
Abstract
Cis-dioxidomolybdenum(VI) complexes [MoO2L1(DMSO)] (1), [MoO2L2(H2O)]·DMF (2) and [MoO2L3(DMF)] (3) were synthesized by solution based reactions of aroylhydrazones (H2L1 = 5-chloro-2-hydroxybenzaldehyde nicotinoylhydrazone, H2L2 = 2-hydroxy-5-iodobenzaldehyde nicotinoylhydrazone and H2L3 = 3,5-dichloro-2-hydroxybenzaldehyde nicotinoylhydrazone) with bis(acetylacetonato)dioxidomolybdenum(VI) complex, [MoO2(acac)2] in presence of DMSO or DMF. The synthesized aroylhydrazones and their molybdenum complexes were characterized by elemental analysis, spectroscopic techniques (FT-IR, UV–Vis, 1H NMR) and conductivity measurements. Finally the three dimensional structures of the complexes were confirmed by single crystal X-ray diffraction studies. Crystal structures of 1, 2 and 3 were solved by direct methods and refined with full-matrix least-squares calculations using the SHELXS97 and SHELXL2014 software programs respectively. Complex 1 got crystallized in monoclinic space group, P21/c with Z = 4, whereas complexes 2 and 3 in triclinic space group, P\(\overline {1}\) with Z = 2. The ligands, H2L1−3 showed coordination to the metal ion in a dibasic tridentate manner through deprotonated phenolate oxygen, azomethine nitrogen and enolate oxygen.
Graphical Abstract

All the three dioxidomolybdenum(VI) complexes exhibit a distorted octahedral geometry around molybdenum atom. ORTEP plot of [MoO2L2(H2O)]·DMF. (Displacement ellipsoids are drawn at 50% probability)
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Aroylhydrazones are significant class of ONO donor ligands which have played an important role in the development of coordination chemistry of molybdenum. Furthermore the study of oxidomolybdenum complexes coordinated with such tridentate ligands have drawn considerable attention due to their similarity in the active site of majority of molybdoenzymes [1,2,3] as well as for the involvement of molybdenum(VI) compounds as catalyst in several industrial processes, such as epoxidation of olefin [4], olefin metathesis [5] and isomerization of allylic alcohol [6]. In this context, dioxidomolybdenum(VI) complexes have been extremely well investigated [7,8,9,10,11,12,13,14], particularly with respect to the catalytic role of transferase enzymes like nitrate reductase in which their active sites consist of a cis-dioxidomolybdenum moiety [15,16,17]. In recent times, a number of aroylhydrazones obtained by the reactions of aromatic acidhydrazides with o-hydroxycarbonyl compounds have been used in molybdenum chemistry. These type of ligands are of particular interest because the MoO2L or MoOL type of complexes possess one or two “open” coordination sites that can be utilized for substrate binding properties [18,19,20,21] and hence these species have frequently been considered as models for enzymatic reactions and catalytic sites. As a part of our studies on aroylhydrazone complexes [22,23,24], this article reports the synthesis and characterization of three new mononuclear cis-dioxidomolybdenum(VI) complexes, [MoO2L1(DMSO)] (1), [MoO2L2(H2O)]·DMF (2) and [MoO2L3(DMF)] (3) with aroylhydrazones derived from nicotinic acid hydrazide and substituted salicylaldehydes as primary ligands with special reference to their crystal structures. In order to investigate the effect of substituents on the stabilization of the overall crystal structure, salicylaldehydes substituted with different halogens were selected.
Experimental
General Procedures
The starting materials, 5-chloro-2-hydroxybenzaldehyde, 2-hydroxy-5-iodobenzaldehyde, 3,5-dichloro-2-hydroxybenzaldehyde, nicotinic acidhydrazide, bis(acetylacetonato)dioxomolybdenum(VI) complex were supplied by Sigma Aldrich chemical company. Reagent grade solvents were dried and distilled prior to use. All other chemicals were of reagent grade, available commercially and used without further purification.
