
Abstract
C-type lectins are a superfamily of Ca2+-dependent carbohydrate-recognizing proteins that often serve as pattern recognition receptors (PRRs), which bind with specific molecular patterns known as pathogen-associated molecular patterns (PAMPs) of certain microorganisms. A new C-type lectin specific for highly branched mannans (GYLman) was isolated from the hemolymph of the mollusk Glycymeris yessoensis. GYLman was an oligomer with a molecular mass of more than 250 kDa by SDS-PAGE and showed a single band of approximately 70 kDa in reducing conditions. Lectin hemagglutination activity was best inhibited by highly branched yeast mannan with α-1,6-linked mannopyranose backbone and α-1,2-linked mannopyranose side chains. The mannan structure was established using 1H and 13C NMR spectroscopy. GYLman activity was reversibly inhibited by EDTA, was maximal in a pH range of 7–9, and was completely abolished by heating at 90°C. Acting as a PRR, GYLman specifically interacted with various PAMPs, including Escherichia coli lipopolysaccharide (LPS) O111:B4, α-D-mannan, β-1,3-glucan, and peptidoglycan, and bound to both Gram-positive (Staphylococcus aureus and Bacillus subtilis) and Gram-negative (E. coli and Vibrio proteolyticus) bacteria and the yeast Candida albicans, suggesting a broad PAMP recognition spectrum.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Lectins are classical carbohydrate-binding proteins and interact with various glycoconjugates with high affinity [1]. By their structures, animal lectins are classified into 13 lectin families (www.imperial.ac.uk/research/animallectins/). Lectins of at least seven groups have been identified in mollusks, the set including C-type, P-type, F-type, and I-type lectins; galectins; ficolins; and chitinase-like lectins [2, 3]. C-type lectins are a superfamily of Ca2+-dependent carbohydrate-recognizing proteins, which usually have an oligomeric structure and often act as pattern recognition receptors (PRRs). These lectins play a crucial role in invertebrate innate immunity and perform the vital function of eliminating pathogens by recognizing conserved carbohydrate structures known as pathogen-associated molecular patterns (PAMPs), which are broadly expressed on the surfaces of microorganisms [4]. More than 30 C-type lectins have been identified and characterized in mollusks [5]. The lectins act as key factors in carbohydrate-mediated recognition of infectious agents in invertebrates. The lectins are involved in a variety of processes, including immobilization, phagocytosis, clearance, infectious agent encapsulation, direct antimicrobial activity, etc. [6].
Invertebrate lectins are more diverse in structure and functions as compared with vertebrate lectins according to the available data. Although the amino acid sequences have been established for many C-type lectins of invertebrates, their functions are still poorly understood.
Mannan-binding lectins (MBLs) have been studied most comprehensively among C-type lectins. MBLs act as secreted receptors for pathogenicity patterns and are responsible for biological recognition of the PAMPs that have mannose and fucose terminal residues [7]. The structures and functions of invertebrate MBLs are poorly understood as compared with vertebrate MBLs. MBLs play a role in the ancient defense system that forms a basis of nonspecific immunity in invertebrates and is a precursor of complex immunity of vertebrates. Studies of the invertebrate nonspecific immunity system and, in particular, MBLs are of importance for understanding how the functions of this component of the defense system arose and evolved in Metazoa.
The bivalve Glycymeris yessoensis is widespread in the Russian Far East and forms vast colonies in waters of southern Primorskii krai. A series of our preliminary experiments has shown that the G. yessoensis hemolymph contains lectin activity directed toward mannans, which are polysaccharides composed of mannose residues. In this work, we isolated a new lectin and demonstrated its function as a PRR.
RESULTS AND DISCUSSION
A primary screening of the G. yessoensis hemolymph and organ extracts for lectin activity was performed by a direct hemagglutination assay (HA) with trypsin-treated human erythrocytes of various blood groups and trypsin-treated rabbit erythrocytes. The results summarized in Table 1 gave grounds to use the HA with trypsin-treated rabbit blood erythrocytes to monitor the lectin isolation and purification.
Preliminary experiments with the G. yessoensis hemolymph showed that certain mannans, including commercial Saccharomyces cerevisiae mannan, most efficiently prevent agglutination in the HA. The result showed that MBLs were present in the hemolymph. On the other hand, a proper mannan was chosen as a ligand to obtain an affinity chromatography resin on the basis of the above results because affinity chromatography provides the most efficient means to isolate a lectin [8]. To obtain the ligand in sufficient amounts, mannan was isolated from the S. cerevisiae culture medium and characterized.
