
Abstract
Metal-organic 1D coordination polymers of Zn(II) and Cd(II), [{Zn(3-Bphz)(H2O)4}(3-Bphz)(NO3)2]n (I), [Zn(3-Bphz)I2]n (II), [Cd(3-Bphz)I2]n (III), [Cd(4-Bphz)(CH3COO)2(H2O)]n (IV), and [Cd(4-Bphz)(NO3)2(H2O)2]n (V), containing azines of the N,N' type, 1,2-bis(pyridin-3-ylmethylene)hydrazine (3-Bphz) and 1,2-bis(pyridin-4-ylmethylene)hydrazine (4-Bphz), as bridging ligands are synthesized. The compositions and structures of the compounds are confirmed by the data of elemental analysis, IR and NMR spectroscopy, and single-crystal X-ray diffraction analysis (CIF files CCDC nos. 1812634–1812638 for I–V). Coordination polymers I–III have zigzag structures. The octahedral environment of the Zn2+ ion in compound I is formed by two 3-Bphz ligands and four water molecules. The external sphere contains nitrate anions and uncoordinated 3-Bphz molecules. In isomorphous compounds II and III, the tetrahedral environment of the metal is formed by two nitrogen atoms of two bridging 3-Bphz ligands and two iodine atoms. Coordination polymers IV and V are linear. The coordination polyhedron of the Cd2+ ion in compound IV is a pentagonal bipyramid, two vertices of which are occupied by the nitrogen atoms of two 4-Bphz molecules, and the equatorial plane is formed by two bidentate-chelating acetate anions and one water molecule. In compound V, the octahedral environment of the Сd2+ ion is formed by two molecules of the 4-Bphz ligand, two monodentate nitrate anions, and two water molecules. All complexes are weak luminophores emitting in the blue-green spectral range.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
INTRODUCTION
Coordination polymers [1, 2] including 3D coordination frameworks [3] are among the most important representatives of polynuclear complexes and remain in the center of attention of the modern coordination chemistry. One of the most efficient synthetic approaches has previously been applied by us and other researchers in order to obtain discrete binuclear coordination compounds and coordination polymers. The approach is based on joining building blocks by bridging ligands such as inorganic anions [4] and nitrogen-containing bipyridine [5–7] or dicarboxyl ligands [8, 9]. Owing to the stability and polydenticity, the N-donor ligands containing two and more functional nitrogen atoms in one molecule are widely used for assembling coordination polymers of various dimensionality [10–16]. The latter have potentially useful properties including catalysis, optical activity, conductance, luminescence, magnetism, and porosity [17–31]. The molecular structure and denticity of organic ligands, the electron configuration and coordination capacity of the ion of the metal-complexing agent, the solvent nature, and the metal to ligand molar ratio should be mentioned among the most significant factors affecting the dimensionality of the coordination network and topology of the coordination polymer [23–35].
Along with the 3D coordination frameworks [3], the low-dimensional (1D and 2D) coordination polymers as objects of the study by materials science and crystal engineering [36, 37] demonstrate examples of molecular ferromagnets, synthetic conductors, nonlinear optical materials, and segnetoelectrics, whose crystal structures and useful properties can be varied by fitting the components of their molecules [38, 39]. The examples of coordination polymers containing azines of the N,N' type, 1,2-bis(pyridin-3-ylmethylene)hydrazine (3-Bphz) and 1,2-bis(pyridin-4-ylmethylene)hydrazine (4-Bphz), as bridging ligands are published [14, 17, 21–28]. These long aromatic molecules with the conjugated system of alternating С=N and N–N bonds can coordinate transition metal ions by the nitrogen atoms of both terminal pyridine and azine fragments [40] to form new coordination polymers and can participate in hydrogen bonds in the presence of strong proton–donor agents [41]. The luminescence properties of the coordination polymers of Zn(II) and Cd(II) with these ligands were mentioned [17, 42, 43] together with their diverse architecture, and the structure–property correlation with allowance for fine packing effects was demonstrated for a number of the Cd(II) compounds with 4-Bphz [42].