IR spectra were recorded on a JASCO FT-IR-5300 spectrometer in the 4000–400 cm−1 range using KBr discs. 1H NMR spectra were recorded using a Bruker AMX 400 FT-NMR Spectrometer in DMSO-d6 by using TMS as the reference. UV–Vis spectroscopic data were recorded in DMF using Thermo Scientific Evolution 220 model UV–Vis spectrophotometer in the 200–900 nm range. Molar conductivity measurements were carried out at room temperature on a Systronic model 303 direct reading conductivity meter. Microanalysis studies were done on a Vario EL III CHNS analyzer.
Synthesis
Synthesis of the Aroylhydrazones
The aroylhydrazones were prepared by the condenzation reaction of the nicotinic acid hydrazide with the corresponding substituted 2-hydroxybenzaldehydes in a 1:1 molar ratio (Fig. 1).

Reaction scheme for the synthesis of the aroylhydrazones and their molybdenum complexes
A hot methanol solution (30 mL) of nicotinic acid hydrazide (1 mmol, 0.137 g) and 1 mmol of the corresponding substituted 2-hydroxybenzaldehydes (5-chloro-2-hydroxybenzaldehyde (H2L1), 2-hydroxy-5-iodobenzaldehyde (H2L2) or 3,5-dichloro-2-hydroxybenzaldehyde (H2L3)) were mixed and refluxed for 3 h. The resulting yellow solution was allowed to cool to room temperature. The compounds were separated as pale yellow crystalline solids.
H2L1: Total yield = 0.179 g (65%). Anal. Calc. for C13H10ClN3O2: C, 56.64; H, 3.66; N, 15.24. Found: C, 56.51; H, 3.72; N, 15.10%. IR (νmax/cm− 1):ν(C=O) 1658s; ν(N–H) 3051 m; ν(O–H) 3345 m. 1H NMR (δ ppm): 8.79 (1H, HC=N), 13.32 (1H, N–NH), 9.10 (1H, phenolic OH), 6.96–8.64 (7H, aromatic).
H2L2: Total yield = 0.264 g (72%). Anal. Calc. for C13H10IN3O2: C, 42.53; H, 2.75; N, 11.45. Found: C, 42.57; H, 2.72; N, 11.10%. IR (νmax/cm− 1):ν(C=O) 1659s; ν(N–H) 3045 m; ν(O–H) 3027 m. 1H NMR (δ ppm): 8.78 (1H, HC=N), 12.294 (1H, N–NH), 9.09 (1H, phenolic OH), 6.79–8.78 (7H, aromatic).
H2L3: Total yield = 0.245 g (79%). Anal. Calc. for C13H9Cl2N3O2: C, 50.35; H, 2.92; N, 13.55. Found: C, 50.12; H, 2.86; N, 13.51%. IR (νmax/cm− 1):ν(C=O) 1653s; ν(N–H) 3027 m; ν(O–H) 3399 m. 1H NMR (δ ppm): 8.81 (1H, HC=N), 12.542 (1H, N–NH), 9.10 (1H, phenolic OH), 7.59–8.79 (6H, aromatic).
Synthesis of Dioxidomolybdenum(VI) Complexes
All the dioxidomolybdenum(VI) complexes were prepared by the reaction of [MoO2(acac)2] with the corresponding aroylhydrazones in the presence of donor solvents DMSO and DMF. The complexes were prepared by the following methods.
[MoO2L1(DMSO)] (1)
To a methanolic solution of the aroylhydrazone, H2L1 (1.0 mmol, 0.275 g), methanolic solution of [MoO2(acac)2] (1.0 mmol, 0.326 g) was added. The solution mixture was refluxed with vigorous stirring for half an hour to get an orange colored precipitate. To this solution, few drops of DMSO was added and refluxed for another 3 h. An orange colored solution thus obtained was allowed to stand at room temperature. Orange block shaped crystals were isolated after 3 days. The crystals were separated, washed with methanol and dried in air. Total yield = 0.258 g (54%). Anal. Calc. for C14H12ClMoN5O4S: C, 37.59; H, 2.89; N, 8.69; S, 6.56 Found: C, 37.55; H, 2.94; N, 8.76; S, 6.68%. IR (νmax/cm−1):ν(C=N) 1550; ν(Caro–O) 1422; ν(N–N) 1143; ν(C–O) 1244; ν(Mo=O) 902, 932. 1H NMR (δ ppm): 9.15 (1H, HC=N), 6.96–8.64 (7H, aromatic). Conductivity (10− 3 M, DMF): 4.9 mho cm2 mol− 1.