The monosaccharide composition was studied by gas-liquid chromatography of acetylated polyols and acetylated 2-(S)-octylglycosides obtained after complete acid hydrolysis. D-Mannose (D-Man) was identified as the only monosaccharide residue, and the polysaccharide was consequently identified as D‑mannan.
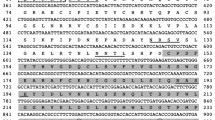
Signals of various intensities were observed in 1H and 13C NMR spectra (Fig. 1), suggesting an irregular structure for D-mannan. The 1H NMR spectrum included signals of anomeric protons in a region of 4.91–5.29 ppm and signals of monosaccharide ring protons in a region of 3.65–4.22 ppm.

(a) 1H and (b) 13C NMR spectra of yeast S. cerevisiae α-D-mannan (this work).
The 13C NMR spectrum included variable-intensity signals of anomeric carbons in a region of 99.6–103.3 ppm, two signals of hydroxymethyl groups at 62.4 and 67.0 ppm (C-6 of D-Man residues on evidence of a DEPT-135 experiment), and signals of carbon atoms of monosaccharide rings in a region of 67.8–79.9 ppm. Signals from non-anomeric carbon atoms were absent in the region of a field weaker than that corresponding to 82.0 ppm, indicating that all monosaccharide residues were in a pyranoside form. A Gated Decoupling experiment showed that α-glycoside bonds connected all monosaccharide residues.
A complete assignment of the signals detected in the 1H and 13C NMR spectra of the polysaccharide was achieved using data from two-dimensional homonuclear 1H,1H-COSY, 1H,1H-TOCSY, and heteronuclear 1H,13C-HSQC, 1H, 13C-HSQC-TOCSY, and 1H,13C-HMBC experiments and published data on the mannan structure [9]. As a result, we identified three spin systems corresponding to the terminal D‑Man residues, two spin-spin systems corresponding to 2,6-disubstituted D-Man residues, and one spin-spin system for each of the 2-, 3-, and 6-substituted D‑Man residues. Chemical shifts of atoms are summarized in Table 2 for each spin–spin system.
The sequence and substitution order of monosaccharide residues in the polysaccharide were determined in a 1H, 13C-HMBC experiment by the presence of cross peaks between anomeric protons and transglycoside carbons. Correlations between H-1/C-6 at δ 4.91/67.0, 5.09/67.0, and 5.12/67.0 ppm were observed for a residue of 6-substituted D-Man and two residues of 2,6-disubstituted D-Man, indicating that the polysaccharide backbone is a polymer of →6)-α-Manp-(1→ residues.
Additional correlations in the 1H, 13C-HMBC spectrum were observed between H-1 of terminal D-Man and С-2 2,6-disubstituted D-Man at δ 5.05/79.9 ppm, between H-1 of terminal D-Man and С-2 2-substituted D-Man at δ 5.06/79.6 ppm, between H-1 of terminal D-Man and С-3 3-substituted Man at δ 5.15/79.2 ppm, between H-1 2-substituted D-Man and С-2 2,6-disubstituted D-Man at δ 5.29/79.9 ppm, and between H-1 3-substituted D-Man and С-2 2-substituted Man at δ 5.05/79.9 ppm. The data indicate that side substituents of the backbone include single D-Man residues, α-D-Manp-(1 → 2)-α-D-Manp-(1 → disaccharides, and α-D-Manp-(1 → 3)-α-D-Manp-(1 → 2)-α-D-Manp-(1→ trisaccharides. A possible schematic structure of the mannan is shown in Fig. 2.

Schematic structure of yeast S. cerevisiae α-D-mannan (this work).
The 1H and 13C NMR data (Table 2, Fig. 1) showed that the mannan isolated in this work is a highly branched homopolysaccharide that consists of α-1,6- and α-1,2-linked D-Man residues (Fig. 2) and is structurally similar to the well-known yeast S. cerevisiae mannan.
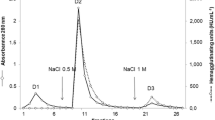
A fraction with Ca2+-dependent MBL activity was isolated from the hemolymph of the bivalve G. yessoensis via anion exchange chromatography on DEAE-Toyopearl 650M, being eluted with 1 M NaCl (Fig. 3). The fraction was used for further lectin purification by affinity chromatography.

Anion exchange chromatography of GYLman on a DEAE-Toyopearl 650М column. Adsorbed proteins were eluted with a stepwise gradient of NaCl concentration (0.15, 0.25, 0.5, 0.75, and 1 M) in TB-Ca2+. The elution profile (a solid line) was obtained by measuring absorption at 280 nm for each fraction. The hemagglutination titer (a dashed line) was obtained as the inverse maximal dilution that caused hemagglutination.