Continuing our studies of the coordination polymers of Zn and Cd with the luminescence properties [4, 44–46], we synthesized two isomeric azine ligands 3-Bphz and 4-Bphz using somewhat modified procedure compared to the published one [26] and also synthesized four new 1D coordination polymers with these ligands: [{Zn(3-Bphz)(H2O)4}(3-Bphz)-(NO3)2]n (I), [Cd(3-Bphz)I2]n (II), [Zn(3-Bphz)I2]n (III), and [Cd(4-Bphz)(CH3COO)2(H2O)]n (IV). The structures and emission properties of the latter were studied. The X-ray diffraction results for 1D coordination polymer [Cd(4-Bphz)(NO3)2(H2O)2]n (V), which was synthesized using a similar procedure, were improved compared to the earlier published data [17] and deposited with the Cambridge Crystallographic Data Centre (CIF file CCDC no. 1812638).
EXPERIMENTAL
Synthesis of 3-Bphz. A weighed sample of hydrazine sulfate (6.5 g, 0.05 mol) was dissolved in water (250 mL). The obtained solution was heated to 70°C on a magnetic stirrer. 3-Pyridinecarboxaldehyde (9.4 mL, 0.1 mol) was added to the solution. Then a solution of NaOH was added to neutral pH, and the mixture was stirred for ~2 h. The obtained mixture was filtered, washed with water to remove sodium sulfate, and dissolved in ethanol (50 mL). The solution was left to stay for slow evaporation at room temperature for crystallization. The yield was ~70%.
IR (ν, cm–1): 3087, 3038, 3012, 1625, 1586, 1571, 1511.
Synthesis of 4-Bphz was carried out using a similar procedure as that for 3-Bphz, but 3-pyridinecarbaoxaldehyde was replaced by 4-pyridinecarboxaldehyde. The yield was ~70%.
IR (ν, cm–1): 3072, 3054, 3038, 3030, 3018, 1628, 1594, 1551, 1496.
Synthesis of complex I. A weighed sample of Zn(NO3)2 · 6H2O (0.03 g, 0.1 mmol) was dissolved with stirring in methanol (10 mL). After dissolution, 3-Bphz (0.042 g, 0.2 mmol) was added. The mixture was heated at 70°C for 5 min on a magnetic stirrer, and DMF (1 mL) was added. The obtained solution was filtered and left to stay for slow evaporation at room temperature. Yellow crystals suitable for X-ray diffraction analysis precipitated from the solution in 2 weeks. The yield was ~35%. The substance is soluble in alcohols, DMF, DMSO, and water.
For C36H38N14O10Zn | |||
Anal. calcd., % | C, 48.27 | H, 4.12 | N, 21.89 |
Found, % | C, 48.43 | H, 4.26 | N, 21.97 |
IR (ν, cm–1): 3085, 3039, 3021, 1627, 1595, 1578, 1412, 1395, 1227, 1223, 1033, 1029.
Synthesis of complex II. A weighed sample of ZnI2 (0.031 g, 0.1 mmol) was dissolved in DMF (6 mL) followed by the addition of 3-Bphz (0.021 g, 0.1 mmol). The solution was stirred at 70°С for 10 min after which was filtered and left to stay at room temperature for crystallization. Yellow crystals precipitated from the solution in 3 weeks. They were filtered, washed with ethanol, and dried at room temperature. The yield was ~23%. The substance is insoluble in water and alcohols and soluble in DMF and DMSO.
For C12H10N4I2Zn | |||
Anal. calcd., % | C, 26.97 | H, 1.82 | N, 10.34 |
Found, % | C, 27.22 | H, 1.90 | N, 10.58 |
IR (ν, cm–1): 3080, 3026, 1630, 1600, 1580.
Synthesis of complex III. A weighed sample of CdI2 (0.036 g, 0.1 mmol) was dissolved in water (4 mL). The solution was transferred to a tube, and a water–ethanol (1 : 1) mixture (2 mL) was slowly added with a pipette. Then an ethanol solution (4 mL) containing 3-Bphz (0.021 g, 0.1 mmol) was added. The tube was closed and left to stay at room temperature. Yellow crystals were formed in the solution in a week. They were filtered and dried at room temperature. The yield was ~30%. The substance is soluble in DMF and DMSO and insoluble in water and alcohols.
