diffractometer using a graphite monochromator MoKα radiation (λ = 0.71073 Å) at 298(2) K. The data were corrected with SADABS programs and refined on F2 with Siemens SHELXL software [16, 17]. The structures were solved by direct methods and difference Fourier syntheses. All non-hydrogen atoms were refined anisotropically. The methanol hydrogen atoms were located from difference Fourier maps and refined isotropically, with O–H distances restrained to 0.85(1) Å. The remaining hydrogen atoms were placed in calculated positions and included in the last cycles of refinement. Crystal data and details of the data collection and refinement are listed in Table 1. Selected coordinate bond lengths and angles are listed in Table 2.
Supplementary material for structures has been deposited with the Cambridge Crystallographic Data Centre (CCDC nos. 1433352 (I) and 1433318 (II); deposit@ccdc.cam.ac.uk or http://www.ccdc.cam. ac.uk).
Catalytic epoxidation process. A mixture of cyclooctene (2.76 mL, 20 mmol), acetophenone (internal reference) and the complexes as catalysts (0.05 mmol) was stirred and heated up to 80°C before addition of aqueous tert-butyl hydroperoxide (TBHP; 70% w/w, 5.48 mL, 40 mmol). The mixture is initially an emulsion, but two phases become clearly visible as the reaction progresses, a colorless aqueous one and a yellowish organic one. The reaction was monitored for 5 h with withdrawal and analysis of organic phase aliquots (0.1 mL) at required times. Each withdrawn sample was mixed with 2 mL of diethylether, treated with a small quantity of MnO2 and then filtered through silica and analyzed by GC.
RESULTS AND DISCUSSION
The aroylhydrazone Schiff bases and their complexes were synthesized in a facile and essentially identical way (Scheme 1).

Scheme 1 .
The aroylhydrazone Schiff bases act as tridentate dianionic ONO donor ligands toward the MoO22+ core. Both Mo(VI) complexes were obtained from a refluxing mixture of the respective ligand and MoO2(Acac)2 in 1 : 1 molar proportion in methanol. Complexes of the general formula [MoO2LMeOH] were isolated as orange single crystals from the reaction mixture by slow evaporation at room temperature. The complexes are stable in room temperature and are found to be fairly soluble in most of the common organic solvents such as methanol, ethanol, acetonitrile, DMF and DMSO. The low molar solution conductance of the complexes in acetonitrile, indicating the complexes have non-electrolyte behavior.
IR spectra of the aroylhydrazone Schiff bases show bands at about 3230 cm–1 for ν(N–H), 3350–3450 cm–1 for ν(O–H), and 1650 cm–1 for ν(C=O) [18]. The peaks attributed to ν(N–H) and ν(C=O) are absent in the spectra of the complexes as each ligand binds in binegative form losing proton from carbohydrazide group. Strong bands observed at 1605 cm–1 for I and 1607 cm–1 for II are attributed to ν(C=N), which are located at lower frequencies as compared to the free aroylhydrazone Schiff bases, viz. 1610 cm–1 for H2L1 and 1613 cm–1 for H2L2 [19]. The two molybdenum complexes exhibit two characteristic bands at 950 and 861–850 cm–1 for asymmetric and symmetric stretching of cis-\({\text{MoO}}_{{\text{2}}}^{{{\text{2 + }}}}\) core, respectively [20, 21].
Electronic spectra of the complexes recorded in acetonitrile solution display strong and medium absorption bands in the regions 390–440 and 290–320 nm. These peaks are assigned as charge transfer transitions of the type N(pπ)–Mo(dπ) LMCT and O(pπ)–Mo(dπ) LMCT, respectively [22, 23], as the ligand-based orbitals are either N or O donor types. The slight change of λmax values within each set of peaks may be due to the difference of electron donating capacity of the ligands.
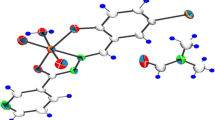
The perspective view of complexes I and II together with the atom numbering scheme are shown in Fig. 1. The coordination geometry around the molybdenum(VI) atom in the complexes reveals a distorted octahedral environment with NO5 chromophore. The aroylhydrazone Schiff base behaves as dianionic tridentate ligand binding through the phenolate oxygen, the enolate oxygen and the imine nitrogen, and occupies three positions in the equatorial plane. One oxo group is located trans to the imine nitrogen in the equatorial plane. The other oxo group and the methanol oxygen are located at the axial positions. The two terminal oxo groups are hence cis to each other and exhibit typical Mo=O double bond distances [24, 25]. The molybdenum is found to be deviated from the mean equatorial plane defined by the four donor atoms by 0.325(1) Å for I and 0.330(1) Å for II. The Mo–Omethanol bond lengths in the complexes are longer than the normal single bond lengths (2.34–2.36 Å against 1.9–2.0 Å). This shows that the methanol molecule is loosely attached to the Mo(VI) center [23]. The remaining Mo–O bond lengths are similar to other molybdenum(VI) complexes [26, 27]. The bond between Mo and the azomethine nitrogen in the complexes are about 2.24 Å which is comparatively longer than the normal Mo–N single bonds. This is due to the trans effect generated by the oxo group trans to the Mo–N bond [23]. The C(8)–O(2) bond lengths in the complexes are 1.30–1.32 Å, which are closer to single bond length rather than C=O double bond length. However, the shorter length compared to C–O single bond may be attributed to extended electron delocalization in the ligands [23]. Similarly shortening of C(8)–N(2) bond length (1.30–1.31 Å instead of normal 1.38 Å) together with the elongation of N(1)–N(2) bond length also supports the electron cloud delocalization in the ligand system [23]. The aroylhydrazone Schiff base ligand forms a five-membered and a six-membered chelate rings with the Mo center. The five-membered metallacycle ring is thus rather planar, but the six-membered metallacycle ring is clearly distorted. The dihedral angle between the two benzene rings is 5.6(3)° for I and 10.5(3)° for II. The trans angles between the donor atom of the methanol ligand and the oxo atom are 171.72(12)° for I and 170.05(11)° for II, indicating considerable distortion of the coordination octahedron around the Mo(VI) center.