To obtain affinity sorbent, α-D-mannan (this work) was conjugated with divinyl sulfone-activated Sepharose CL-4B according to a published protocol [10]. The Ca2+-dependent lectin from the fraction obtained via anion exchange chromatography was totally bound with mannan-Sepharose. A buffer containing 20 mM EDTA was used for elution. The lectin, which was designated GYLman, was thus purified to homogeneity according to SDS-PAGE. GYLman was a high-molecular-weight oligomer of more than 250 kDa and consisted of several monomeric subunits, which were approximately 70 kDa in molecular weight and were linked via disulfide bonds (Fig. 4). The exact molecular weight of the subunits was found to be 69 189.6 Da by MALDI-TOF/TOF spectrometry.

SDS-PAGE to check the homogeneity of the lectin isolated. Gel was stained with Coomassie Brilliant Blue R-250. GYLman was examined (1) in nonreducing conditions and (2) in reducing conditions (in the presence of DTT); (lane 3) molecular weight markers.
Using HA, we found that GYLman shows maximal activity in a pH range of 7–9, is totally inactivated upon heating to 90°C, and is Ca2+ dependent as characteristic of many lectins of marine invertebrates [11, 12].
To quantitatively study the amino acid composition, GYLman was hydrolyzed (Table 3). Glycine, aspartic acid, and glutamic acid were identified as the most prevalent. The GYLman molecule is hydrophilic and has a low negative charge. Aromatic amino acid residues account for only 3.6% of the protein, the circumstance possibly resulting in low optical density at 280 nm.
Fine carbohydrate specificity is an important characteristic of a lectin. To study the specificity for GYLman, we obtained the GYLman-HRP conjugate and developed the enzyme-linked lectin assay (ELLA).. The specificity was studied with a set of monosaccharides, oligosaccharides, and polysaccharides; the PSM glycoprotein; and lipopolysaccharide (LPS) of E. coli O111:B4.
The following substances caused no inhibition when used at 10 mg/mL: D-galactose, N-acetyl-D-galactosamine, D-glucose, N-acetyl-D-glucosamine, L-fucose, lactose, raffinose, D-talose, sucrose, melibiose, N-glycolylneuraminic acid, N-acetylneuraminic acid, D-mannose, N-acetyl-D-mannosamine, tetramannoside α-Man-(1 → 2)-[Man-(1 → 2)]2-Man, hexamannoside α-Man-(1 → 2)-[Man-(1 → 2)]4-Man, heptamannoside α-Man-(1 → 2)-[Man-(1 → 2)]5-Man, and fucoidan.
As is seen from Table 4, the majority of monosaccharides and oligosaccharides, including mannose, did not inhibit GYLman activity. The lectin showed the highest activity toward highly branched yeast mannans with an α-1,6-linked mannopyranosyl backbone and short α-1,2-linked mannopyranosyl side chains, as is characteristic of MBLs of marine invertebrates [10, 13]. It should be noted that lectin inhibition increased in the following order: disaccharide < trisaccharide < pentasaccharide. Man9 and Man5 have been found in the carbohydrate moiety of gp120 expressed on the HIV surface. These mannosides play a substantial role in HIV binding to cell surface receptors, such as CD4. Certain mannose-specific lectins inhibit HIV binding with CD4-positive cells by interacting with gp120 [14]. A similar biological effect is possible to assume for GYLman.
Marine organisms have recently attracted increasing attention as potential sources of substances possessing a broad range of biological activities, including antivirus, antibacterial, and fungicidal activities [15–17]. Interacting with carbohydrates exposed on target cells, lectins may recognize the surface carbohydrate determinants of various pathogens. The process plays an important role in protective reactions of innate immunity in various animals and is especially significant for invertebrates, which lack an acquired immune system. Recognition of PAMPs, which are groups of molecules that are characteristic of pathogens and lacking in the host organism, is a main recognition process of innate immunity.
To check whether GYLman acts as a PRR, we studied its binding with several PAMPs, including E. coli O111:B4 LPS, S. aureus peptidoglycan, S. cerevisiae α-D-mannan, Euglena gracilis β-1,3-glucan, which are major glycoconjugates of the bacterial cell wall. LPS acts as a main antigen and virulence factor in Gram-negative bacteria. Commercial E. coli O111:B4 LPS was used as a model because core oligosaccharides are mostly targeted by MBLs and the core is relatively conserved [18].
As was shown by ELLA, GYLman bound with all PAMPs in a concentration-dependent manner, the binding decreasing in the following order: α-D-mannan > peptidoglycan > LPS > β-1,3-glucan (Fig. 5).

GYLman binding with PAMPs as assessed by ELLA. Experiments were performed in triplicate; the results are presented as mean ± SD.