For C12H10N4I2Cd | |||
Anal. calcd., % | C, 24.83 | H, 1.41 | N, 9.47 |
Found, % | C, 25.00 | H, 1.74 | N, 9.71 |
IR (ν, cm–1): 3083, 3024, 1629, 1597, 1578.
Synthesis of complex IV. A weighed sample of Cd(CH3COO)2 ∙ 2H2O (0.026 g, 0.1 mmol) was dissolved in ethanol (10 mL) with stirring (70°C). The solution was added by 4-Bphz (0.021 g, 0.1 mmol), which was accompanied by precipitation. After water (2 mL) was added, the precipitate dissolved and the solution turned transparent. The obtained solution was filtered and left to stay for slow evaporation at room temperature. Yellow crystals suitable for X-ray diffraction analysis precipitated from the solution in a week. The yield was ~42%. The substance is soluble in DMF and DMSO and insoluble in alcohols.
For C16H18N4O5Cd | |||
Anal. calcd., % | C, 41.73 | H, 3.69 | N, 11.94 |
Found, % | C, 41.89 | H, 3.95 | N, 12.21 |
IR (ν, cm–1): 3088, 3035, 1630, 1605, 1551, 1485, 1410, 681, 679.
Synthesis of complex V. A weighed sample of Cd(NO3)2 ∙ 6H2O (0.035 g, 0.1 mmol) was dissolved in methanol (10 mL) with stirring (70°C). The solution was added by 4-Bphz (0.021 g, 0.1 mmol), which was accompanied by precipitation. After DMF (3 mL) was added, the precipitate dissolved and the solution turned transparent. The transparent solution was filtered and left to stay for slow evaporation at room temperature. Yellow crystals suitable for X-ray diffraction analysis precipitated from the solution in 3 weeks. The yield was ~37%. The substance is soluble in DMF and DMSO and insoluble in alcohols.
For C12H14CdN6O8 | |||
Anal. calcd., % | C, 23.92 | H, 2.11 | N, 13.87 |
Found, % | C, 24.21 | H, 2.37 | N, 14.12 |
IR (ν, cm–1): 3099, 3043, 1652, 1611, 1558, 1423, 1386, 1342, 1304, 1021.
The compositions and structures of the compounds were determined from the data of elemental analysis and IR and NMR spectroscopy. IR spectra were recorded on a FT-IR Perkin-Elmer Spectrum100 spectrometer in Nujol in a range of 4000–400 cm–1, and the spectra in the attenuated total reflectance (ATR) mode were measured in a range of 4000–650 cm–1. NMR spectra were detected on a Bruker Avance III spectrometer. The crystal structures of compounds I–V were determined by single-crystal X-ray diffraction analysis. Solid state luminescence spectra were recorded using a pulse nitrogen laser (λexc = 337.1 nm) at 300°С. The excitation pulse duration was 15 ns, the repetition frequency was 50 Hz, and the pulse energy was 0.2 mJ. The emission was detected with a FEU-79 instrument (multialkaline photocathode Sb(Na2K) with the adsorbed cesium layer on the surface, characteristic of the S20 type). The intrinsic time of the detecting system was 20 ns. The afterglow duration (at 300°С) for all studied compounds was shorter than the time resolution of the detecting system.