[MoO2 L2 (H2 O)]·DMF (2)
Methanolic solution of the aroylhydrazone, H2L2 (1.0 mmol, 0.367 g) was added to a methanolic solution of [MoO2(acac)2] (1.0 mmol, 0.326 g) and the resultant mixture was refluxed with vigorous stirring for about half an hour to get clear yellow colored solution. To this solution, few drops of DMF was added and refluxed for another 3 h resulting in a yellow colored solution which was allowed to stand at room temperature. Yellow block shaped crystals were isolated on the following day. The mixture was filtered, washed with methanol and dried in air. Total yield = 0.304 g(52%). Anal. Calc. for C16H17IMoN4O6: C, 32.70; H, 2.86; N, 9.45. Found: C, 32.75; H, 2.81; N, 9.50%. IR (νmax/cm− 1):ν(C=N) 1569; ν(Caro–O) 1412; ν(N–N) 1197; ν(C–O) 1256; ν(Mo=O) 920, 939. 1H NMR (δ ppm): 8.941 (1H, HC=N), 6.79–8.77 (7H, aromatic). Conductivity (10− 3 M, DMF): 5.1 mho cm2 mol− 1.
[MoO2L3(DMF)] (3)
To a methanolic solution of the aroylhydrazone, H2L3 (1.0 mmol, 0.310 g), methanolic solution of [MoO2(acac)2] (1.0 mmol, 0.326 g) was added and refluxed with stirring for half an hour to get clear yellow colored solution. To this solution, few drops of DMF was added and reflux was continued for another 2 h. A yellow colored solution thus obtained was allowed to stand at room temperature. Yellow block shaped crystals were isolated after a week. The crystals were separated, washed with methanol and dried in air. Total yield = 0.300 g (59%). Anal. Calc. for C16H14Cl2MoN4O5: C, 37.79; H, 2.75; N, 11.24. Found: C, 37.75; H, 2.71; N, 11.10%. IR (νmax/cm− 1):ν(C=N) 1580; ν(Caro–O) 1442; ν(N–N) 1198; ν(C–O) 1263; ν(Mo=O) 903, 927. 1H NMR (δ ppm): 8.936 (1H, HC=N), 7.61–8.79 (7H, aromatic). Conductivity (10− 3 M, DMF): 3.7 mho cm2 mol− 1.
Structure Determination Procedures
Single Crystal Data Analysis
Single crystals of the compounds 1–3 with suitable dimensions were selected for X-ray diffraction measurements and mounted on a Bruker SMART APEXII CCD diffractometer, equipped with a graphite crystal, incident-beam monochromator and a fine focus sealed tube with Mo Kα (λ = 0.71073 Å) radiation as the X-ray source. The unit cell dimensions were measured and the data collection was performed. The programs SAINT and XPREP were used for data reduction and APEX2 and SAINT were used for cell refinement [25]. Absorption corrections were carried out using SADABS based on Laue symmetry using equivalent reflections [26]. The structure was solved by using SHELXS-97 direct methods and refined by full matrix least-squares refinement on F2 using SHELXL-2014/7 [27] on a WinGX software package [28]. The molecular and crystal structures were plotted using ORTEP-3 [28] and DIAMOND version 3.2 g [29].
In all the complexes, anisotropic refinements were performed for all non-hydrogen atoms and all H atoms on C atoms were placed in calculated positions, guided by difference maps, with C–H bond distances of 0.93–0.96 Å. H atoms were assigned as Uiso = 1.2 Ueq (1.5 for Me). The hydrogen atoms, H(5A) and H(5B) attached to the oxygen atom of the coordinated water molecule in 2 were located from difference maps and their distances were restrained using DFIX and DANG instructions with distance restraint of O–H = 0.86 ± 0.01 and H⋯H = 1.36 ± 0.02 Å followed by refinement of their displacement parameters.