Binding with bacteria, rather than their cell membrane components, was additionally assessed by ELLA. GYLman was found to specifically bind to both Gram-positive (S. aureus and B. subtilis) and Gram-negative (E. coli and V. proteolyticus) bacteria and the yeast C. albicans, suggesting a broad PAMP recognition spectrum (Fig. 6).

GYLman binding with microorganisms as assessed by ELLA. BSA (50 μg/mL) was used as a negative control; PSM (50 μg/mL), as a positive control of binding. Experiments were performed in triplicate; the results are presented as mean ± SD.
Similar activities have been reported for lectins isolated from the oyster Crassostrea gigas [19] and the scallops Chlamys farreri [20] and Argopecten irradians [21]. However, the highest binding activity of GYLman was observed with E coli, rather than with C. albicans, although mannan most efficiently interacted with GYLman among all PAMPs examined and mannoproteins are known to be a main component of the cell wall in yeasts. More intricate mechanisms are thought to sustain the recognition of pathogens because the glycocalyx can be structurally complex in microbial cells.
Experiments with binding inhibition showed that a lectin pathway mediates the lectin interaction with microorganisms (Table 5).
To verify the results, agglutination of bacterial and yeast cells was studied in the presence of GYLman. The lectin agglutinated the microorganisms, tightly binding them to produce aggregates and partly preventing their further growth (Fig. 7 ).
Thus, GYLman was shown to act as a PRR and to specifically bind with various microorganisms, potentially playing an important role in immune recognition. Lectins of the kind are thought to be suitable for designing new antimicrobial drugs [22]. Systematic comparative studies of various PRRs are necessary in order to better understand functional differentiation of lectins. Continuous exposure of pathogenic microorganisms to antibiotics naturally leads to the formation of resistant strains, and intense and promiscuous use of these drugs may accelerate the process. Resistant bacterial strains are insensitive to the majority of standard treatments, the circumstance increasing risk for a spreading of certain diseases [23]. New antibacterial agents are consequently designed at an increasing rate, and natural objects provide a potential source of new compounds active toward various microorganisms [24].
EXPERIMENTAL
Materials. We used Tween-20, Tris, sodium dodecyl sulfate (SDS), sodium dihydrophosphate (Helicon, United States), trypsin 1 : 250 (PanEko, Russia), sodium chloride, sodium acetate, sodium carbonate, sulfuric acid, hydrochloric acid, acetic acid, ethanol, methanol, dimethyl sulfoxide (DMSO), hydrogen peroxide, NaBH4 (Khimmed, Russia), BSA, sodium hydrocarbonate (ICN Biomedicals, United States), (Acros Organics, Spain), PSM, E. coli O111:B4 LPS, S. aureus peptidoglycan, S. cerevisiae mannan, Euglena gracilis β-1,3-glucan, sodium periodate, Bradford reagent, horseradish peroxidase, 3,3',5,5'-tetramethylbenzidine (TMB) (Sigma, United States), an electrophoretic protein standard kit (Thermo Fisher Scientific, Sweden), Coomassie R‑250, tetramethylbenzidine (TMB) (Aldrich, United States), monosaccharides (Merk, Darmstadt, Germany), and glycerol (Serva, Germany).
α-D-Mannan from P. atlantica (strain IAM 14165), α-D-mannan from C. albicans (strain KMM 455), and a set of mannosides were kindly provided by Prof. Wei Li (Dalian Fisheries University, China).
Bacterial strains S. aureus КММ 434, B. subtilis ATCC 6633, E. coli VKPM V 7335, and V. proteolyticus CCUG 20302T and yeast strain C. albicans KMM 455 were kindly provided by O.I. Nedashkovskaya (Laboratory of Microbiology, Elyakov Pacific Institute of Bioorganic Chemistry).
Human red cell mass was obtained from a blood transfusion station (Vladivostok); rabbit erythrocytes were collected in a breeding facility of the Elyakov Pacific Institute of Bioorganic Chemistry.
Hemolymph isolation. Glycymeris yessoensis mollusks were collected in the Peter the Great Bay (Troitsa Bight, Sea of Japan). Hemolymph samples were drawn from adductor muscles with a sterile syringe, pooled, and centrifuged at 2000 rpm for 30 min to precipitate hemocytes. The cell-free supernatant was dialyzed against the TB-Ca2+ buffer (0.01 M Tris-HCl, 10 mM CaCl2, pH 8), centrifuged at 14 000 rpm for 30 min, and used for lectin isolation. Sodium azide and PMSF were added to prevent bacterial growth and to inhibit protease activity, respectively.