ORTEP plots (30% probability level) and numbering scheme for I (a) and II (b).
The crystal packing diagrams of the complexes are quite similar (Fig. 2). The adjacent two complex molecules are linked by two O–H···N hydrogen bonds (I: O(6)–H(6) 0.85(1), H(6)···N(2)i 1.94(1), O(6)···N(2)i 2.782(4) Å, O(6)–H(6)···N(2)i 177(5)°; II: O(6)–H(6) 0.85(1), H(6)···N(2)ii 1.95(1), O(6)···N(2)ii 2.791(4) Å, O(6)–H(6)···N(2)ii 170(5)°; symmetry codes: i 1 – x, 2 – y, –z; ii 1 – x, 1 – y, –z), to form dimer.

Hydrogen bond (dashed lines) linked structure of I (a) and II (b), viewed down the b axis.
After addition of aqueous TBHP at 80°C, the molybdenum complexes dissolve completely in the organic phase. The aqueous phase of the complexes was colorless and the organic one yellowish, indicating that the catalyst is mainly confined in the organic phase. TBHP is mainly transferred into the organic phase under those conditions, and for that reason the reactant and products in the organic layer were analyzed. Cyclooctene and cyclooctene oxide are not significantly soluble in water, therefore the determination of the epoxide selectivity (epoxide formation/cyclooctene conversion) is expected to be accurate. The obtained results are given in Table 3. For the cyclooctene epoxidation by using aqueous TBHP with no extra addition of organic solvents, the present study shows effective activity. Kinetic profiles of the molybdenum complexes as catalysts are presented in Fig. 3. No induction time was observed. The cyclooctene conversion for the molybdenum complexes is high after 5 h, viz. 92% for I and 86% for II.

Kinetic monitoring of cis-cyclooctene epoxidation with TBHP–H2O in the presence of the molybdenum complexes I (⚫), II (◼).
Possible mechanistic consideration involves coordination of TBHP as a neutral molecule with the hydrogen bond O–H···O (Scheme 2). Even though high conversion of cyclooctene is observed, selectivity towards cyclooctene oxide is not high, with 52% for I and 57% for II.