The sulfur atom of the coordinated DMSO molecule in complex 1 was disordered over two closely positioned sets of sites (C14 and C15) with site occupation factor of about 0.570(3) for the major occupied and 0.430(3) for the minor occupied sites. The hydrogen atoms attached to the aforementioned carbon atoms and their disordered counterparts were fixed using HFIX instructions. The distance restrained to be equal to SADI instructions. The connectivity array was restrained using DELU instructions. Similarly the coordinated DMF molecule of complex 3 was also disordered over its two carbon atoms (C(14) and C(15)) with a site occupation factor of 0.51(4) for the major occupied and 0.49(4) for the minor occupied sites. The reflections (1 0 0) in 1, (0 1 1), (0 1 0) and (1 0 1) in 2 and (0 1 0), (0 0 1) in 3 were omitted owing to bad agreement. The details of the crystal structure and data refinement are given in Table 1.
Hirshfeld Surface Calculations
Hirshfeld surface analysis serves as a powerful tool for gaining additional insight into the various intermolecular interactions of molecular crystals. Hirshfeld surface analyses were carried out and finger print plots were plotted using the software CrystalExplorer 17.5 [30,31,32,33]. Surfaces in 3D (dnorm) that have been mapped over a range of − 0.328 to 1.5807 Å for 1, − 0.682 to 1.3509 Å for 2 and − 0.1624 to 1.2845 Å for 3, shape index (− 1.0 to 1.0 Å) and curvedness (− 4.0 to 0.4 Å). The asphericity (Ω) [34] and globularity values [35] signifying the degree of anisotropy of objects and deviation from spherical surface, respectively are also tabulated in Table 1. The expanded 2D fingerprint plots [36] are displayed in the range of 0.6–2.6 Å viewed with the de and di distance scales displayed on the graph axes where, de and di are the nearest distances to the nuclei from outside and inside the surface to the Hirshfeld surface, respectively.
The red spots over the surface indicate the intercontacts involved in hydrogen bond [37]. The dark-red spots on the dnorm surface arise as a result of the short interatomic contacts, i.e., strong hydrogen bonds, while the other intermolecular interactions appear as light-red spots. The intermolecular and hydrogen bond interactions like H⋯H, O⋯H and C⋯H appear as two distinct spikes of almost equal lengths overlaid on grey color 2D plots of all atoms appear as blue patches over grey color 3D plots.
Results and Discussion
The reaction of [MoO2(acac)2] with three ONO donor aroylhydrazone ligands yielded three new dioxidomolybdenum complexes. The complexes are stable at room temperature and are insoluble in most of the organic solvents, but are readily soluble in DMF and DMSO. Molar conductivity values suggest the non-conducting nature of the complexes.
Spectral Analysis
Infrared Spectra
IR spectral analysis gave an insight into the structure of the compounds by providing valuable information regarding the various functional groups present. The prominent IR bands in the aroylhydrazones and their respective complexes are depicted in Table 2.
The IR spectra of the aroylhydrazones exhibited ν(O–H) bands corresponding to phenolic group in the range 3244–3399 cm− 1 and ν(N–H) bands around 3050 cm− 1. All the aroylhydrazones exhibited a strong band around 1650 cm− 1 indicating the presence of a C=O group. This band confirms the existence of the ligand in the amido form in the solid state [38]. The C–O stretching band was observed around 1275 cm− 1, whereas the ν(N–N) stretching vibrations were found between the region 1130–1185 cm− 1.
In comparison to the IR spectra of aroylhydrazones, the spectra of complexes were devoid of the bands corresponding to the C=O, N–H and O–H, demonstrating the enolization and deprotonation of the hydrazones prior to coordination to the MoO22+ core. This was further confirmed by the shift of azomethine band of acylhydrazone observed in the 1604–1611 cm− 1 range to lower frequency in the spectra of complexes, indicating the coordination of azomethine nitrogen to the metal centre [39]. The increase in the ν(N–N) frequency in complexes, due to the increase in double bond character is another proof for the coordination of aroylhydrazone through the azomethine nitrogen. The ν(C–O) bands present in the ligands are shifted to lower frequency in complexes suggesting the coordination of phenolic oxygen. Similarly the presence of a new band around 1250 cm− 1 in the complexes is assignable to ν(Cenolic–O) stretch. All the molybdenum complexes exhibited two strong bands in the region, 900–950 cm− 1 assigned to symmetric and asymmetric vibrations of cis-MoO22+ core [40].