Lectin activity assays. Direct hemaggnutination assay (HAs) were carried out using trypsin-treated human erythrocytes of blood groups A, B, AB, and 0 and trypsin-treated rabbit blood erythrocytes. A series of twofold dilutions was prepared with 25 μL of an initial lectin solution and an equal volume of the TBS-Ca2 + buffer (0.01 M Tris-HCl, 0.15 M NaCl, 10 mM CaCl2, pH 8) in wells of a U-shaped-bottom non-binding microplate (Nunc, Netherlands). Each well was supplemented with 25 μL of 2% erythrocyte suspension; erythrocytes were preliminarily washed with the same buffer. The plate was incubated at room temperature for 30 min, and the hemagglutination titer was measured as the maximal dilution of the lectin preparation at which hemagglutination was visible. HA inhibition was studied in the TBS-Ca2+ buffer. An optimal lectin concentration was preliminarily established in HA as the dilution of the initial lectin preparation in the second to last well where agglutination was distinct. A lectin solution with the given concentration was used to obtain twofold dilutions of monosaccharides, oligosaccharides, and polysaccharides. Samples were incubated at room temperature for 1 h, each well was supplemented with 25 μL of 2% erythrocyte suspension, and the minimal concentration that abolished hemagglutination was established for the respective inhibitor.
Yeast mannan isolation and properties. To isolate glycopolymers, the culture medium of a yeast S. cerevisiae X2180 culture was autoclaved with 50 mM Na-citrate (pH 7.5) for 1 h, ethanol precipitation was carried out. The precipitate was dissolved in water, and charged substances were removed via cetavlon precipitation. Mannan was precipitated with 1% borate, dissolved in 2% AcOH, and precipitated with ethanol. The resulting material was digested with proteinase K (50°C, 3 h), dialyzed, purified by anion exchange chromatography on a DEAE TSK 650M column with elution using a 0–1 M NaCl concentration gradient, and dialyzed [9].
Anion exchange chromatography was performed using a TOYOPEARL DEAE-650M column (40 × 130 mm) equilibrated with the TB-Ca2+ buffer (a start buffer). Proteins bound to the sorbent were eluted using a stepwise NaCl concentration gradient in the start buffer (0.15, 0.25, 0.5, 0.75, and 1 M). The fractions were tested via HA and HA inhibition assay. Elution of a mannan-binding lectin was observed at 1 M NaCl. The respective fraction was dialyzed against TBS-Ca2+ and used for affinity chromatography.
Affinity chromatography. To synthesize the affinity sorbent, extracellular mannan isolated from the culture medium of a yeast S. cerevisiae X2180 culture was immobilized on Sepharose CL-4B (Рharmacia, Sweden). Sepharose (40 mL) was washed with water and then with 0.5 M Na2CO3 (pH 11) and dried in air. Gel was suspended in 80 mL of 0.5 M Na2CO3 (pH 11), combined with 4 mL of divinyl sulfone, and incubated with continuous agitation at room temperature for 70 min. Activated Sepharose was washed with water and vacuum dried until moist. Then, activated Sepharose was combined with 80 mL of 0.5 M Na2CO3 (pH 10) containing 400 mg of mannan, and the mixture was incubated with continuous agitation at 4°C for 48 h. The sorbent was washed with water, suspended in 0.5 M Na2CO3 (pH 8.5) supplemented with 1.6 mL of β-mercaptoethanol, incubated with continuous agitation at room temperature for 3 h, and washed consecutively with water and TBS-Ca2+ [10].
Batch affinity chromatography was used. The sorbent was combined with a lectin solution in a glass at a 1 : 4 ratio. The mixture was incubated for 1 h, and a complete binding of the lectin was checked by the HA. Activity was not detected in the nonbound fraction after 1-h incubation; i.e., the mannan-specific lectin was totally bound.
The sorbent was washed with TBS- Ca2+ until zero absorption at 280 nm. The Ca2+-dependent lectin was eluted with TBS containing 20 mM EDTA (pH 8), concentrated, dialyzed against distilled water, and lyophilized.
SDS-PAGE was carried out in 15% gel according to Laemmli [25], in reducing (in the presence of dithiothreitol (DTT)) or nonreducing conditions. Proteins were visualized by Coomassie Brilliant Blue R-250 staining. Protein molecular weights were estimated against a set of marker proteins with known molecular weights.
Ca2+ dependence of lectin activity. A lectin solution (0.5 mL) was dialyzed consecutively against 0.01 M TBS in the presence of 0.02 M EDTA at 4°C overnight and 0.01 M TBS for 1 day. Serial twofold dilutions were prepared in a plate using 25-μL samples and 0.01 M TBS supplemented with 0, 1, 5, 10, of 20 mM CaCl2. The plate was incubated at room temperature for 1 h, and lectin activity was estimated by HA.