X-ray diffraction analysis. The structural data for compounds I–V were obtained at room temperature on an Xcalibur ССD Oxford Diffraction diffractometer (MoKα radiation, λ = 0.71073 Å, graphite monochromator, ω scan mode). The unit cell parameters were refined over the whole set of experimental data. The crystal structures were solved by direct methods and refined by least squares in the anisotropic full-matrix approximation for non-hydrogen atoms (SHELX-97) [47]. The positions of the hydrogen atoms of the water molecules were revealed from difference Fourier syntheses, and positions of other hydrogen atoms were calculated geometrically and refined isotropically in the rigid body model. The set of experimental diffraction data for compound II was obtained from the nonmerohedral two-component twin crystal. Twinning resolution was performed in the CrysAlisPro program package, the twin matrix was [0.9992 0.0148 0.0196–0.0255 0.9987 0.0093–0.0668–0.0162 1.0011], and the block ratio was 0.524 : 0.476. The refinement was conducted for the F 2 set of structural data in the SHELX HKLF 5 format. The crystallographic data and experimental characteristics for compounds I–IV are presented in Table 1. Selected interatomic distances and bond angles are given in Table 2. The positional and thermal parameters for the structures of compounds I–V were deposited with the Cambridge Crystallographic Data Centre (CIF files CCDC nos. 1812634–1812638; deposit@ ccdc.cam.ac.uk or http://www.ccdc.cam.ac.uk).
RESULTS AND DISCUSSION
Ligands 3-Bphz and 4-Bphz were synthesized by the condensation of the corresponding pyridinecarboxaldehyde with hydrazine sulfate (Scheme 1). The structures of the obtained ligands were confirmed by the data of IR and NMR spectroscopy.

Scheme 1.
The IR spectra of the 3-Bphz and 4-Bphz ligands contain bands at 1625.8 and 1628.3 cm–1 corresponding to the ν(C=N)ar vibrations. The spectrum of complex I exhibits the bands characteristic of nitrate anions: 1412, 1395 νas(NO2); 1227, 1223 νs(NO2); and 1029, 1033 ν(NO) cm–1. The bands caused by the bound ν(OH) group are observed at 3500–2800 cm–1, and there are bands at 3021, 3039, 3085 ν(CH); 1627 ν(C=N)ar; and 1595, 1578 ν(C=C)ar cm–1. The IR spectra of complexes II and III exhibit the following absorption bands (cm–1): 1629 and 1630 ν(C=N)ar; 1597, 1578, and 1600, 1580 ν(C=C)ar; and 3024, 3083 and 3026, 3080 ν(CH), respectively. The acetate ions in the spectrum of complex IV are characterized by the absorption bands at 1605, 1551 νas(C–O); 1410, 1485 νs(C–O); and 681, 679 ν(O–C–O) cm–1. The bands at 3400–3000 cm–1 can be assigned to the ν(OH) vibrations of the bound group. The bands at 3035, 3088 ν(CH); 1630 ν(C=N)ar; and 1604 ν(C=C)ar cm–1 are attributed to the bridging ligand. The IR spectrum of compound V contains the following bands (cm–1): 3500–2800 ν(OH) (bound group); 3043, 3099 ν(CH); 1652 ν(C=N)ar; 1611, 1558 ν(C=C)ar; 1423, 1386 νas(NO2); 1342, 1304 νs(NO2); and 1021 ν(NO). These values confirm the monodentate coordination mode of the nitrate anion.
The different point symmetries of the ligands (Ci for 3-Bphz and Cs for 4-Bphz) determine the differences in their 1H and 13С NMR spectra. The 3-Bphz ligand is characterized by five signals corresponding to five types of nonequivalent hydrogen atoms: 8.79, 9.02, 8.70, and 8.27 ppm. Only three signals are detected in the 1H NMR spectrum of the 4-Bphz ligand: 8.67, 7.80, and 8.73 ppm. The 13C NMR spectrum of the 3-Bphz ligand exhibits six characteristic signals for six types of nonequivalent carbon atoms: 160.1, 129.9, 124.6, 124.6, 135.3, and 150 ppm. The 4-Bphz ligand is characterized by four signals: 160, 140.8, 122.5, and 150.9 ppm. The 1H and 13С NMR signals of 3-Bphz and 4-Bphz are presented in Scheme 2.

Scheme 2.