Electronic Spectra
The electronic spectra of the hydrazones and their complexes were recorded in DMF and the electronic spectral assignments are given in Table 3. The electronic spectra of the hydrazones exhibited three spectral bands in the region 300–410 nm corresponding to the n → π*/π → π* transitions. All these bands appear in the spectra of the complexes with slight variation. In addition, they also exhibit a low intensity band above 400 nm due to charge transfer from phenolate oxygen to an empty orbital of the molybdenum atom (LMCT) [41]. The absence of d-d transition absorption bands in the visible region confirms the 4d0 electronic configuration of molybdenum(VI).
1H NMR Spectra
The 1H NMR spectra of the hydrazones and their molybdenum(VI) complexes were recorded in DMSO. The 1H NMR spectra of the free hydrazones displayed singlets in the δ = 12.29–13.32 ppm range and δ = 9.09–9.10 ppm range due to the iminolic proton (HN-N) and phenolic OH proton (OHphenolic) respectively. Himine–C=N protons resonate at δ = 8.78–8.81 ppm range (Table 4).
The OH and NH proton signals disappeared upon complexation with molybdenum, indicating the deprotonation of the corresponding protons and subsequent coordination of the respective oxygen atoms to the molybdenum centre. The participation of the imine nitrogen in complexation was indicated by an appreciable downfield shift of the azomethine proton signal in the corresponding complexes. All the aromatic proton signals of the hydrazones and their complexes were observed in the expected range of δ = 6.79–8.77 ppm.
Molecular and Crystal Structures of Molybdenum Complexes
Complex 1 got crystallized in monoclinic space group, P21/c whereas complexes 2 and 3 crystallized into triclinic space group, P\(\overline {1}\). The asymmetric units of all the complexes consist of a single molecule. In crystal structure of complex 2, one dimethylformamide molecule was present without any coordination to the metal centre.
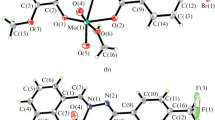
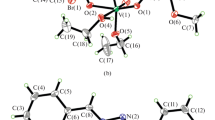
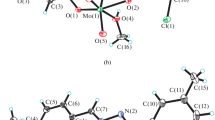
The coordination geometry around molybdenum(VI) in complexes revealed a distorted octahedral environment with an NO5 coordination sphere (Figs. 2, 3, 4) where the dianionic tridentate aroylhydrazone ligands, H2L1–3 coordinate to the molybdenum ion through phenolate oxygen, O(1), azomethine nitrogen, N(1) and enolate oxygen, O(2). The equatorial plane was formed by the aforementioned donor atoms and one of the terminal oxido oxygen atom, O(4) (in the case of complexes 1 and 2) or O(5) (in the case of complex 3). The equatorial plane in all the three complexes are slightly distorted with respect to the planarity (mean deviation from planarity: 0.0252(2) Å for O1/N1/O2/O4 in complex 1, 0.0283(4) Å for O1/N1/O2/O4 in complex 2 and 0.0263(2) Å for O1/N1/O2/O5 in complex 3). Finally the oxygen atom of a neutral monodentate solvent molecule, DMSO (in 1) or H2O (in 2) or DMF (in 3) completes the coordination sphere of the distorted octahedron. The oxygen atoms of these coordinated solvent molecules and the other oxido oxygen atom, O(3) (in the case of complexes 1 and 2) and O(4) (in the case of complex 3) occupies the axial positions. Moreover the axial positions defined by these donor atoms forms O(3)–Mo(1)–O(5) bond angle of 169.51(2)° and 170.95(14)° for complexes 1 and 2 respectively and O(3)–Mo(1)–O(4) bond angle of 169.8(9)° for complex 3. The cis configuration acquired by the MoO22+ moiety can be confirmed from the O(3)–Mo(1)–O(4) bond angle of 105.2(2)° for complex 1, O(4)–Mo(1)–O(3) bond angle of 106.3(2)° for complex 2 and O(4)–Mo(1)–O(5) bond angle of 105.9(1)° for complexes 3. The tridentate hydrazone ligands,[L1−3]2− bind to the molybdenum atom in a meridional fashion forming a five membered and a six membered metallocycle involving the MoO22+ moiety.