Thermal stability assay of GYLman. Aliquots (0.05 mL) of a GYLman preparation were incubated in at temperatures of 4, 20, 37, 50, 60, 70, 80, 90, and 95°C for 30 min, chilled, and tested for lectin activity in HA.
pH dependence of GYLman activity. Aliquots (0.05 mL) of a GYLman preparation were dialyzed against 50 mL of a buffer without agitation at room temperature for 1 h. The following buffers were used: 0.05 M citrate (pH 4 and 5), 0.05 M PBS (pH 7.5 and 8), and 0.05 M TBS (pH 9 and 10). Then the aliquots were dialyzed against 0.01 M TBS (pH 8) at 4°C for 24 h. Lectin activity was assayed via HA.
Amino acid composition of GYLman. GYLman (100 μg) was combined with 0.5 mL of 6 N HCl and hydrolyzed in an argon atmosphere at 100°C for 24 h. After hydrolysis, HCl was evaporated, and the composition was established on a Hitachi 835 automated amino acid analyzer (Japan).
Lectin conjugation with an enzymatic label. GYLman was conjugated with horseradish peroxidase (HRP) by periodate oxidation according to Nakane [26], yielding GYLman-HRP. HRP (2 mg; Sigma, RZ: 3,2) was dissolved in 0.5 mL of distilled water and combined with 0.1 mL of 0.1 M NaIO4. The mixture was incubated in the dark at room temperature for 30 min. Oxidized HRP was dialyzed against 1 L of 0.001 acetate buffer (pH 4.4). The HRP preparation was combined with 2 mg of the lectin in 0.5 mL of 0.05 M carbonate buffer (pH 9.5), pH was adjusted to 9.5. The mixture was incubated at room temperature for 2 h, combined with 50 μL of 4 mg/mL NaBH4 (the solution was prepared immediately before use), and incubated at room temperature for 2 h. To store the conjugate at –18°C, the preparation was combined with glycerol at a 1 : 1 ratio.
Fine carbohydrate specificity of the lectin. To study the lectin specificity, we developed an enzyme-linked lectin assay (ELLA) and selected the optimal assay conditions. To immobilize porcine stomach mucin (PSM) on a solid surface, 50 μg/mL PSM in 0.01 M TBS-Ca2+ (pH 8) was added to wells of a 96-well plate (Nunc, Denmark) at 100 μL per well. The plate was incubated at 4°C overnight and washed with a washing buffer (0.01 M TBS-Ca2 +, pH 8, 0.05% Tween-20). To prevent nonspecific binding, the same buffer was added at 300 μL per well and the plate was incubated with continuous agitation at 37°C for 1 h and washed three times with the washing buffer. Potential inhibitors were dissolved in 0.01 M TBS- Ca2+ (pH 8) to necessary concentrations. Serial twofold dilutions were prepared in triplicate (50 μL per well), and 4 μg/mL GYLman-HRP (a constant concentration) in the washing buffer was simultaneously added at 50 μL per well. The plate was incubated with continuous agitation at 37°C for 1 h and washed as above. The substrate TMB was added at 100 μL per well, the plate was incubated in the dark for 5 min, and the reaction was terminated by adding 50 μL of 5% H2SO4. The optical density at 450 nm was measured using a µQuant spectrophotometer (BioTek Instruments, United States).
Lectin interactions with various PAMPs. The following substances were immobilized on 96-well plates (Nunc, Denmark): E. coli O111:B4 LPS, S. aureus peptidoglycan, S. cerevisiae mannan, and E. gracilis β‑1,3-glucan. The substances were used at 50 μg/mL in 0.01 M TBS-Ca2+ (pH 8) and were added in triplicate at 100 μL per well. The plates were incubated at 4°C overnight and at 60°C for 2 h and washed three times with the washing buffer. To prevent nonspecific binding, the same buffer was added at 300 μL per well and the plates were incubated with continuous agitation at 37°C for 1 h and washed three times with the washing buffer. Serial twofold dilutions of GYLman-HRP (initial concentration 10 μg/mL) in the washing buffer were added to wells at 100 μL per well. The plates were incubated with continuous agitation at 37°C for 1 h and washed as above. Enzymatic activity was assayed and the optical density measured as above.