The spectrum of compound IV contains five proton signals, three of which are assigned to the 4-Bphz ligand, and no significant shifts are observed. One signal is attributed to the CH3 group of the acetate anion (1.83 ppm). The 1H NMR signal of the water molecule coordinated to the metal coincides with the signal of water in DMSO-d (3.4 ppm). The 13C NMR spectrum exhibits six carbon signals: four signals at the same values as those in the spectrum of the 4-Bphz ligand and two characteristic signals for the carbon atoms of the acetate anion: δ(CH3) 22.13 and δ(COО–) 178.39 ppm.
No substantial shifts of the signals compared to the values of the 3-Bphz and 4-Bphz ligands were observed for the 1H and 13C NMR spectra of compounds I–III and V.
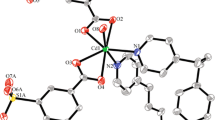
Compound I contains the out-of-sphere nitrate anions and solvate 3-Bphz molecules along with the positively charged polymer chain [Zn(3-Bphz)(H2O)4\(]_{n}^{{2 + }}\), the fragment of which is shown in Fig. 1. The zinc atom and coordinated 3-Bphz ligand occupy positions in the inversion centers. The coordination polyhedron of the metal atom is a distorted octahedron formed by a set of donor atoms (N2О4), which is formed by four water molecules determining the equatorial plane and two 3-Bphz ligands almost perpendicular to this plane, and the dihedral angle between the corresponding planes is 87.46°. The distances are Zn–N 2.208(2), Zn–О 2.036(2), and 2.097(2) Å (Table 2). In the polymer chain, the distance between the zinc atoms separated by the bidentate-bridging 3-Bphz ligand is equal to 14.44 Å.

Coordination environment of the zinc atom in compound I with the numeration of the basis atoms.
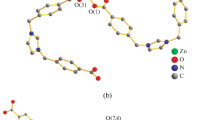
Unlike the centrosymmetric planar ligand coordinated to the metal, the out-of-sphere 3-Bphz molecule (in the general position) is characterized by the twist conformation with the dihedral angle between the pyridine fragments equal to 38.81°. This is due to the supramolecular architecture of the crystal formed by the hydrogen bonds ОН···N involving the coordinated water molecules and out-of-sphere ligands. The 3-Bphz ligands are added to the cationic polymer chain via the chelate mode by two hydrogen bonds: O(4)–H(4A)···N(3) (x + 1, y, z) (O···N 2.713(3), H···O 1.87(2) Å, angle OHN 172(3)°) and O(4)–H(4B)···N(6) (x, y, z – 1) (O···N 2.733(3), H···O 1.89(2) Å, angle ОHN 176(3)°) (Fig. 2a). In turn, the out-of-sphere nitrate anions serve as bridges between the adjacent chains due to the О–Н···О hydrogen bonds with the water molecules coordinated to the metal: O(5)–H(5A)···O(2) (О···О 2.840(3), H···O 2.01(2) Å, angle OHO 166(3)°), O(5)–H(5A)···O(3) (О···О 3.218(4), H···O 2.54(3) Å, angle OHO 138(3)°), and O(5)–H(5B)···O(2) (1 – x, 2 – y, 1 – z) (О···О 3.102(4), H···O 2.47(3) Å, angle OHO 132(3)°) to form the supramolecular 2D layer (Fig. 2b).

Packing fragments in compound I: (a) cationic coordination chains surrounded by the out-of-sphere ligands 3-Bphz and (b) joining of the coordination chains into the supramolecular layer by the out-of-sphere nitrate anions; nonfunctional hydrogen atoms are omitted.
The mononuclear complex [Zn(NO3)2(H2O)2(3-Bphz)2] [48] and 1D coordination polymer {[Zn(NO3)2(H2O)(3-Bphz)] . H2O}n [49] can be mentioned among the closest compositional and structural analogs of compound I. In both cases, the azine ligand occupies two coordination sites in the octahedral polyhedron of the metal, whereas four other positions are occupied by the water molecules and nitrate anions in the 2 : 2 and 1 : 2 ratio in the first and second cases, respectively.