ORTEP plot of [MoO2L1(DMSO)] (1) along with atom numbering scheme of the non-hydrogen atoms. Displacement ellipsoids are drawn at 50% probability

ORTEP plot of [MoO2L2(H2O)]·DMF (2) along with atom numbering scheme of the non-hydrogen atoms. Displacement ellipsoids are drawn at 50% probability

ORTEP plot of [MoO2L3(DMF)] (3) along with atom numbering scheme of the non-hydrogen atoms. Displacement ellipsoids are drawn at 50% probability
The hydrazone ligands coordinate to the metal centre in their dianionic iminolate form which is evident from the C(8)–O(2) bond length values (1.353(7) Å for complex 1, 1.306(4) Å for complex 2 and 1.316(3) Å for complex 3) which are closer to the C–O single bond distance of 1.311 Å [42] rather than the reported C=O distance of 1.22(2) for a similar compound [43]. This fact was further substantiated by the C(8)–N(2) bond length values of the complexes [1.293(6) Å for 1, 1.303(5) Å for 2 and 1.290(4) Å for 3]. The Mo–Ooxido bond distances were closer to the already reported Mo=O bond distances in these type of complexes [44]. The longer Mo=O bond (1.700(4) Å vs 1.692(4) Å for complex 1, 1.700(3) Å verses 1.683(3) Å for complex 2 and 1.696(2) Å vs 1.681(2) Å for complex 3) are found to be trans to the neutral monodentate ligands like DMSO, H2O and DMF in complexes 1, 2 and 3 respectively [45].
Another significant observation regarding the crystal structures of these complexes is that, similar to other reported molybdenum complexes [44, 46,47,48,49], the oxygen atom of the respective coordinated neutral monodentate ligands in these complexes are attached to molybdenum metal ion at a greater bond length (2.314(3) Å for complex 1, 2.329(3) Å for complex 2 and 2.340(3) Å for complex 3) compared to the other Mo–O bonds such as enolate and phenolate distances indicating a weak coordination of these ligands to the metal centre which can act as a potential substrate binding site in catalytic reactions. Among the three complexes, complex 3 can serve as an appropriate candidate for this purpose since it has got the weakest (or longest) Mo–O distance.
The six membered metallocycles (Mo1/O1/C1/C6/C7/N1) in the complexes are puckered with puckering amplitudes of Q = 0.138(3) Å, 0.314(3) Å and 0.3446(18) Å and φ = 4(2)°, 23.5(8)° and 30.5(5)° respectively for complexes 1, 2 and 3 respectively. Among the various classical and weak non-classical hydrogen bonding interactions present in these complexes, the most significant one is that exhibited by the molecules of complex 2 (Fig. 5) in which the oxygen atom of the DMF molecule, O(6) present outside the coordination sphere is involved in bifurcated hydrogen bonding interaction with the hydrogen atom of the coordinated water molecule, H(5B) and the aldehydic proton, H(7). This hydrogen bonding interactions connect two independent molecules along with the solvent, DMF. The crystal packing of the complexes were further stabilized by various types of π-π interactions which are presented in Figs. 4, 6, 7 and 8. The interaction parameters for the hydrogen bonding interactions and π-π interactions are summarized in the Tables 5, 6 and 7.

Various types of hydrogen bonding interactions in the molecules of [MoO2L2(H2O)]·DMF (2)

Diagram showing various hydrogen bonding and π–π interactions in the complex [MoO2L1(DMSO)] (1)

π–π interactions present in the molecules of [MoO2L2(H2O)]·DMF (2)

π–π interactions present in the molecules of [MoO2L3(DMF)] (3)
Hirshfeld Surface Analysis
Hirshfeld surfaces 2D fingerprint, 3D dnorm, shape index, curvedness were plotted using Crystal Explorer Version 17.5 [30,31,32,33]. 2D fingerprint plots portray the molecular hydrogen bonds in the crystals as two distinct spikes of almost equal lengths (Fig. 10). Bright red shaded area in the dnorm and in shape index (Fig. 9) represents the major hydrogen bonding interactions like C–H⋯O and C–H⋯N in the crystals and the contributions made by individual molecular contacts to the 2D-fingerprint plots are as follows. C⋯H (10.9% for 1, 5.5% for 2 and 6.9% for 3) exhibits the maximum and C⋯O (1.2% for 1, 2.1% for 2 and 2.6% for 3) exhibits the minimum 2D surfaces generated in the carbon and other elements interactions of a molecule (Fig. 10).