Scheme 2 .
In summary, the present paper reports a very common but facile way to synthesize two new Mo(VI) complexes with aroylhydrazone Schiff bases. In the complexes, the aroylhydrazone Schiff base ligands differ only in the terminal substitute groups of the phenyl rings, viz. Br for I and CF3 for II. The complexes are efficient catalysts for the epoxidation of cyclooctene.
REFERENCES
Heffern, M.C., Reichova, V., Coomes, J.L., et al., Inorg. Chem., 2015, vol. 54, no. 18, p. 9066.
Meghdadi, S., Amirnasr, M., Majedi, M., et al., Inorg. Chim. Acta, 2015, vol. 437, p. 64.
Qu, D., Niu, F., Zhao, X., et al., Bioorg. Med. Chem., 2015, vol. 23, no. 9, p. 1944.
Zafar, H., Ahmad, A., Khan, A.U., et al., J. Mol. Struct., 2015, vol. 1097, p. 129.
Zhao, X., Chen, X., Li, J., et al., Polyhedron, 2015, vol. 97, p. 268.
Mohamed, R.G., Elantabli, F.M., Helal, N.H., et al., Synth. React. Inorg. Met.-Org. Nano-Met. Chem., 2015, vol. 45, no. 12, p. 1839.
Oliveira, T.S.M., Gomes, A.C., Lopes, A.D., et al., Dalton Trans., 2015, vol. 44, no. 31, p. 14139.
Koellner, C.A., Piro, N.A., Kassel, W.S., et al., Inorg. Chem., 2015, vol. 54, no. 15, p. 7139.
Alemohammad, T., Safari, N., Rayati, S., et al., Inorg. Chim. Acta, 2015, vol. 434, p. 198.
Bagherzadeh, M., Ghanbarpour, A., and Khavasi, H.R., Catal. Commun., 2015, vol. 65, p. 72.
Liu, H.Y., Zang, L.Q., and Lv, J.L., Russ. J. Coord. Chem., 2015, vol. 41, no. 7, p. 451. doi 10.1134/S1070328415070052
Morales-delaRosa, S., Campos-Martin, J.M., Terreros, P., et al., Top. Catal., 2015, vol. 58, nos. 4−6, p. 325.
Baskaran, T., Kumaravel, R., Christopher, J., et al., New J. Chem., 2015, vol. 39, no. 5, p. 3758.
Amir, M. and Shikha, K., Eur. J. Med. Chem., 2004, vol. 39, no. 6, p. 535.
Chen, G.J.J., McDonald, J.W., and Newton, W.E., Inorg. Chem., 1976, vol. 15, no. 11, p. 2612.
Sheldrick, G.M., SADABS, Siemens Analytical X-ray Instrument Division, Madison, 1995.
Sheldrick, G.M., SHELXS-97, Program for Solution of Crystal Structures, Göttingen: Univ. of Göttingen, 1997.
Ye, Y.-T., Niu, F., Sun, Y., et al., Chin. J. Inorg. Chem., 2015, vol. 31, no. 5, p. 1019.
You, Z.-L., Xian, D.-M., and Zhang, M., CrystEngComm, 2012, vol. 14, no. 21, p. 7133.
Lal, R.A., Chakrabarty, M., Choudhury, S., et al., J. Coord. Chem., 2010, vol. 63, no. 1, p. 163.
Glowiak, T., Jerzykiewicz, L., Sobczak, J.M., et al., Inorg. Chim. Acta, 2003, vol. 356, p. 387.
Hahn, R. and Kusthardt, U., Inorg. Chim. Acta, 1993, vol. 210, no. 2, p. 177.
Gupta, S., Barik, A.K., Pal, S., et al., Polyhedron, 2007, vol. 26, no. 1, p. 133.
Bagherzadeh, M., Amini, M., Parastar, H., et al., Inorg. Chem. Commun., 2012, vol. 20, p. 86.
Dinda, R., Sengupta, P., Ghosh, S., et al., Dalton Trans., 2002, no. 23, p. 4434.
Dinda, R., Ghosh, S., Falvello, L.R., et al., Polyhedron, 2006, vol. 25, no. 12, p. 2375.
Vrdoljak, V., Prugovecki, B., Matkovic-Calogovic, D., et al., Cryst. Growth Des., 2011, vol. 11, no. 4, p. 1244.
ACKNOWLEDGMENTS
This work was supported by the Collaborative Innovation Plan of Hubei Province for Key Technology of Eco-Ramie Industry.
Author information
Authors and Affiliations
Corresponding author
Additional information
The article is published in the original.
Rights and permissions
About this article

Cite this article
Cui, Y.M., Liu, W.T. & Yan, W.X. Two cis-Dioxomolybdenum(VI) Complexes with Tridentate ONO Aroylhydrazone Schiff Bases: Syntheses, Characterization, Crystal Structures, and Catalytic Epoxidation Property. Russ J Coord Chem 45, 222–229 (2019). https://doi.org/10.1134/S1070328419030023
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1070328419030023