The isomorphous crystals of compounds II and III differ by the metal cation only (Fig. 3). All components occupy partial positions: the metal atom and iodide anions lie on the m plane, and the 3-Bphz ligand is localized in the inversion center. The tetrahedral configuration of the metal atom N2I2 is formed by two iodide anions and two 3-Bphz ligands (Zn–I 2.544(2) and 2.557(3), Zn–N 2.086(9) Å, and the corresponding bond angles range from 106.2(3)° to 123.79(8)°). The corresponding distances in complex III are insignificantly elongated because of a larger radius of the Cd atom compared to Zn (Table 2). The dihedral angle between the planar coordinated ligands is equal to 72.09° in complex II and 76.26° in complex III.

Coordination environment of the zinc atom in compound II with the numeration of the basis atoms.
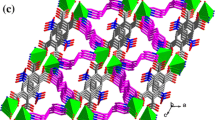
The zigzag coordination chains are parallel to the crystallographic axis b in the crystals of compounds II and III (Fig. 4), and the distances between the metal atoms separated by the bidentate-bridging ligand 3‑Bphz are 14.217 and 14.622 Å in compounds II and III, respectively. The antiparallel chains are packed along the crystallographic axis а with a substantial overlapping of the pyridine cycles of the ligand, which is indicated by the distances between the centroids of these cycles equal to 3.497 and 3.483 Å in complexes II and III, respectively.

Packing fragments in compounds II and III.
The crystals of compounds II and III are isomorphous to the earlier studied Hg(II) compounds, [HgI2(3-Bphz)2]n and [HgBr2(3-Bphz)2]n [49, 50].
The structure of compound IV is the 1D chain. The cadmium atom and water molecule occupy positions on the twofold axis passing also through the middle of the N–N bond of the 4-Bphz ligand. The coordination polyhedron of the cadmium atom is a pentagonal bipyramid (Fig. 5) in which two axial positions are occupied by the 4-Bphz ligands and the water molecule and two acetate anions coordinated via the bidentate-chelating mode lie in the equatorial plane. The distances are Cd–N 2.413(2) and Cd–O 2.332(3)–2.439(2) Å (Table 2).The symmetry of the twofold axis of the 4-Bphz ligand determines its twist conformation with the dihedral angle between the pyridine cycles equal to 71.64°.

Coordination environment of the cadmium atom in compound IV with the numeration of the basis atoms.
In the crystal of compound IV, the linear coordination chains are parallel to the direction [110] (Fig. 6), and the distance between the metal atoms separated by the bidentate-bridging 4-Bphz ligand is equal to 15.94 Å. The chains are associated in the crystals by hydrogen bonds O–H···O involving the water molecules and acetate anions (O(3)–H(3A)···O(2) (x + 1/2, y, 1 – z) (О···О 2.681(3), H···O 1.89 Å, angle OHO 150.9°), due to which the supramolecular 2D layer parallel to the crystallographic plane ab is formed. Only van der Waals interactions occur between the layers in the crystal of compound IV.

Packing of coordination chains in the crystal of compound IV; nonfunctional hydrogen atoms are omitted.
The closest compositional analog of compound IV is the 1D coordination polymer of the ladder type, [Zn2(4-Bphz)2(CH3COO)4. 2(MeOH)2]n, in which the bipyridine ligand and one of the acetate ligands perform the bidentate-bridging function [17].
Although both Schiff bases 3-Bphz and 4-Bphz used in the syntheses of coordination polymers I–V have an extended conjugated system including the aromatic fragments, which is a necessary prerequisite for the detection of the emission properties [51], only restricted data on the compounds based on the 4-Bphz ligand are published [17, 42, 43]. This situation stimulated our studies. The photoluminescence spectra (PL) for the initial 3-Bphz and 4-Bphz ligands (Fig. 7a) and coordination polymers I–V (Figs. 7b, 7c) were detected in the solid state at room temperature. The PL spectra are complicated, indicating a superposition of several radiation processes. All coordination polymers demonstrate the emission intensity from moderate (for I–III, Fig. 7b) to weak (for IV and V, Fig. 7c) in the blue-green spectral range with the emission maxima at 530 (I), 425 (II), 440 (III), 515 (IV), and 520 and 580 nm (V). The emission intensity decreases in the order III > I\( \gg \)II > IV > V. The same profiles of the spectra for compounds II and III are consistent with the identity of the crystal structures of these compounds. As compared to the corresponding ligands, the spectra of compounds I–IV demonstrate the nontrivial for coordination polymers pronounced hypsochromic shift of the emission maxima, which can be explained (by analogy to literature data [42]) by a substantial contribution of the destabilizing π–π-stacking interactions between the pyridine rings of the adjacent ligands. The most probable mechanism of the radiation processes is based on the intraligand π*–π transitions because of the closed d10-electron configurations of the Zn2+ and Cd2+ cations.