Hirshfeld surfaces for 1, 2 and 3 mapped with a dnorm over a fixed color scale of − 0.16 (red) to 1.58 Å (blue) b shape index and c curvedness. ‘Bow-tie’ pattern (marked by black circles) on the HS’s indicating the relative percentage of π⋯π interactions. (Color figure online)

2D finger print plots with de and di ranging from 0.6 to 2.6 Å for 1–3 and their major decomposition plots
Similarly the hydrogen and other elements contribution H⋯H (31.0% for 1, 34.1% for 2 and 27.6% for 3), O⋯H (23.4% for 1, 25.1% for 2 and 20.4% for 3) and N⋯H (5.7% for 1, 4.9% for 2 and 5.7% for 3) 2D surfaces are observed. Among the various interaction between the same elements, maximum contribution was made by C⋯C (6.8% for 1, 6.7% for 2 and 4.7% for 3) on comparison to all atoms 2D surface of the molecule. These interactions appear as bow-tie patterns in the shape index (Fig. 9). The Hirshfeld surface analysis was also helpful in visualizing the prominent hydrogen bonding interaction present in the crystal structure of complex 2 which is portrayed in Fig. 11.

a Classical bifurcated hydrogen bonding interaction present in 2 involving oxygen atom (O6) of DMF molecule and hydrogen atom of coordinated water (H5B) and aldehydic proton (H7) b viewing the same short contact in Hirshfeld surface with external molecules
The asphericity and globularity values obtained for these complexes (Table 8) indicate that they deviate from symmetry and spherical surface to some extent.
Conclusion
Three new dioxidomolybdenum(VI) complexes of three different aroylhydrazones derived from nicotinic acid hydrazide were successfully synthesized and spectrally characterized (FT-IR, UV–Vis, 1H NMR). The crystal structures of the synthesized compounds were confirmed from single crystal X-ray diffraction methods. All the three complexes hold a labile sixth coordination site that can be utilized for substrate binding.
Supplementary Materials
CCDC numbers 1854447-1854449 contain the supplementary crystallographic data for this paper. These data can be obtained, free of charge, at http://www.ccdc.cam.ac.uk/conts/retrieving.html, or from the Cambridge Crystallographic Data Centre (CCDC) at 12 Union Road, Cambridge CB2 1EZ, UK. Fax: +44(0) 1223 336 033 and e-mail: deposit@ccdc.cam.ac.uk.
References
Rajan OA, Chakravorty A (1981) Inorg Chem 20:660
Abu-Omar MM, Loaiza A, Hontzeas N (2005) Chem Rev 105:2227
Topich J (1981) Inorg Chem 20:3704
Judmaier ME, Holzer C, Volpe M, Mosch-Zanetti NC (2012) Inorg Chem 51:9956
Maksimowski P, Skupinski W (1991) J Mol Catal 65:187
Gusina L, Bulhac I, Dragancea D, Sinonov YA, Shova S (2011) Rev Roum Chim 56:133
Shylesh SP, Jia M, Seifert A, Adappa S, Ernst S, Thiel WR (2009) New J Chem 33:717
Gago S, Neves P, Monteiro B, Pessego M, Lopes AD, Valente AA, Paz FAA, Pillinger M, Moreira J, Silva CM, Gonçalves IS (2009) Eur J Inorg Chem 4528
Neves P, Gago S, Pereira CCL, Figueiredo S, Lemos A, Lopes AD, Goncalves IS, Pillinger M, Silva CM, Valente AA (2009) Catal Lett 132:94
Chai FL, Su HL, Wang XY, Tao JC (2009) Inorg Chim Acta 362:3840
Bagherzadeh M, Tahsini L, Latifi R, Woo LK (2009) Inorg Chim Acta 362:3698
Alonso JC, Neves P, da Silva MJPD, Quintal S, Vaz PD, Silva C, Valente AA, Ferreira P, Calhorda MJ, Felix V, Drew MGB (2007) Organometallics 26:5548