Lectin interactions with microorganisms. Gram-positive (S. aureus and B. subtilis) and Gram-negative (E. coli and V. proteolyticus) bacteria and the yeast C. albicans were immobilized in triplicate in wells of 96-well plates (Nunc, Denmark) by adding 100 μL of a microbial suspension (OD600 0.7–1.00) to each well and incubating the plates at 4°C overnight. The plates were heated at 80°C for 45 min to fix bacteria. The well contents were discarded carefully. To prevent nonspecific binding, the washing buffer was added at 300 μL per well and the plates were incubated at 37°C for 1 h and washed three times with the washing buffer. Serial twofold dilutions of GYLman-HRP (initial concentration 10 μg/mL) in the washing buffer were added to wells at 100 μL per well. The plates were incubated at 37°C for 1 h and washed as above. Enzymatic activity was assayed and the optical density measured as above.
Inhibition of lectin binding with bacteria. Bacteria were immobilized on a 96-well plate (Nunc, Denmark) as above. Inhibitors (10 mg/mL D-galactose or D-glucose or 1 mg/mL S. cerevisiae α-D-mannan (Sigma)) were prepared in 0.01 M TBS- Ca2+ (pH 8). Serial twofold dilutions were added at 50 μL per well. All wells were simultaneously supplemented with 50 μL of 4 μg/mL GYLman-HRP in the washing buffer. The plates were incubated with continuous agitation at 37°C for 1 h and washed as above. Enzymatic activity was assayed and the optical density measured as above.
Bacterial agglutination was carried out according to a published protocol [27]. A cell suspension (25 μL, OD600 0.7–1.0) in sterile TBS-Ca2 + (pH 8) was combined with 25 μL of a lectin solution in the same buffer to a final concentration of 0.2 mg/mL. The plate was incubated at 20°C for 1 h. Cells suspended in sterile TBS-Ca2+ (pH 8) were used as a control. Agglutination results were assessed under an AxioCam MRc light microscope (Zeiss); magnification ×400 or ×1000.
Statistical analyses were carried out using the Microsoft Excel spreadsheets.

Agglutination of microorganisms by the lectin. Microorganisms were incubated with (a, c, e, g, i) TBS-Ca2+ or (b, d, f, h, j) GYLman.
REFERENCES
Sharon, N., J. Biol. Chem., 2007, vol. 282, pp. 2753–2764. https://doi.org/10.1074/JBC.X600004200
Zelensky, A.N. and Gready, J.E., FEBS J., 2005, vol. 272, pp. 6179–6217. https://doi.org/10.1111/j.1742-4658.2005.05031.x
Wang, L., Wang, L., Huang, M., and Zhang, H., ISJ, 2011, vol. 8, pp. 241–246.
Cambi, A., Koopman, M., and Figdor, C.G., Cell. Microbiol., 2005, vol. 7, pp. 481–488. https://doi.org/10.1111/j.1462-5822.2005.00506.x
Wang, W., Song, X., Wang, L., and Song, L., Int. J. Mol. Sci., 2018, vol. 19, p. 721. https://doi.org/10.3390/ijms19030721
Gerdol., M., Gomez-Chiarri, M., Castillo, M.G., Figueras, A., Fiorito, G., Moreira, R., Novoa, B., Pallavicini, A., Ponte, G., Roumbedakis, K., Venier, P., and Vasta, G.R., in Advances in Comparative Immunology, Cooper, E.L., Ed., Springer Int. Publ., AG, part of Springer Nature, 2018, pp. 225–341. https://doi.org/10.1007/978-3-319-76768-0_11
Medzhitov, R., Nature, 2007, vol. 449, pp. 819–826. https://doi.org/10.1038/nature06246
Nascimento, K.S., Cunha, A.I., Nascimento, K.S., Cavada, B.S., Azevedo, A.M., and Aires-Barros, M.R., J. Mol. Recognit., 2012, vol. 25, pp. 527–541. https://doi.org/10.1002/jmr.2200
Vinogradov, E., Petersen, B., and Bock, K., Carbohydr. Res., 1998, vol. 307, pp. 177–183.
Bulgakov, A.A., Eliseikina, M.G., Petrova, I.Yu., Nazarenko, E.L., Kovalchuk, S.N., Kozhemyako, V.B., and Rasskazov, V.A., Glycobiology, 2007, vol. 17, pp. 1284–1298. https://doi.org/10.1093/glycob/cwm093
Garcia-Maldonado, E., Cano-Sanchez, P., and Hernandez-Santoyo, A., Fish Shellfish Immunol., 2017, vol. 66, pp. 564–574.