PL spectra of (a) ligands 3-Bphz and 4-Bphz, (b) complexes I–III, and (c) complexes IV and V.
REFERENCES
Janiak, C., Dalton Trans., 2003, p. 2781.
Robin, A.Y. and Fromm, K.M., Coord. Chem. Rev., 2006, vol. 250, p. 2127.
Rowsell, J.L.C. and Yaghi, O.M., Microporous Mesoporous Mater., 2004, vol. 73, p. 3.
Croitor, L., Coropceanu, E., Masunov, A., et al., J. Phys. Chem., 2014, vol. 118, p. 9217.
Coropceanu, E., Rija, A., Lozan, V., et al., Cryst. Growth Des., 2016, vol. 16, p. 814.
Croitor, L., Coropceanu, E., Chisca, D., et al., Cryst. Growth Des., 2014, vol. 14, p. 3015.
Coropceanu, E.B., Croitor, L., Wicher, B., et al., Inorg. Chim. Acta, 2009, vol. 362, p. 2151.
Croitor, L., Coropceanu, E., Petuhov, O., et al., Dalton Trans., 2015, vol. 44, p. 7896.
Chisca, D., Croitor, L., Coropceanu, E., et al., Cryst-EngComm, 2016, vol. 18, p. 384.
Chen, C.-L., Kang, B.-S., and Su, C.-Y., Aust. J. Chem., 2006, vol. 59, p. 3.
Robinson, F. and Zaworotko, M.J., Chem. Commun., 1995, p. 2413.
Sailaja, S. and Rajasekharan, M.V., Inorg. Chem., 2000, vol. 39, p. 4586.
Du, M., Bu, X.-H., Huang, Z., et al., Inorg. Chem., 2003, vol. 42, p. 552.
Gao, E.-Q., Cheng, A.-L., Xu, Y.-X., et al., Cryst. Growth Des., 2005, vol. 5, p. 1005.
MacGillivray L.R., Groeneman, R.H., and Atwood, L., J. Am. Chem. Soc., 1998, vol. 120, p. 2676.
Liu, Y.-Y., Yi, L., Ding, B., et al., Inorg. Chem. Commun., 2007, vol. 10, p. 517.
Zhang, G.-Q., Yang, G.-Q., and Ma, J.-S., Cryst. Growth Des., 2006, vol. 6, p. 1897.
Kumar, D.K., Das, A., and Dastidar, P., Cryst. Growth Des., 2006, vol. 6, p. 1903.
Huang, X.-C., Zhang, J.-P., and Chen, X.-M., Cryst. Growth Des., 2006, vol. 6, p. 1194.
Oh, M., Stern, C.L., and Mirkin, C.A., Inorg. Chem., 2005, vol. 44, p. 2647.
Ciurtin, D.M., Dong, Y.-B., Smith, M.D., et al., Inorg. Chem., 2001, vol. 40, p. 2825.
Diskin-Posner, Y., Patra, G.K., and Goldberg, I., Dalton Trans., 2001, p. 2775.
Gao, E.-Q., Cheng, A.-L., Xu, Y.-X., et al., Inorg. Chem., 2005, vol. 44, p. 8822.
Withersby, M.A., Blake, A.J., Champness, N.R., et al., Inorg. Chem., 1999, vol. 38, p. 2259.
Carlucci, L., Ciani, G., and Proserpio, D.M., J. Chem. Soc., Dalton Trans. (1972–1999), 1999, p. 1799.