Maiti SK, Dinda S, Gharah N, Bhattacharyya R (2006) New J Chem 30:479
Maiti SK, Banerjee S, Mukherjee AK, Malik KMA, Bhattacharyya R (2005) New J Chem 29:554
Iran S, Abdolreza R, Niaz M, Samira K (2009) Polyhedron 28:733
Sigel A, Sigel H (2002) Met Ions Biol Syst 39:856
Hille R (1996) Chem Rev 96:2757
Dinda R, Sengupta P, Ghosh S, Sheldrick WS (2003) Eur J Inorg Chem 363
Seena EB, Kurup MRP (2007) Polyhedron 26:3595
Purohit S, Koley AP, Prasad LS, Manoharan PT, Ghosh S (1989) Inorg Chem 283:735
Dinda R, Ghosh S, Falvello LR, Tomas M, Mak TCW (2006) Polyhedron 25:2375
Mathew N, Kurup MRP (2011) Spectrochim Acta Part A 78:1424
Sudheer R, Sithambareshan M, Sajitha NR, Manoj E, Kurup MRP (2005) ActaCryst. E71:702
Aravindakshan AA, Joseph B, Kala UL, Kurup MRP (2017) Polyhedron 123:206
SMART and SAINT, Area Detector Software Package and SAX Area Detector Integration Program, Bruker Analytical X-ray, Madison, 1997
SADABS, Area Detector Absorption Correction Program; Bruker Analytical Xray; Madison, 1997
Sheldrick GM (2015) Acta Crystallogr C 71:3
Farrugia LJ (2012) J Appl Crystallogr 45:849
Brandenburg K (2011) Diamond version 3.2 g. Crystal Impact GbRBonn, Germany
Wang A, Craven BM (1979) J Pharm Sci 68:361
Spackman MA, Byrom PG (1997) Chem Phys Lett 267:215
Spackman MA, Jayatilaka D (2009) CrystEngComm 11:19
Seth SK, Sarkar D, Jana AD, Kar T (2011) Cryst Growth Des 11:4837
Meyer AY (1986) Chem Soc Rev 15:449
Devi PP, Kalaivania D (2016) Acta Cryst. E72:570
Wolff SK, Grimwood DJ, McKinnon JJ, Jayatilaka D, Spackman MA (2012) Crystal Explorer, Version 3.1 University of Western Australia, Perth
Kumara K, Shivalingegowda N, Mahadevaswamy LD, Kariyappa AK, Lokanath NK (2017) Chem Data Collect 9:251
Kuriakose M, Kurup MRP, Suresh E (2007) Spectrochim Acta Part A 66:353
Mangalam NA, Sivakumar S, Sheeja SR, Kurup MRP, Tiekink ERT (2009) Inorg Chim Acta 362:4191
Nakamoto K (1997) Infrared and Raman spectra of inorganic and coordination compounds, 5th edn. Wiley, New York
Maurya MR, Khurana S, Schulzke C, Rehder D (2001) Eur J Inorg Chem 779–788
Pasayat S, Dash SP, Saswati, Majhi PK, Patil YP, Nethaji M, Dash HR, Das S, Dinda R (2012) Polyhedron 38:198
Bakir M, Brown O (2002) J Mol Struct 609:129
Ebrahimipour SY, Khabazadeh H, Castro J, Sheikhshoaie I, Crochet A, Fromm KM (2015) Inorg Chim Acta 427:52
Ding SB, Li WH (2013) J Coord Chem 66:2023
Asha TM, Kurup MRP (2018) Inorg Chim Acta 483:44
Ngan NK, Lo KM, Wong CSR (2011) Polyhedron 30:2922
Ngan NK, Lo KM, Wong CSR (2012) Polyhedron 33:235
Shoeili ZM, Zare M, Bagherzadeh M, Kubicki M, Davar MB (2015) J Coord Chem 68:548
Acknowledgements
The authors thank the Cochin University of Science and Technology, Kochi, India for financial assistance and thankful to the Sophisticated Analytical Instrumentation Facility, Cochin University of Science and Technology, Kochi, India for elemental ananlysis, 1H NMR and Single crystal X-ray diffraction measurements.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Asha, T.M., Kurup, M.R.P. Synthesis, Spectral Characterization and Crystal Structures of Dioxidomolybdenum(VI) Complexes Derived from Nicotinoylhydrazones. J Chem Crystallogr 49, 219–231 (2019). https://doi.org/10.1007/s10870-018-0756-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10870-018-0756-9