Singh, R.S., Walia, A.K., and Kennedy, J.F., Int. J. Biol. Macromol., 2019, vol. 128, pp. 124–131. https://doi.org/10.1016/j.ijbiomac.2019.01.103
Bulgakov, A.A., Eliseikina, M.G., Kovalchuk, S.N., Petrova, I.Yu., Likhatskaya, G.N., Shamshurina, E.V., and Rasskazov, V.A., Mar. Biotechnol., 2013, vol. 15, pp. 73–86. https://doi.org/10.1007/s10126-012-9460-5
Leal, R.B., Pinto-Junior, V.R., Osterne, V.J.S., Wolin, I.A.V., Nascimento, A.P.M., Neco, A.H.B., Araripe, D.A., Welter, P.G., Neto, C.C., Correia, J.L.A., Rocha, C.R.C., Nascimento, K.S., and Cavada, B.S., Int. J. Biol. Macromol., 2018, vol. 114, pp. 64–76.
Marine Proteins and Peptides: Biological Activities and Applications, Se-Kwon Kim, Ed., UK: Wiley, 2013.
Luk’yanov, P.A., Chernikov, O.V., Kobelev, S.S., Chikalovets, I.V., Molchanova, V.I., and Li, W., Russ. J. Bioorg. Chem., 2007, vol. 33, pp. 161–169. https://doi.org/10.1134/S1068162007010190
Chikalovets, I.V., Mizgina, T.O., Molchanov, V.I., Ovcharenko, Yu.S., and Chernikov, O.V., Chem. Nat. Compd., 2017, vol. 53, pp. 717–721. https://doi.org/10.1007/s10600-017-2098-9
Man-Kupisinska, A., Swierzko, A.S., Maciejewska, A., Hoc, M., Rozalski, A., Siwinska, M., Lugowski, C., Cedzynski, M., and Lukasiewicz, J., Front. Immunol., 2018, vol. 9, art. 1498. https://doi.org/10.3389/fimmu.2018.01498
Li, H., Zhang, H., Jiang, S., Wang, W., Xin, L., Wang, H., Wang, L., and Song, L., Fish Shellfish Immunol., 2015, vol. 44, pp. 566–575. https://doi.org/10.1016/j.fsi.2015.03.011
Yang, J.L., Wang, L.L., Zhang, H.A., Qiu, L.M., Wang, H., and Song, L.S., PLoS One, 2011, vol. 6, e17089. https://doi.org/10.1016/j.fsi.2015.03.011
Huang, M.M., Song, X.Y., Zhao, J.M., Mu, C.K., Wang, L.L., Zhang, H., and Song, L., Gene, 2013, vol. 531, pp. 31–38. https://doi.org/10.1016/j.gene.2013.08.042
Garderes, J., Bourguet-Kondracki, M.-L., Hamer, B., Batel, R., Schroder, H., and Muller, W., Mar. Drugs, 2015, vol. 13, pp. 5059–5101. https://doi.org/10.3390/md13085059
Critically Important Antimicrobials for Human Medicine, 5th rev., Geneva: World Health Organization, 2017, Licence CC BY-NC-SA 3.0 IGO.
Smith, V.J., Desbois, A.P., and Dyrynda, E.A., Mar. Drugs, 2010, vol. 8, pp. 1213–1262. https://doi.org/10.3390/md8041213
Laemmli, U., Nature, 1970, vol. 227, pp. 680–685.
Egorov, A.M., Osipov, A.P., Dzantiev, B.B., and Gavrilova, E.M., Teoriya i praktika immunofermentnogo analiza (Theory and Practice of Enzyme Immunoassay), Vysshaya Shkola, 1991, pp. 182–183.
Ming, L., Chaozheng, L., Chunxia, M., and Haoyang, L., Dev. Comp. Immunol., 2014, vol. 46, pp. 231–240. https://doi.org/10.1016/j.dci.2014.04.014
ACKNOWLEDGMENTS
We are grateful to Prof. Wei Li (Dalian Fishery University, China) for P. atlantica (strain IAM 14165) α-D-mannan, C. albicans, and a set of mannosides; O.I. Nedashkovskaya (Laboratory of Microbiology, Elyakov Pacific Institute of Bioorganic Chemistry) for bacterial and yeast strains; and P.S. Dmitrenok (Laboratory of Instrumental and Radioisotope Analytical Methods, Elyakov Pacific Institute of Bioorganic Chemistry) for help with spectrum recording and interpretation.
Funding
This work was supported by the Russian Foundation for Basic Research (project no. 19-34-90112).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interests
The authors declare that they have no conflict of interest.
This work does not contain any studies involving animals or human subjects performed by any of the authors.
Additional information
Translated by T. Tkacheva
Rights and permissions
About this article

Cite this article
Mizgina, T.O., Chikalovets, I.V., Molchanova, V.I. et al. Lectin of the Bivalve Glycymeris yessoensis as a Pattern Recognition Receptor. Russ J Bioorg Chem 46, 1187–1197 (2020). https://doi.org/10.1134/S1068162020060205
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1068162020060205