Dong, Y.-B., Smith, M.D., Layland, R.C., and zur Loye, H.-C., Chem. Mater., 2000, vol. 12, p. 1156.
Yang, W. and Xiang Lin, X., Inorg. Chem., 2009, vol. 48, no. 23, p. 11067.
Granifo, J., Moreno, Y., Garland, M.T., et al., J. Mol. Struct., 2010, vol. 983, p. 76.
Jung, O.-S., Park, S.H., Kim, K.M., and Jang, H.G., Inorg. Chem., 1998, vol. 37, p. 5781.
Zaman, M.B., Smith, M.D., Ciurtin, D.M., and zur Loye, H.-C., Inorg. Chem., 2002, vol. 41, p. 4895.
Chen, C.-L., Goforth, A.M., Smith, M.D., et al., Inorg. Chem., 2005, vol. 44, p. 8762.
Patra, G.K. and Glodberg, I., Cryst. Growth Des., 2003, vol. 3, p. 321.
Chen, C.-L., Kang, B.-S., and Su, C.-Y., Aust. J. Chem., 2006, vol. 59, p. 3.
Dong, Y.-B., Layland, R.C., Smith, M.D., et al., Inorg. Chem., 1999, vol. 38, p. 3056.
Cui, Y., Ngo, H.L., and Lin, W., Chem. Commun., 2003, p. 1388.
Chen, C.-T. and Suslick, K.S., Coord. Chem. Rev., 1993, vol. 128, p. 293.
Leong, W.L. and Vittal, J.J., Chem. Rev., 2011, vol. 111, p. 688.
Song, Y., Yu, L., Gao, Y., et al., Inorg. Chem., 2017, vol. 56, p. 11603.
Miyasaka, H., Julve, M., Yamashita, M., and Clérac, R., Inorg. Chem., 2009, vol. 48, p. 3420.
Mahmoudi, G., Gurbanov, A.V., Rodríguez-Hermida, S., et al., Inorg. Chem., 2017, vol. 56, p. 9698.
Kennedy, A.R. and Waterson, F.R.N., Acta Crystallogr. Sect. C: Cryst. Struct. Commun., 2003, vol. 59, p. o613.
Calahorro, A.J., San Sebastian, E., Salinas-Castil-lo, A., et al., CrystEngComm, 2015, vol. 17, p. 3659.
Manna, B., Singh, S., and Ghosh, S.K., J. Chem. Sci., 2014, vol. 126, no. 5, p. 1417.
Croitor, L., Coropceanu, E.B., Siminel, A.V., et al., Cryst. Growth Des., 2011, vol. 11, p. 3536.
Croitor, L., Coropceanu, E.B., Masunov, A.E., et al., Cryst. Growth Des., 2014, vol. 14, p. 3935.
Croitor, L., Coropceanu, E.B., Duca, G., et al., Polyhedron, 2017, vol. 129, p. 9.
Sheldrick, G.M., Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, vol. 64, p. 112.
Paulin, S., Kelly, P., Williams, K.B., et al., Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, vol. 63, р. m420.
Wang, Q., Liang, B., Zhang, J.-Y., et al., Z. Anorg. Allg. Chem., 2007, vol. 633, p. 2463.
Mahmoudi, G., Morsali, A., Hunter, A.D., and Zeller, M., CrystEngComm, 2007, vol. 9, p. 704.
Metelitsa, A.V., Burlov, A.S., and Borodkina, I.G., et al., Russ. J. Coord. Chem., 2006, vol. 32, no. 12, p. 858. doi 10.1134/S1070328406120025
Author information
Authors and Affiliations
Corresponding authors
Additional information
Translated by E. Yablonskaya
Rights and permissions
About this article

Cite this article
Lozovan, V., Coropceanu, E.B., Bourosh, P.N. et al. Coordination Polymers of Zn and Cd Based on Two Isomeric Azine Ligands: Synthesis, Crystal Structures, and Luminescence Properties. Russ J Coord Chem 45, 11–21 (2019). https://doi.org/10.1134/S107032841901007X
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S107032841901007X